Action of Clathrodin and Analogues on Voltage-Gated Sodium Channels

 , , ,
, , , 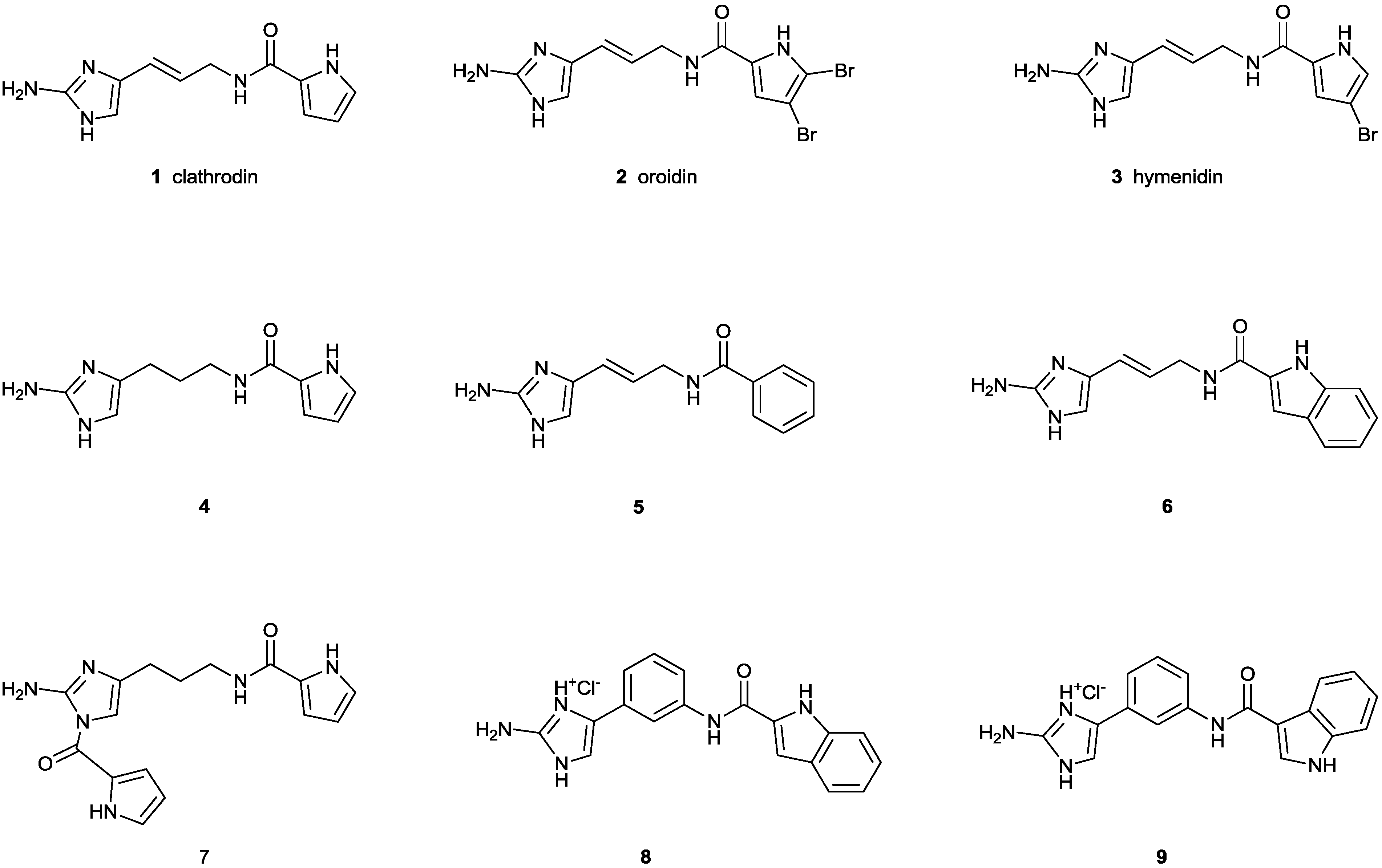{