Modification of Chitin with Kraft Lignin and Development of New Biosorbents for Removal of Cadmium(II) and Nickel(II) Ions






Abstract
:1. Introduction
2. Results and Discussion
2.1. Physicochemical Evaluation
2.1.1. Morphological and Microstructure Characteristics

2.1.2. FT-IR Spectroscopy
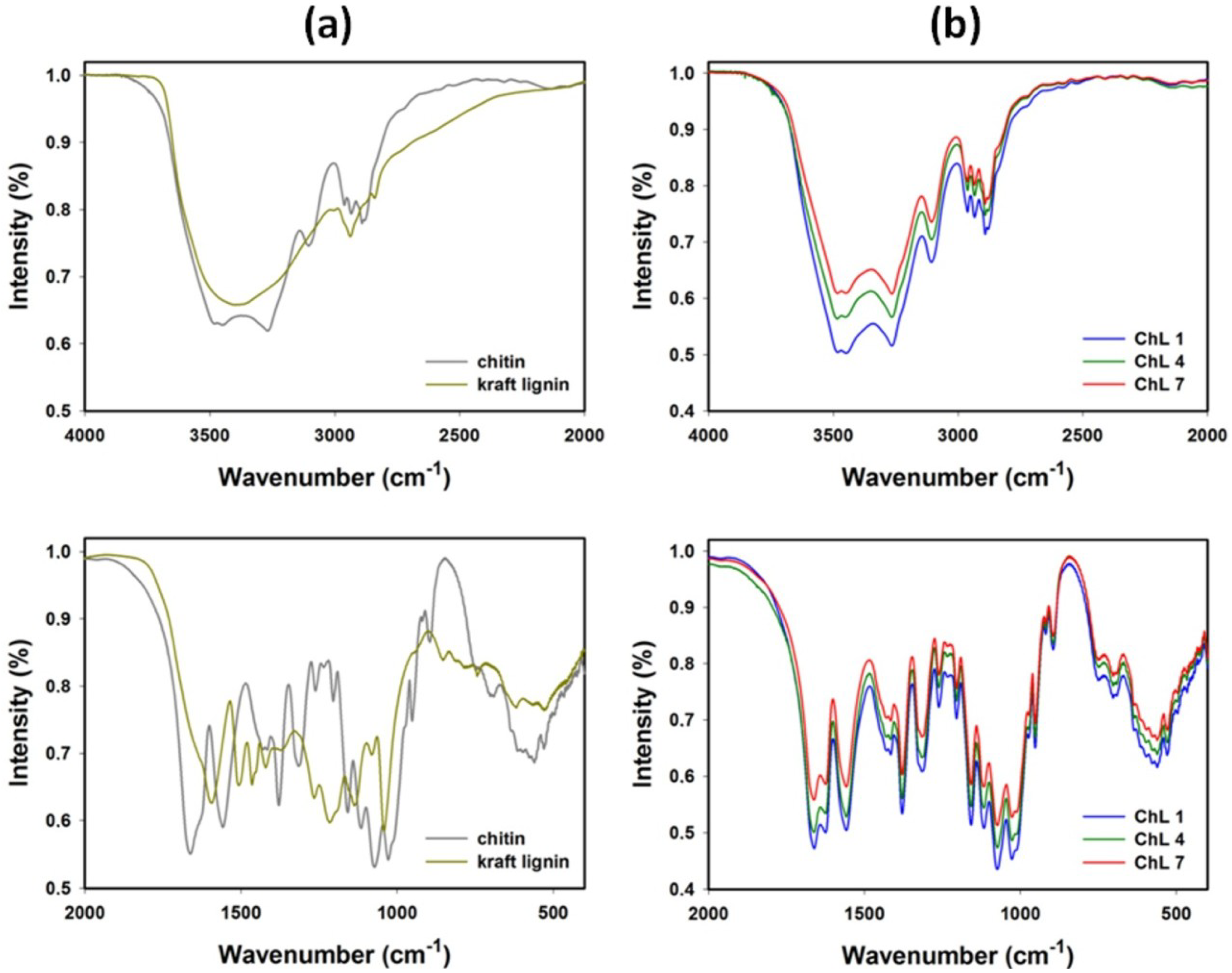

{kind=link}
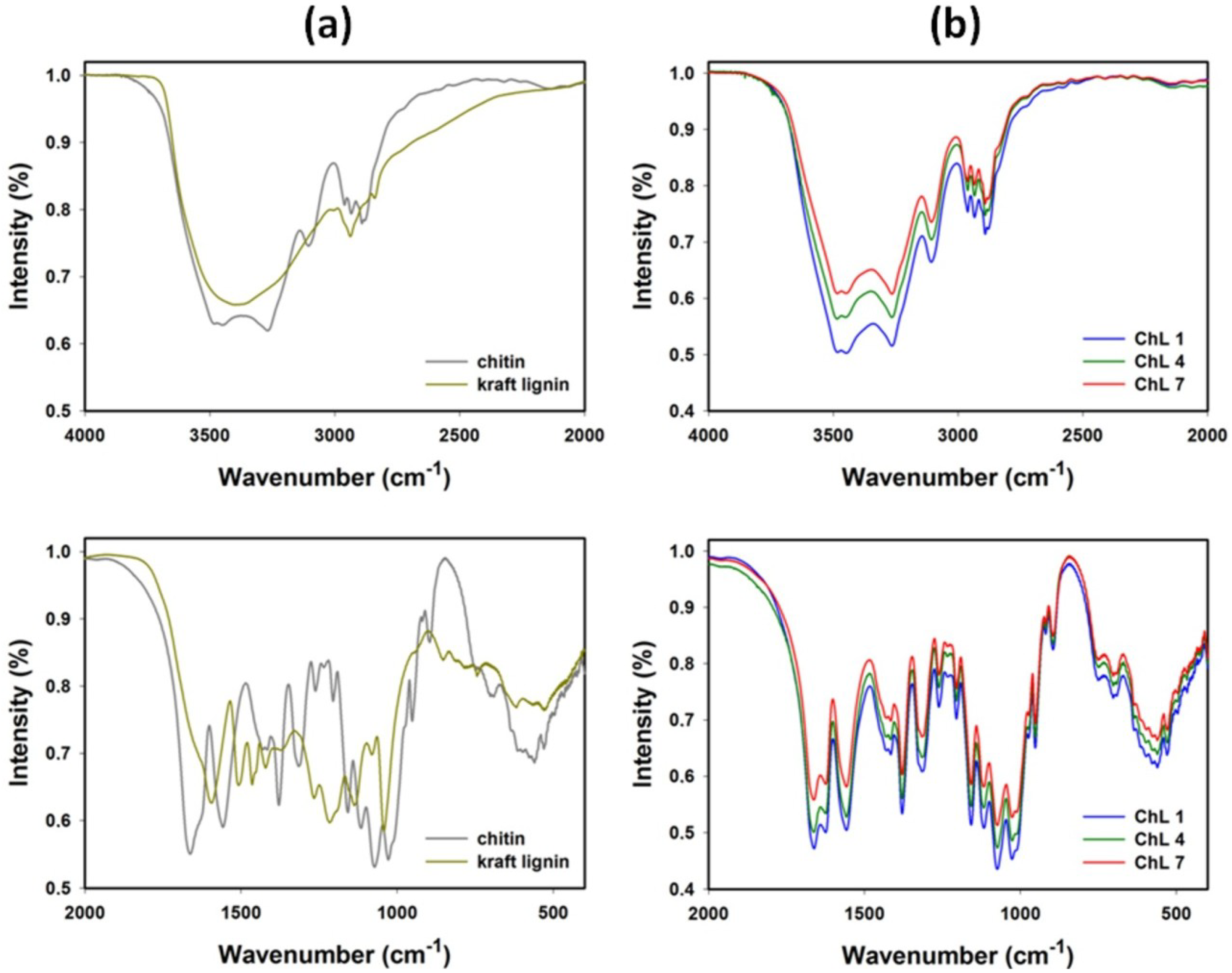{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chitin | Kraft lignin | Chitin/lignin material (ChL 1) | Vibrational assignment |
|---|---|---|---|
| 3445 | 3387 | 3483 | O–H stretching |
| 3285 | - | 3264 | N–H stretching |
| 3107 | - | 3108 | N–H stretching |
| 2963 | - | 2966 | CHx stretching |
| 2932 | 2935 | 2935 | CHx stretching |
| 2875 | - | 2877 | CHx stretching |
| 1663 | - | 1659 | C=O (amide I) stretching |
| 1630 | 1630 | 1624 | C=O stretching |
| - | 1595 | - | C–C (aromatic skeleton) stretching |
| 1558 | - | 1558 | C–N (amide II) bending |
| - | 1505 | - | C–C (aromatic skeleton) stretching |
| - | 1463 | - | C–H, CH3 + CH2 bending |
| 1430 | - | 1436 | CH2 bending |
| - | 1421 | 1415 | C–C (aromatic skeleton) stretching |
| 1378 | - | 1381 | C–H bending |
| - | 1370 | - | O–H (phenolic OH) bending |
| - | 1326 | 1328 | C–O (syringyl unit) streching |
| 1316 | - | - | C–N (amide III) stretching |
| - | 1266 | - | C–O (guaiacyl unit) streching |
| 1261 | - | 1259 | N–H (amide III) bending |
| - | 1216 | - | C–OH (phenolic OH) stretching |
| 1158 | - | 1156 | C–O–C (ring), C–O stretching |
| - | 1136 | - | Aromatic C–H (guaiacyl unit), stretching |
| 1116 | - | 1116 | C–O–C (ring), C–O stretching |
| 1073 | - | 1073 | C–O–C (ring), C–O stretching |
| - | 1040 | - | C–OH + C–O–C (aliphatic OH + ether) stretching |
| 1028 | - | 1028 | C–O–C (ring), C–O stretching |
| 951 | - | 951 | CH3 bending |
| 896 | - | 896 | β-1,4-glycosidic bond |
| - | 863 | 863 | Aromatic C–H(guaiacyl unit), bending |
| - | 745 | 745 | Aromatic C–H(guaiacyl unit), bending |
| 635 | - | 635 | N–H bending |
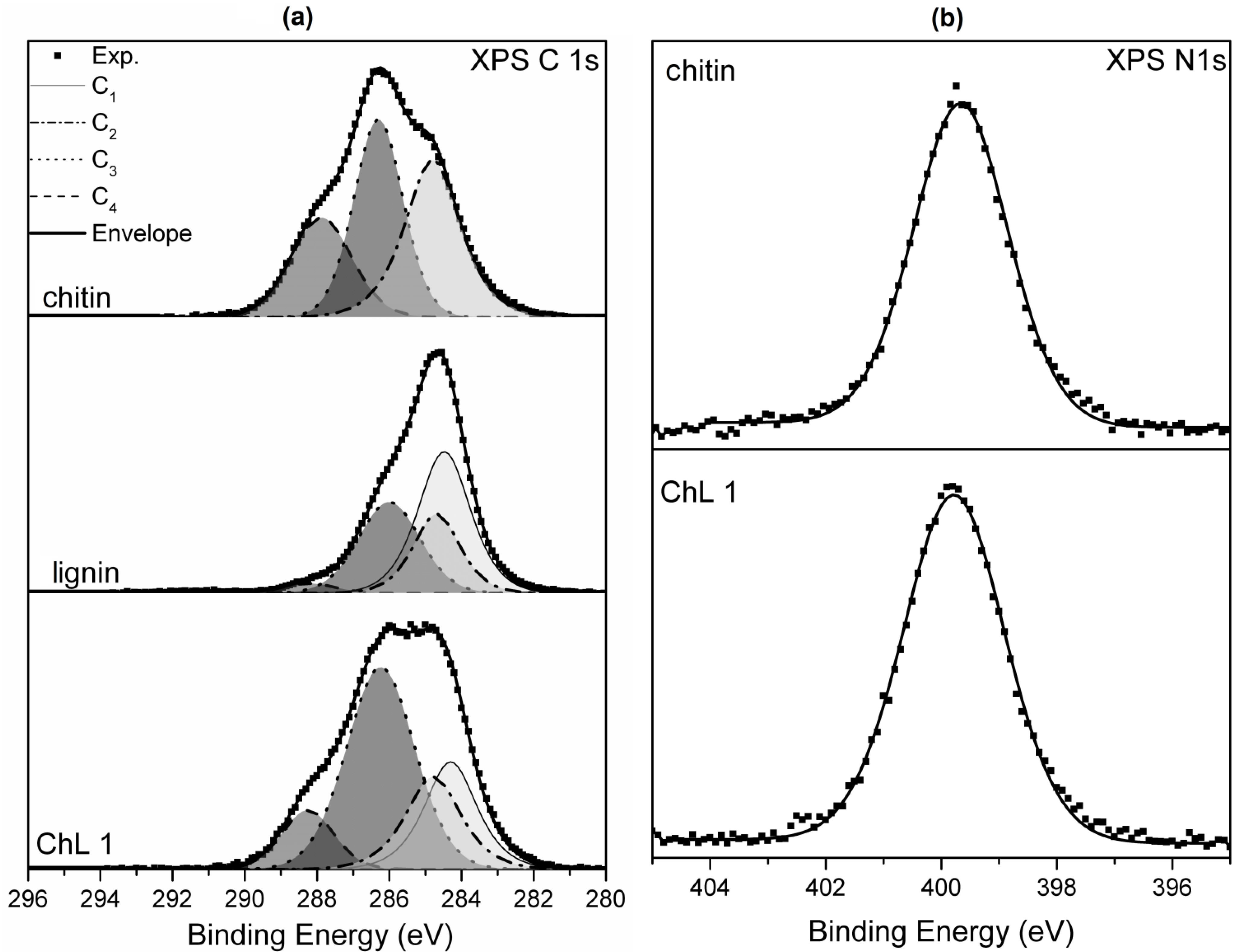
2.1.3. XPS Analysis
| Sample | C | O | N | Na | S | Ca | Cl |
|---|---|---|---|---|---|---|---|
| at. % | |||||||
| ChL 1 | 64.8 | 29.5 | 4.8 | - | - | 0.9 | - |
| chitin | 60.0 | 32.9 | 5.9 | 0.3 | - | 0.6 | 0.3 |
| kraft lignin | 68.0 | 25.0 | - | 5.0 | 2.0 | - | - |

| Sample | C1 | C2 | C3 | C4 |
|---|---|---|---|---|
| ChL1 | 21 | 19 | 49 | 11 |
| chitin | - | 37 | 39 | 24 |
| kraft lignin | 43 | 22 | 32 | 3 |
2.1.4.13C CP MAS NMR Spectroscopy
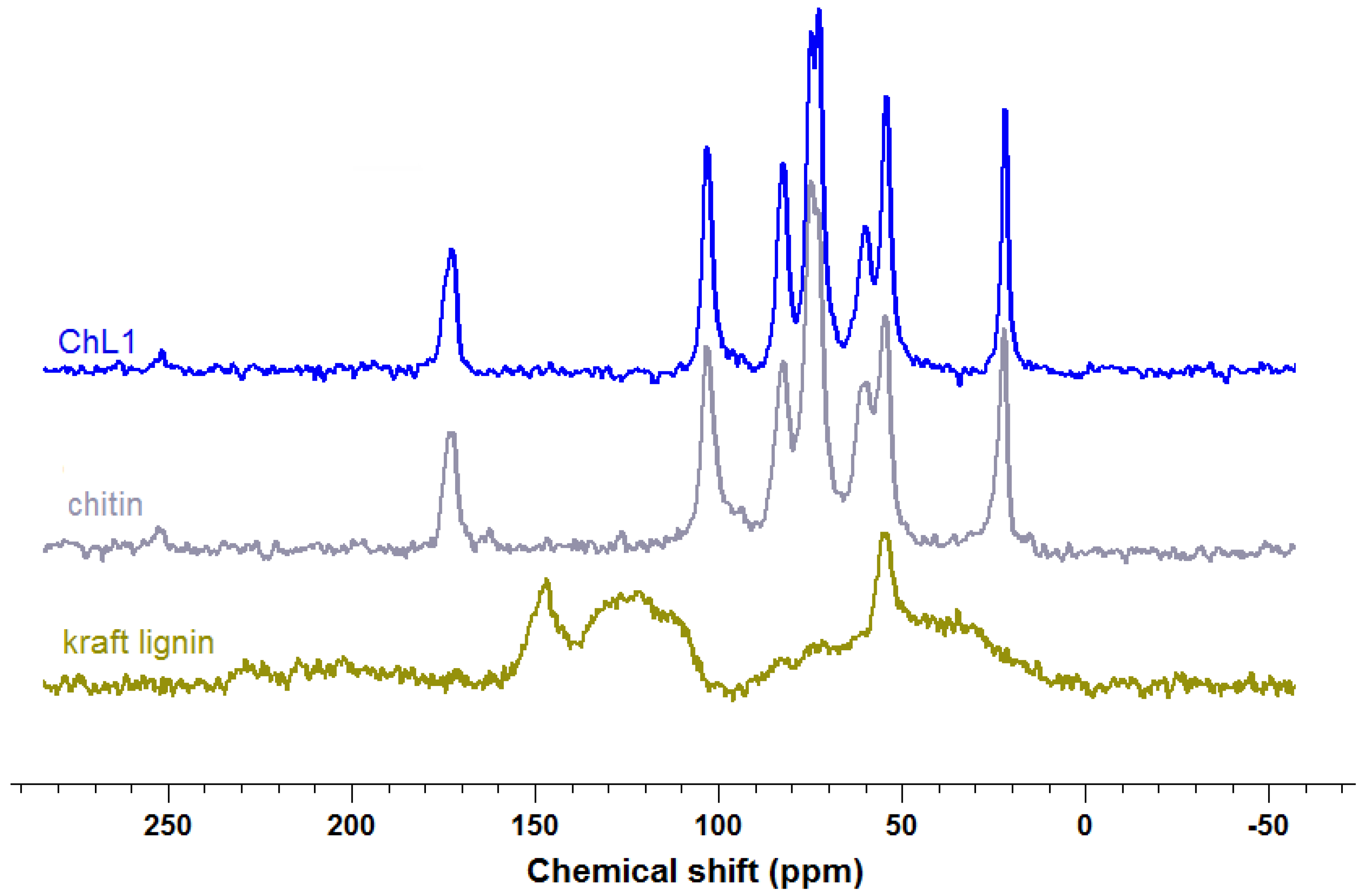
| Chitin | Lignin | ChL 1 | Assignment |
|---|---|---|---|
| - | 13.6 | - | γ–CH3 in n-propyl side chain |
| 22.1 | - | 21.9 | CH3 in acetamide group |
| - | 24.3 | - | CH3 or CH2 group in saturated side chains |
| - | 36.1 | - | CH3 groups, ketones (conj.) or in aliphatic |
| - | 52-54 | 54.2 | C-β in β-5 and β-β units |
| 54.4 | - | 54.2 | C2 in hexose ring |
| - | 55.6 | - | C in Ar–OCH3 |
| 59.6 | - | 59.9 | C6 in hexose ring |
| 73.1 | - | 72.6 | C3 in hexose ring |
| 74.7 | - | 74.7 | C5 in hexose ring |
| - | 74-1 | 72.6 | C-α in guaiacyl type β-0-4 units (threo and erythro) C-γ in β-β, C-γ, β-aryl ether |
| 82.2 | - | 82.4 | C4 in hexose ring |
| - | 85-83 | - | C-β in guaiacyl type β-0-4 units (threo and erythro) |
| 103.3 | - | 103.1 | C1 in hexose ring |
| - | 112-110 | - | C-2 in guaiacyl units |
| - | 117-113 | - | C-5 in guaiacyl units |
| - | 118-119 | - | C-6 in guaiacyl units |
| - | 121.4 | - | C1 and C6 in Ar–C(=O)C–C |
| - | 128.2 | - | C-α and C-β in Ar–CH=CH–CH2OH |
| - | 129.3 | - | C-α and C-β in Ar–CH=CH–CHO |
| - | 143.3 | - | C-4 in ring B of β-5 units, C-4/C-4′ of non–etherified 5-5 units |
| - | 145.8 | 145.5 | C-4 in non-etherified G units |
| - | 146.2 | - | C-3 in non-etherified G units (β-0-4 type) |
| - | 146.8 | - | C-4 in etherified G units |
| - | 169-172 | - | C=O in φ–COOH, Ester C=O in φ–C(=O)OR and R–C(=O)OCH3 |
| 173.4 | - | 172.7 | C=O in acetamide group |
| - | 192-202 | - | C=O in φ–CH=CH–CHO, C=O in φ–C(=O)CH(–O φ)–C– and other carbonyl groups |
2.1.5. Elemental Analysis
| Sample name | Elemental content (%) | |||
|---|---|---|---|---|
| N | C | H | S | |
| ChL 1 | 6.01 | 44.17 | 8.37 | 1.16 |
| ChL 2 | 6.03 | 44.04 | 8.31 | 0.96 |
| ChL 3 | 6.03 | 44.01 | 8.27 | 0.79 |
| ChL 4 | 6.03 | 43.93 | 8.24 | 0.63 |
| ChL 5 | 6.01 | 43.75 | 8.23 | 0.49 |
| ChL 6 | 6.03 | 43.72 | 8.20 | 0.27 |
| ChL 7 | 6.02 | 43.58 | 8.19 | 0.06 |
| chitin | 6.21 | 40.54 | 7.36 | - |
| kraft lignin | - | 42.21 | 5.02 | 3.14 |
2.1.6. Electrokinetic Characteristics
| Sample name | Zeta potential (mV) vs. pH | pHIEP | |||||
|---|---|---|---|---|---|---|---|
| 2 | 4 | 6 | 8 | 10 | 12 | ||
| ChL 1 | −1.3 | −17.5 | −26.5 | −37.0 | −43.2 | −46.3 | 1.8 |
| ChL 2 | 1.7 | −14.2 | −24.2 | −35.0 | −41.0 | −43.1 | 2.2 |