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
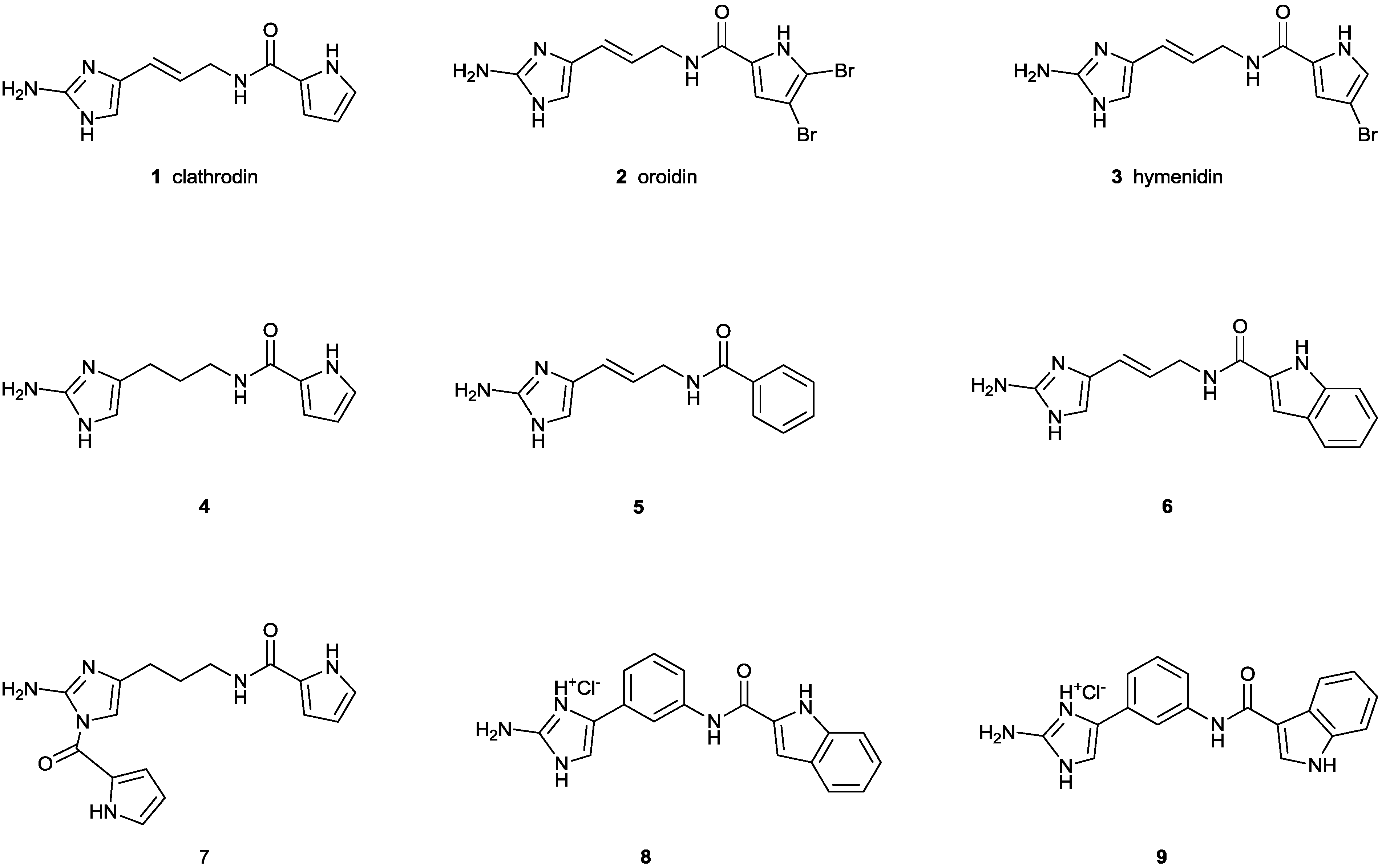