| ChL 3 | 2.1 | −11.1 | −22.0 | −33.9 | −39.0 | −42.0 | 2.7 |
| ChL 4 | 3.9 | −10.0 | −20.9 | −30.1 | −37.9 | −40.9 | 2.7 |
| ChL 5 | 5.2 | −8.0 | −19.5 | −27.9 | −36.5 | −38.2 | 2.8 |
| ChL 6 | 7.1 | −7.5 | −17.3 | −23.0 | −34.2 | −37.9 | 3.1 |
| ChL 7 | 9.3 | −6.5 | −15.1 | −20.5 | −31.9 | −36.0 | 3.4 |
| chitin | 19.9 | −8.5 | −23.6 | −31.4 | −37.2 | −40.1 | 3.5 |
| kraft lignin | −20.1 | −35.2 | −40.2 | −43.8 | −48.3 | −51.2 | - |
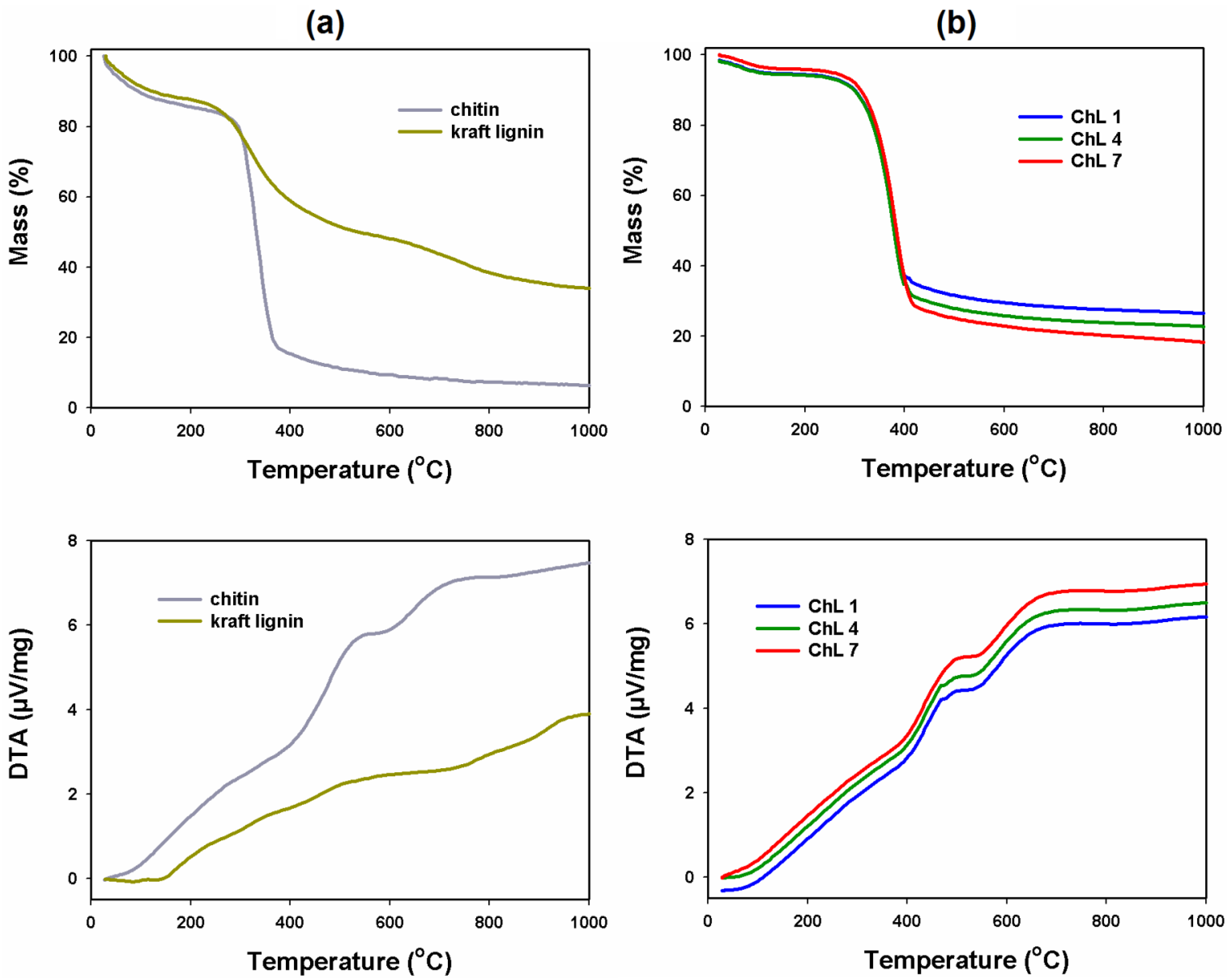
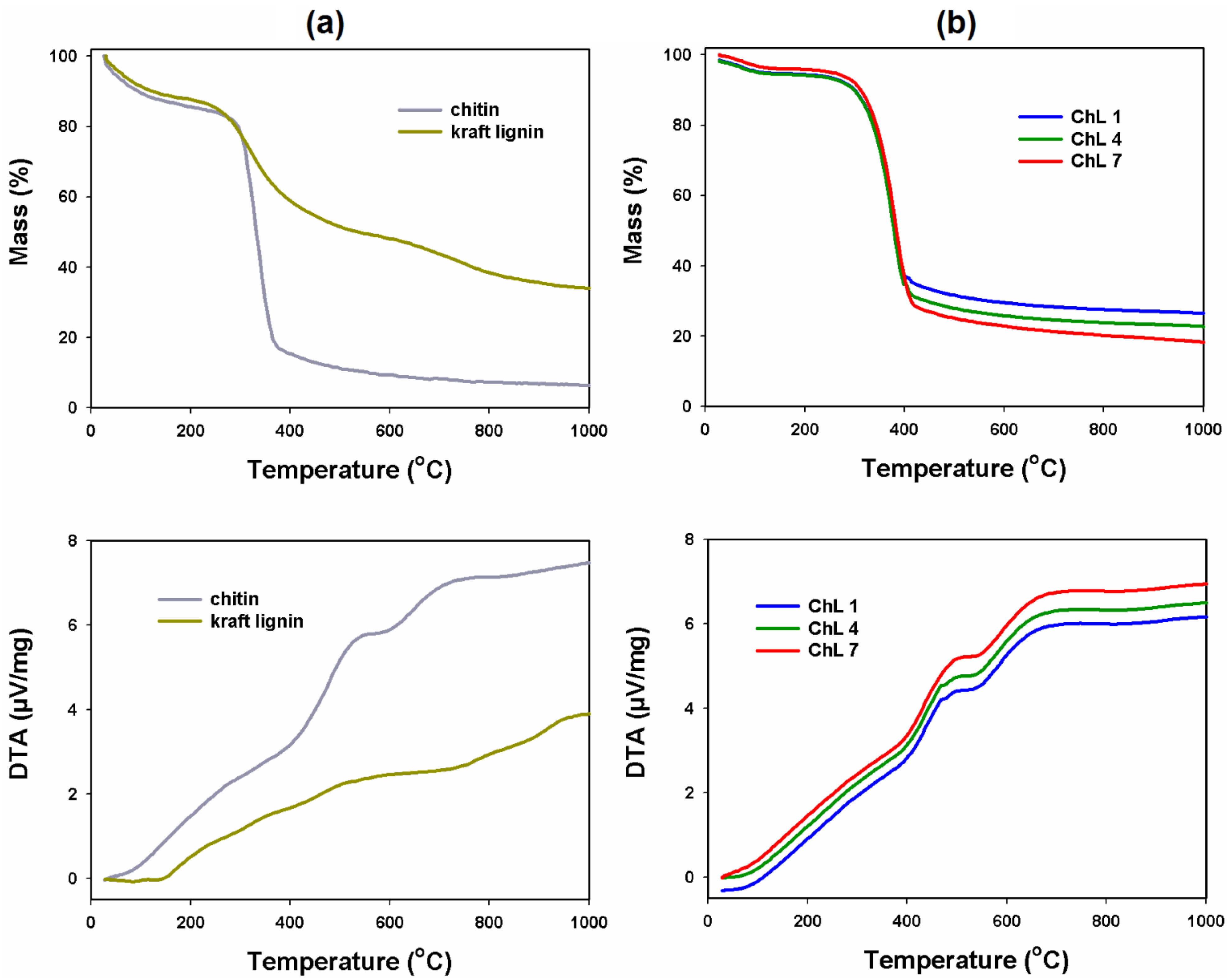
2.1.7. Thermal Stability
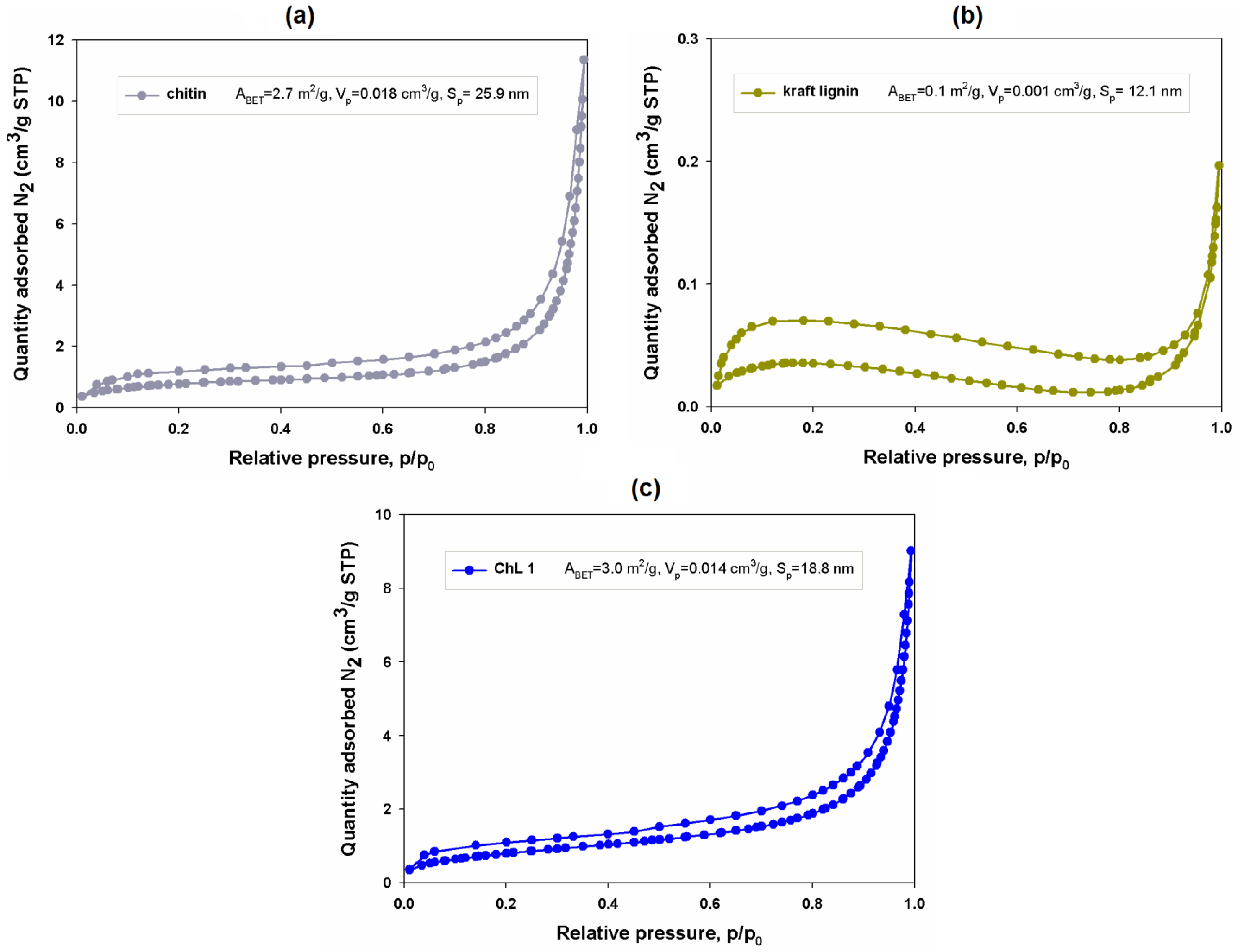
2.1.8. Porous Structure Properties
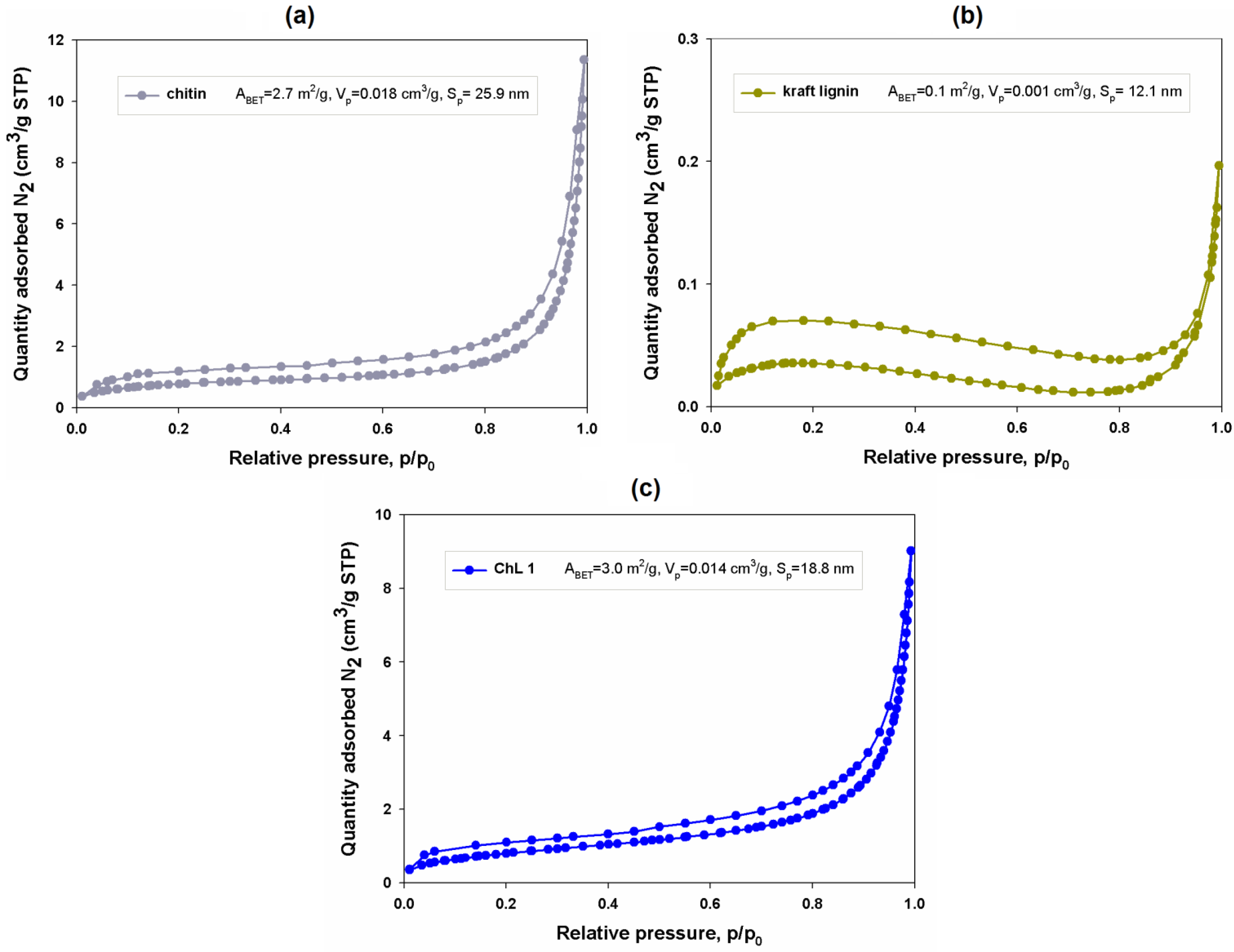
2.2. Batch Adsorption Study
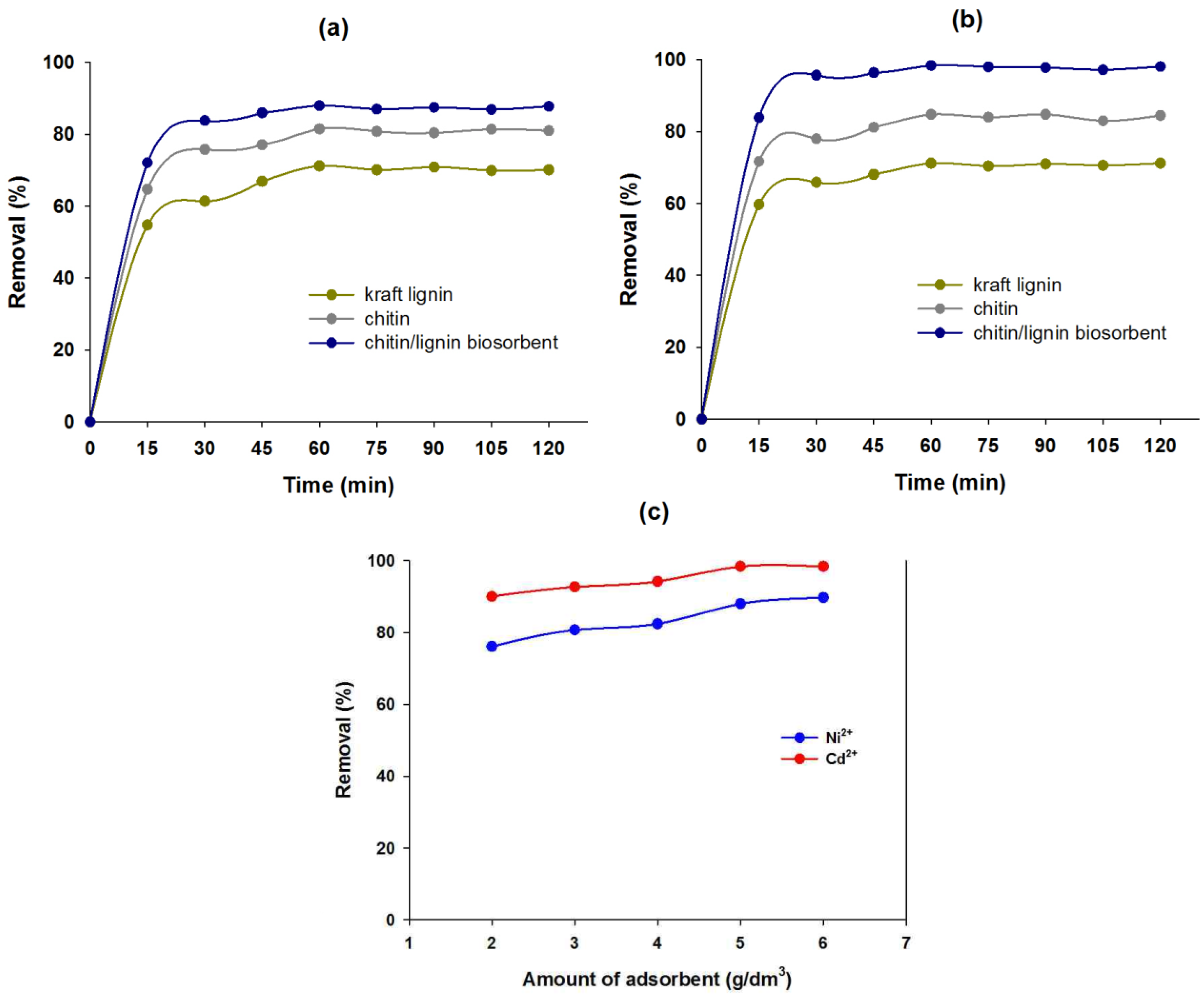
2.2.1. Effect of Contact Time on Sorption Efficiency
2.2.2. Effect of Quantity of Chitin/Lignin Biosorbents on Sorption Efficiency
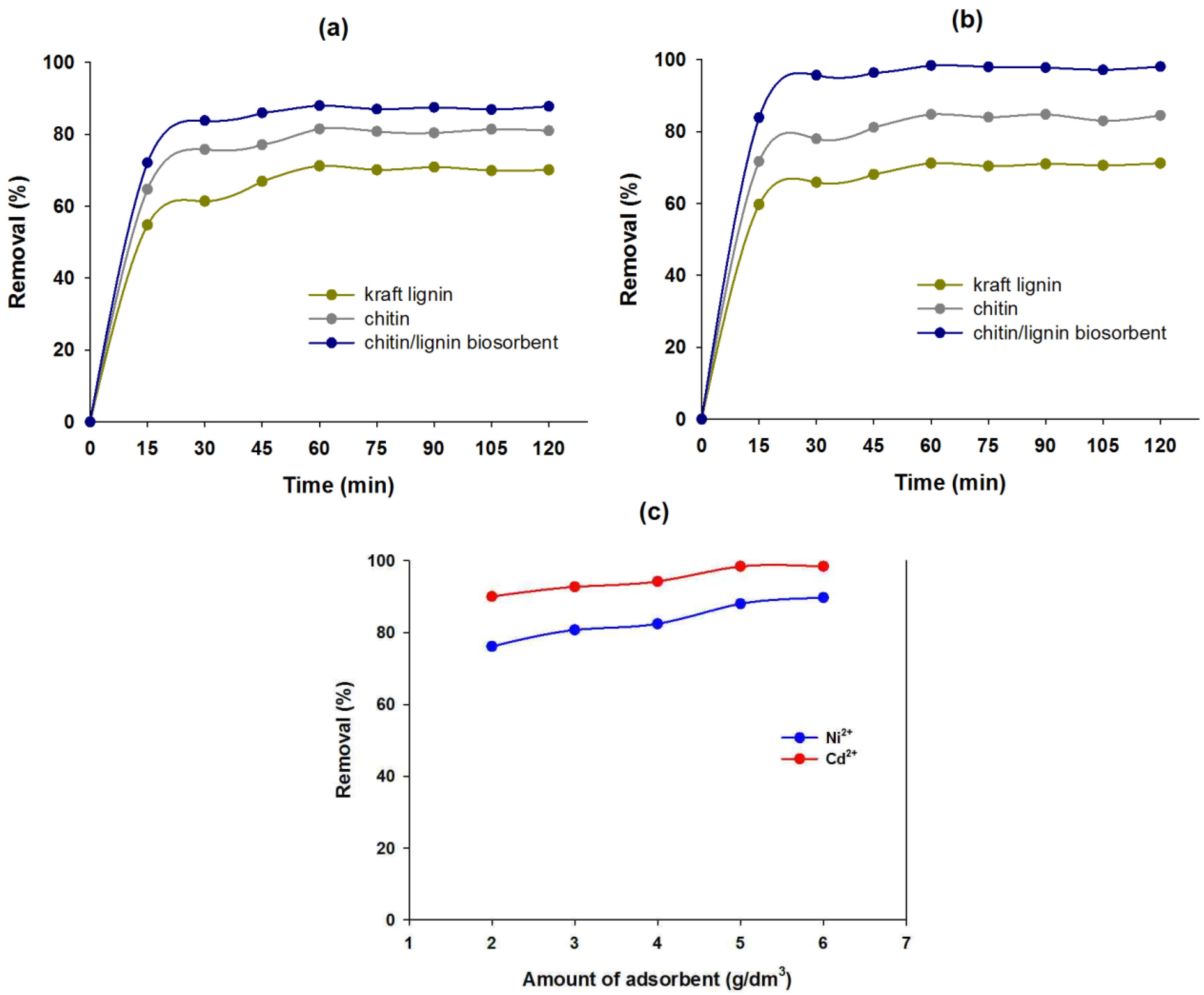
| Kind of sorbents | The amount of metal ions adsorbed at equilibrium (mg/g) | |
|---|---|---|
| Ni2+ | Cd2+ | |
| lignin | 4.27 | 4.28 |
| chitin | 4.89 | 5.09 |
| chitin/lignin biosorbents | 5.28 | 5.90 |
3. Experimental Section
3.1. Materials
3.2. Preparation of Chitin/Lignin Materials
| Sample name | The weight ratio of precursors (chitin:lignin) | Amount of H2O2 (cm3) |
|---|---|---|
| ChL 1 | 1:1 | 100 |
| ChL 2 | 1:0.75 | |
| ChL 3 | 1:0.5 | |
| ChL 4 | 1:0.3 | |
| ChL 5 | 1:0.2 | |
| ChL 6 | 1:0.1 | |
| ChL 7 | 1:0.05 |
3.3. Physicochemical Evaluation
3.4. Adsorption Experiments
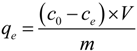
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gładysz-Płaska, A.; Majdan, M.; Pikus, S.; Sternik, D. Simultaneous adsorption of chromium(VI) and phenol on natural red clay modified by HDTMA. Chem. Eng. J. 2012, 179, 140–150. [Google Scholar] [CrossRef]
- Ashrafa, M.A.; Rehmanb, M.A.; Aliasa, Y.; Yusof, I. Removal of Cd(II) onto Raphanussativus peels biomass: Equilibrium, kinetics, and thermodynamics. Desalination Water Treat. 2013, 51, 4402–4412. [Google Scholar]
- Dhir, B.; Srivastava, S. Heavy metal removal from a multi-metal solution and wastewater by Salvinianatans. Ecol. Eng. 2011, 37, 893–896. [Google Scholar] [CrossRef]
- Jiang, M.; Jin, X.; Lu, X.; Chen, Z. Adsorption of Pb(II), Cd(II), Ni(II) and Cu(II) onto natural kaolinite clay. Desalination 2010, 252, 33–39. [Google Scholar] [CrossRef]
- León-Torres, A.; Cuerda-Correa, E.M.; Fernández-González, C.; Gómez-Serrano, V. On the use of a natural peat for the removal of Cr(VI) from aqueous solutions. J. Colloid Interface Sci. 2012, 15, 325–332. [Google Scholar]
- Zhang, A.; Lu, F.; Sun, R.C.; Ralph, J. Isolation of cellulolytic enzyme lignin from wood dissolved in dimethyl sulfoxide/N-methylimidazole. J. Agric. Food Chem. 2010, 58, 3446–3450. [Google Scholar] [CrossRef]
- Monteil-Rivera, F.; Phuong, M.; Ye, M.; Halasz, A.; Hawari, J. Isolation characterization of herbaceous lignins for applications in biomaterials. Ind. Crop. Prod. 2013, 41, 356–364. [Google Scholar] [CrossRef]
- Sarkar, S.; Adhikari, B. Jute felt composite from lignin modified phenolic resin. Polym. Compos. 2001, 22, 518–527. [Google Scholar] [CrossRef]
- Sahoo, S.; Misra, M.; Mohanty, A.K. Enhanced properties of lignin-based biodegradable polymer composites using injection moulding process. Compos. Part A 2011, 42, 1710–1718. [Google Scholar] [CrossRef]
- Carrott, P.J.; Ribeiro Carrott, M.M. Lignin—From natural adsorbent to activated carbon: A review. Bioresour. Technol. 2007, 98, 2301–2312. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, S.; Shan, X. Adsorption of metal ions on lignin. J. Hazard. Mater. 2008, 151, 134–142. [Google Scholar] [CrossRef]
- Šćiban, M.B.; Klašnja, M.T.; Antor, M.G. Study of the biosorption of different heavy metal ions onto Kraft lignin. Ecol. Eng. 2011, 37, 2092–2095. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Single, binary and multi-component adsorption of copper and cadmium from aqueous solutions on Kraft lignin—A biosorbent. J. Colloid Interface Sci. 2006, 297, 489–504. [Google Scholar] [CrossRef]
- Shulga, G.; Shakels, V.; Skudra, S.; Bogdanovs, V. Modified lignin as an environmentally friendly surfactant. Environment. Technology. Resources. In Proceedings of the 8th International Scientific and Practical Conference, Tomsk, Russia, 8–12 April 2002; Volume 1, pp. 276–281.
- Milczarek, G.; Inganäs, O. Renewable cathode materials from biopolymer/conjugated polymer interpenetrating networks. Science 2012, 335, 1468–1471. [Google Scholar] [CrossRef]
- Jesionowski, T.; Klapiszewski, Ł.; Milczarek, G. Kraft lignin and silica as precursors of advanced composite materials and electroactive blends. J. Mater. Sci. 2014, 49, 1376–1385. [Google Scholar] [CrossRef]
- Ehrlich, H. Chitin and collagen as universal and alternative templates in biomineralization. Int. Geol. Rev. 2010, 52, 661–699. [Google Scholar] [CrossRef]