2. Results and Discussion
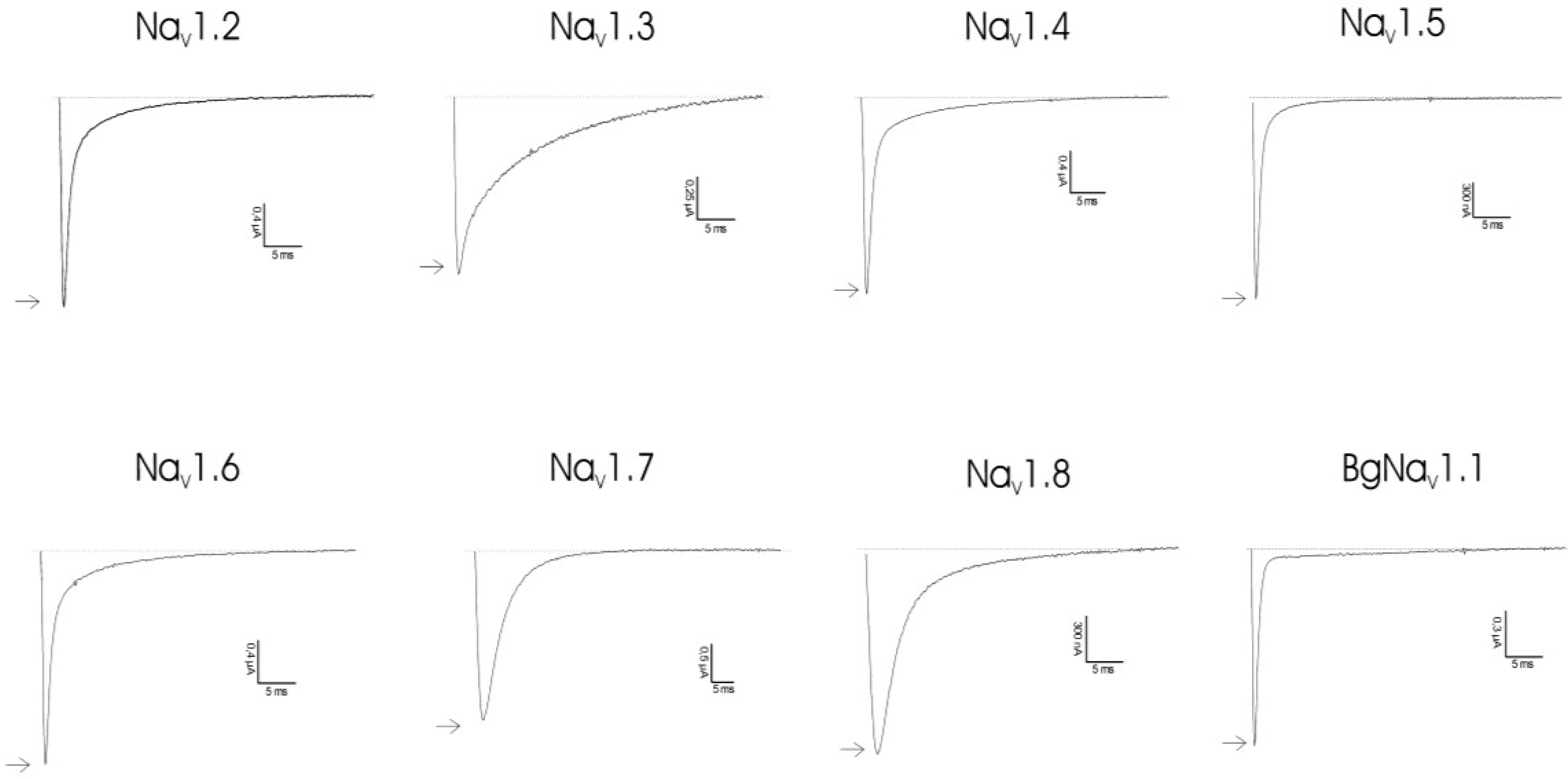