- Brunner, E.; Richthammer, P.; Ehrlich, H.; Paasch, S.; Simon, P.; Ueberlein, S.; van Pée, K.-H. Chitin-based organic networks: An integral part of cell wall biosilica in the diatom Thalassiosira pseudonana. Angew. Chem. Int. Ed. 2009, 48, 9724–9727. [Google Scholar]
- Goodrich, J.D.; Winter, W.T. α-Chitin nanocrystals prepared from shrimp shells and their specific surface area measurement. Biomacromolecules 2007, 8, 252–257. [Google Scholar] [CrossRef]
- Sajomsang, W.; Gonil, P. Preparation and characterization of α-chitin from cicada sloughs. Mater. Sci. Eng. C 2010, 30, 357–363. [Google Scholar] [CrossRef]
- Ehrlich, H.; Maldonado, M.; Spindler, K.D.; Eckert, C.; Hanke, T.; Born, R.; Goebel, C.; Simon, P.; Heinemann, S.; Worch, H. First evidence of chitin as a component of the skeletal fibers of marine sponges. Part I. Verongidae (Demospongia: Porifera). J. Exp. Zool. Part B 2007, 308, 347–356. [Google Scholar]
- Ehrlich, H.; Ilan, M.; Maldonado, M.; Muricy, G.; Bavestrello, G.; Kljajic, Z.; Carballo, J.L.; Schiaparelli, S.; Ereskovsky, A.; Schupp, P.; et al. Three-dimensional chitin-based scaffolds from Verongida sponges (Demospongiae: Porifera). Part I. Isolation and identification of chitin. Int. J. Biol. Macromol. 2010, 47, 132–140. [Google Scholar] [CrossRef]
- Wysokowski, M.; Bazhenov, V.V.; Tsurkan, M.V.; Galli, R.; Stelling, A.L.; Stöcker, H.; Kaiser, S.; Niederschalg, E.; Gärtner, G.; Behm, T.; et al. Isolation and identification of chitin in three-dimensional skeleton of Aplysina fistularis marine sponge. Int. J. Biol. Macromol. 2013, 62, 94–100. [Google Scholar] [CrossRef]
- Ehrlich, H.; Kaluzhnaya, O.V.; Tsurkan, M.V.; Ereskovsky, A.; Tabachnick, K.R.; Ilan, M.; Stelling, A.; Galli, R.; Petrova, O.V.; Nekipelov, S.V.; et al. First report on chitinous holdfast in sponges (Porifera). Proc. Biol. Sci. 2013, 280, 20130339. [Google Scholar] [CrossRef]
- Ehrlich, H.; Kaluzhnaya, O.; Brunner, E.; Tsurkan, M.V.; Ereskovsky, A.; Ilan, M.; Tabachnick, K.R.; Bazenov, V.V.; Paasch, S.; Kammer, M.; et al. Identification and first insights into the structure and biosynthesis of chitin from the freshwater sponge Spongilla lacustris. J. Struct. Biol. 2013, 183, 474–483. [Google Scholar] [CrossRef]
- Yang, T.C.; Zall, R.R. Absorption of metals by natural polymers generated from seafood processing wastes. Ind. Eng. Chem. Prod. Res. Dev. 1984, 23, 168–172. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Chitins and chitosans for the repair of wounded skin, nerve, cartilage and bone. Carbohydr. Polym. 2009, 76, 167–182. [Google Scholar] [CrossRef]
- Jayakumar, R.; Nair, A.; Sanoj Rejinold, N.; Maya, S.; Nair, S.V. Doxorubicin-loaded pH-responsive chitin nanogels for drug delivery to cancer cells. Carbohydr. Polym. 2012, 87, 2352–2356. [Google Scholar] [CrossRef]
- Di Giuseppe, A.; Crucianelli, M.; Passacantado, M.; Nisi, S.; Saladino, R. Chitin- and chitosan-anchored methyltioxorhenium: An innovative approach for selective heterogenous catalytic epoxidations of olefins. J. Catal. 2010, 276, 412–422. [Google Scholar] [CrossRef]
- Filipkowska, U. Desorption of reactive dyes from modified chitin. Environ. Technol. 2008, 29, 681–690. [Google Scholar] [CrossRef]
- Tang, H.; Zhou, W.; Zhang, L. Adsorption isotherms and kinetics studies of malachite green on chitin hydrogels. J. Hazard. Mater. 2012, 209–210, 218–225. [Google Scholar] [CrossRef]
- Schleuter, D.; Günter, A.; Paasch, S.; Ehrlich, H.; Kljajić, Z.; Hanke, T.; Bernhard, G.; Brunner, E. Chitin-based renewable materials from marine sponges for uranium adsorption. Carbohydr. Polym. 2013, 92, 712–718. [Google Scholar] [CrossRef]
- Li, C.B.; Hein, S.; Wang, K. Adsorption of chitin and chitosan. Mater. Sci. Technol. 2008, 24, 1088–1099. [Google Scholar] [CrossRef]
- Lobo-Recio, M.A.; Lapolli, F.R.; Belli, T.J.; Folzke, C.T.; Tarpani, R.R.T. Study of the removal of residual aluminium though the biopolymers carboxymethylcellulose, chitin and chitosan. Desalination Water Treat. 2013, 51, 1735–1743. [Google Scholar]
- Kurita, K. Controlled functionalization of the polysaccharide chitin. Prog. Polym. Sci. 2001, 26, 1921–1971. [Google Scholar] [CrossRef]
- Wang, X.; Xing, B. Importance of structural makeup of biopolymers for organic contaminant sorption. Environ. Sci. Technol. 2007, 41, 3559–3565. [Google Scholar] [CrossRef]
- Klapiszewski, Ł.; Nowacka, M.; Milczarek, G.; Jesionowski, T. Physicochemical and electrokinetic properties of silica/lignin biocomposites. Carbohydr. Polym. 2013, 94, 345–355. [Google Scholar] [CrossRef]
- Cárdenas, G.; Cabrera, G.; Taboada, E.; Miranda, S.P. Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J. Appl. Polym. Sci. 2004, 93, 1876–1885. [Google Scholar] [CrossRef]