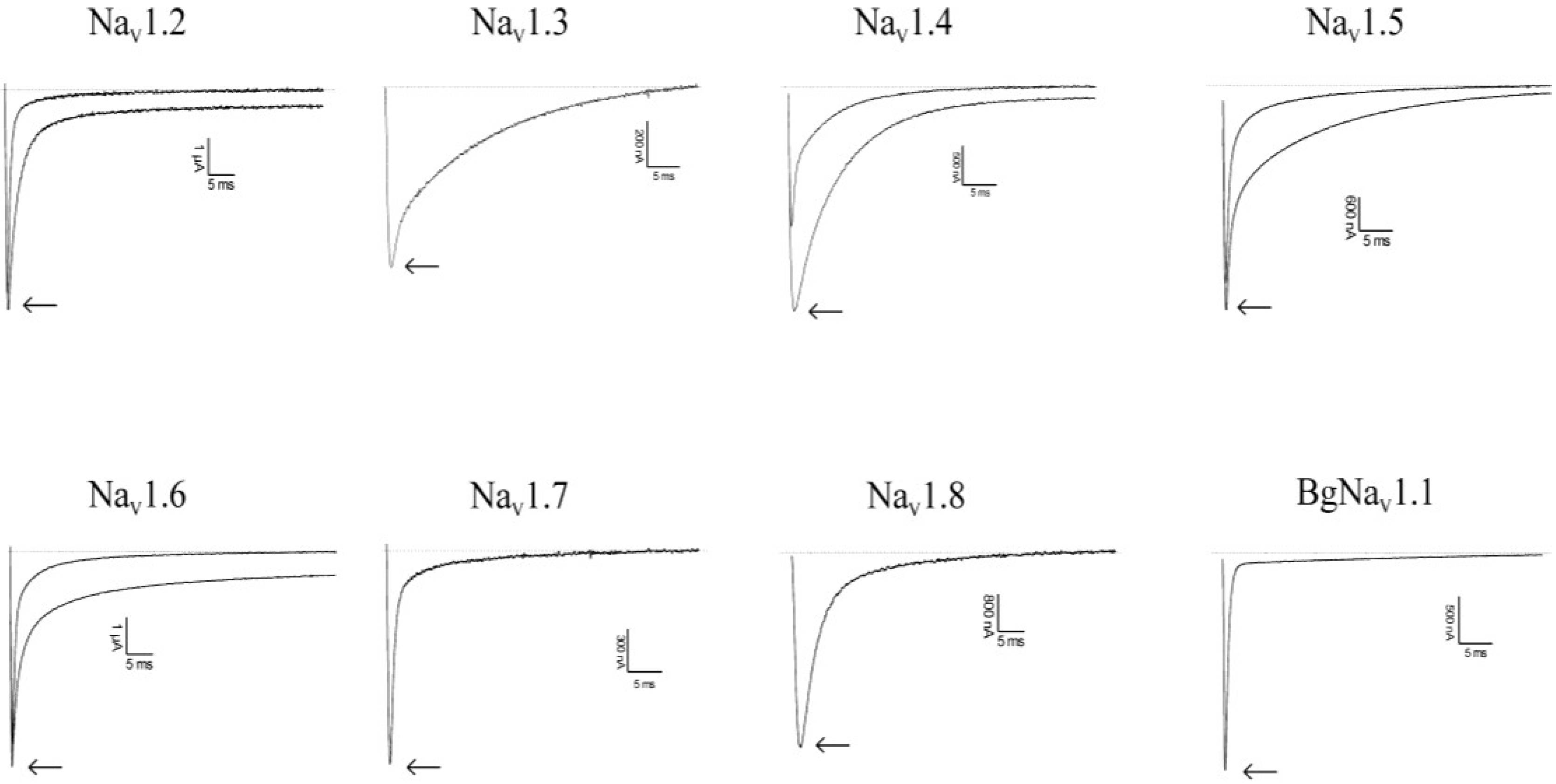
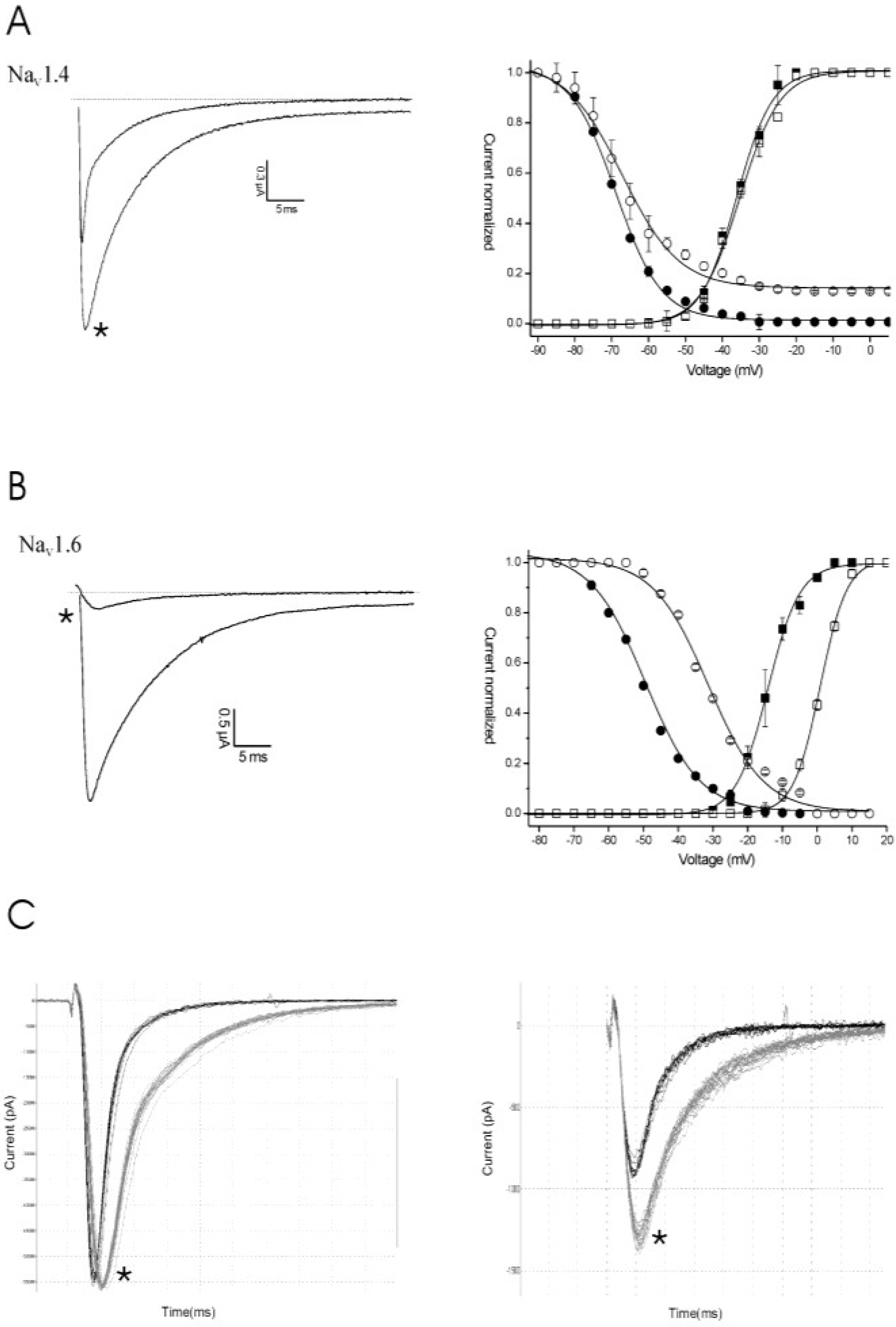