- Ago, M.; Jakes, J.E.; Johansson, L.S.; Park, S.; Rojas, O.J. Interfacial properties of lignin-based electrospun nanofibers and films reinforced with cellulose nanocrystals. ACS Appl. Mater. Interfaces 2012, 4, 6849–6856. [Google Scholar]
- Wang, B.; Li, J.; Zhang, J.; Li, H.; Chen, P.; Gu, Q.; Wang, Z. Thermo-mechanical properties of the composite made of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and acetylated chitin nanocrystals. Carbohydr. Polym. 2013, 95, 100–106. [Google Scholar] [CrossRef]
- Johansson, L.S.; Campbell, J.M.; Koljonen, K.; Stenius, P. Evaluation of surface lignin on cellulose fibers with XPS. Appl. Surf. Sci. 1999, 144–145, 92–95. [Google Scholar] [CrossRef]
- Briggs, D.; Grant, J.T. Surface Analysis by Auger and X-ray Photoelectron Spectroscopy; IM Publications and Surface Spectra Limited: Charlton Manchester, UK, 2003. [Google Scholar]
- Wang, J.; Wang, Z.; Li, J.; Wang, B.; Liu, J.; Chen, P.; Miao, M.; Gu, Q. Chitin nanocrystals grafted with poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and their effects on thermal behavior of PHBV. Carbohydr. Polym. 2012, 87, 784–789. [Google Scholar] [CrossRef]
- Oh, D.; Shin, S.; Lim, C.; Hwang, D. Dopamine-mediated sclerotization of regenerated chitin in ionic liquid. Materials 2013, 6, 3826–3839. [Google Scholar] [CrossRef]
- Haensel, T.; Comouth, A.; Lorenz, P.; Ahmed, S.I.U.; Krischok, S.; Zydziak, N.; Kauffmann, A.; Schaefer, J.A. Pyrolysis of cellulose and lignin. Appl. Surf. Sci. 2009, 255, 8183–8189. [Google Scholar] [CrossRef]
- Lange, P.J.; Mahy, J.W.G. ToF-SIMS and XPS investigations of fibers, coatings and biomedical materials. Fresenius J. Anal. Chem. 1995, 353, 487–493. [Google Scholar] [CrossRef]
- Stevens, J.S.; Luca, A.C.; Pelendritis, M.; Terenghi, G.; Downes, S.; Schroeder, S.L.M. Quantitative analysis of complex amino acids and RGD peptides by X-ray photoelectron spectroscopy (XPS). Surf. Interface Anal. 2013, 45, 1238–1246. [Google Scholar] [CrossRef]
- Spinde, K.; Kammer, M.; Freyer, K.; Ehrlich, H.; Vournakis, J.N.; Brunner, E. Biomimetic silicification of fibrous chitin from diatoms. Chem. Mater. 2011, 23, 2973–2978. [Google Scholar] [CrossRef]
- Wen, J.L.; Sun, S.L.; Xue, B.L.; Sun, R.C. Recent advances in characterization of lignin polymer by solution-state nuclear magnetic resonance (NMR) methodology. Materials 2013, 6, 359–391. [Google Scholar] [CrossRef]
- Li, J.; Revol, J.F.; Naranjo, E.; Marchessault, R.H. Effect of electrostatic interaction on phase separation behaviour of chitin crystallite suspensions. Int. J. Biol. Macromol. 1996, 18, 177–187. [Google Scholar] [CrossRef]
- Stawski, D.; Rabiej, S.; Herczyńska, L.; Draczyński, Z. Thermogravimetric analysis of chitins of different origin. J. Therm. Anal. Calorim. 2008, 93, 489–494. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zheng, Y.; Luo, Z.; Cen, K. Mechanism study of wood lignin pyrolysis by using TG-FTIR analysis. J. Anal. Appl. Pyrolysis 2008, 82, 170–177. [Google Scholar] [CrossRef]
- Shen, D.K.; Gu, S.; Luo, K.H.; Wang, S.R.; Fang, M.X. The pyrolytic degradation of wood-derived lignin from pulping process. Bioresour. Technol. 2010, 101, 6136–6146. [Google Scholar] [CrossRef]
- Singha, B.; Kumar Das, S. Adsorptive removal of Cu(II) from aqueous solution and industrial effluent using natural/agricultural wastes. Colloids Surf. B 2013, 107, 97–106. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wysokowski, M.; Klapiszewski, Ł.; Moszyński, D.; Bartczak, P.; Szatkowski, T.; Majchrzak, I.; Siwińska-Stefańska, K.; Bazhenov, V.V.; Jesionowski, T. Modification of Chitin with Kraft Lignin and Development of New Biosorbents for Removal of Cadmium(II) and Nickel(II) Ions. Mar. Drugs 2014, 12, 2245-2268. https://doi.org/10.3390/md12042245
Wysokowski M, Klapiszewski Ł, Moszyński D, Bartczak P, Szatkowski T, Majchrzak I, Siwińska-Stefańska K, Bazhenov VV, Jesionowski T. Modification of Chitin with Kraft Lignin and Development of New Biosorbents for Removal of Cadmium(II) and Nickel(II) Ions. Marine Drugs. 2014; 12(4):2245-2268. https://doi.org/10.3390/md12042245
Chicago/Turabian StyleWysokowski, Marcin, Łukasz Klapiszewski, Dariusz Moszyński, Przemysław Bartczak, Tomasz Szatkowski, Izabela Majchrzak, Katarzyna Siwińska-Stefańska, Vasilii V. Bazhenov, and Teofil Jesionowski. 2014. "Modification of Chitin with Kraft Lignin and Development of New Biosorbents for Removal of Cadmium(II) and Nickel(II) Ions" Marine Drugs 12, no. 4: 2245-2268. https://doi.org/10.3390/md12042245
APA StyleWysokowski, M., Klapiszewski, Ł., Moszyński, D., Bartczak, P., Szatkowski, T., Majchrzak, I., Siwińska-Stefańska, K., Bazhenov, V. V., & Jesionowski, T. (2014). Modification of Chitin with Kraft Lignin and Development of New Biosorbents for Removal of Cadmium(II) and Nickel(II) Ions. Marine Drugs, 12(4), 2245-2268. https://doi.org/10.3390/md12042245