2.1. Results
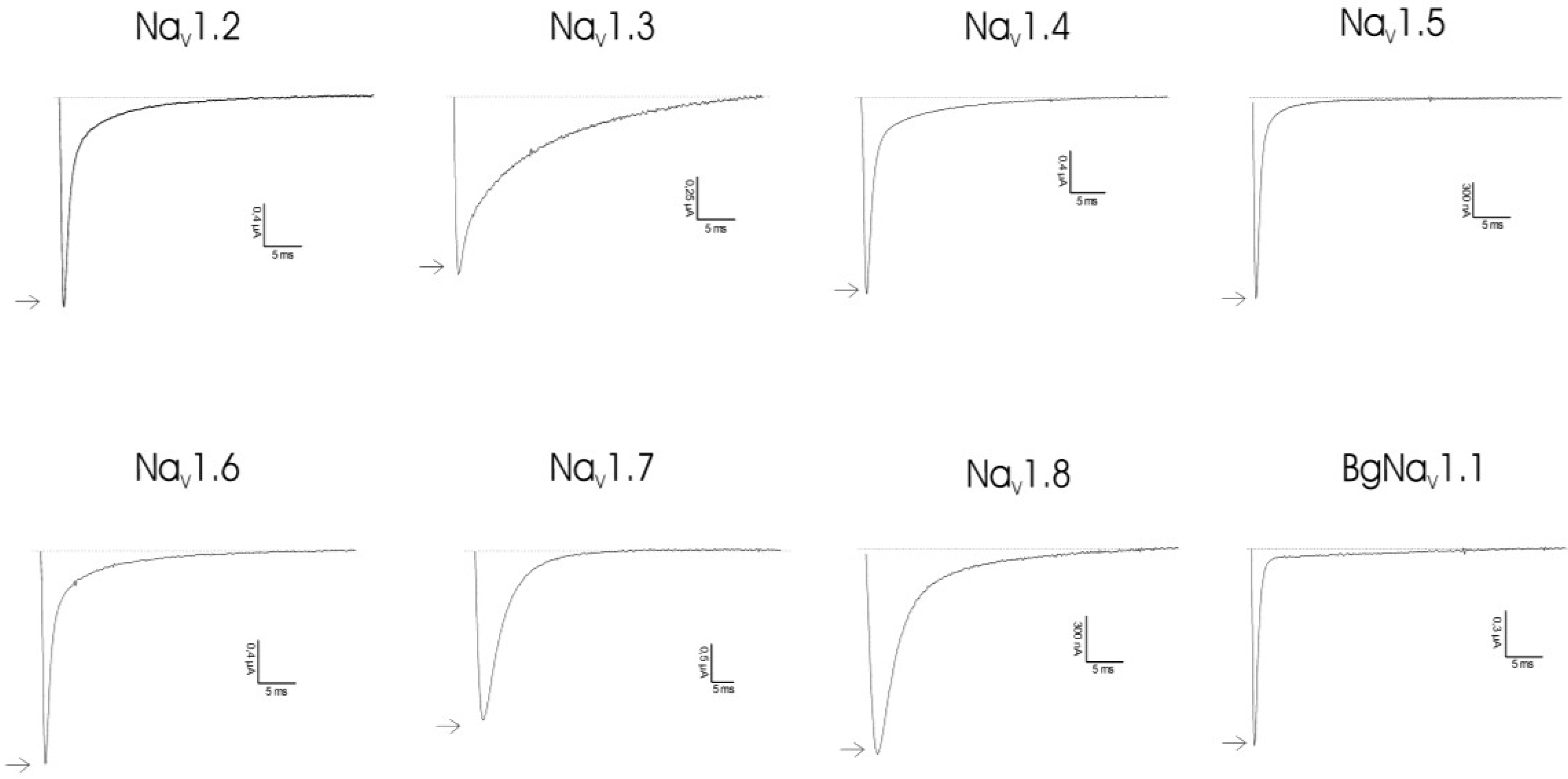
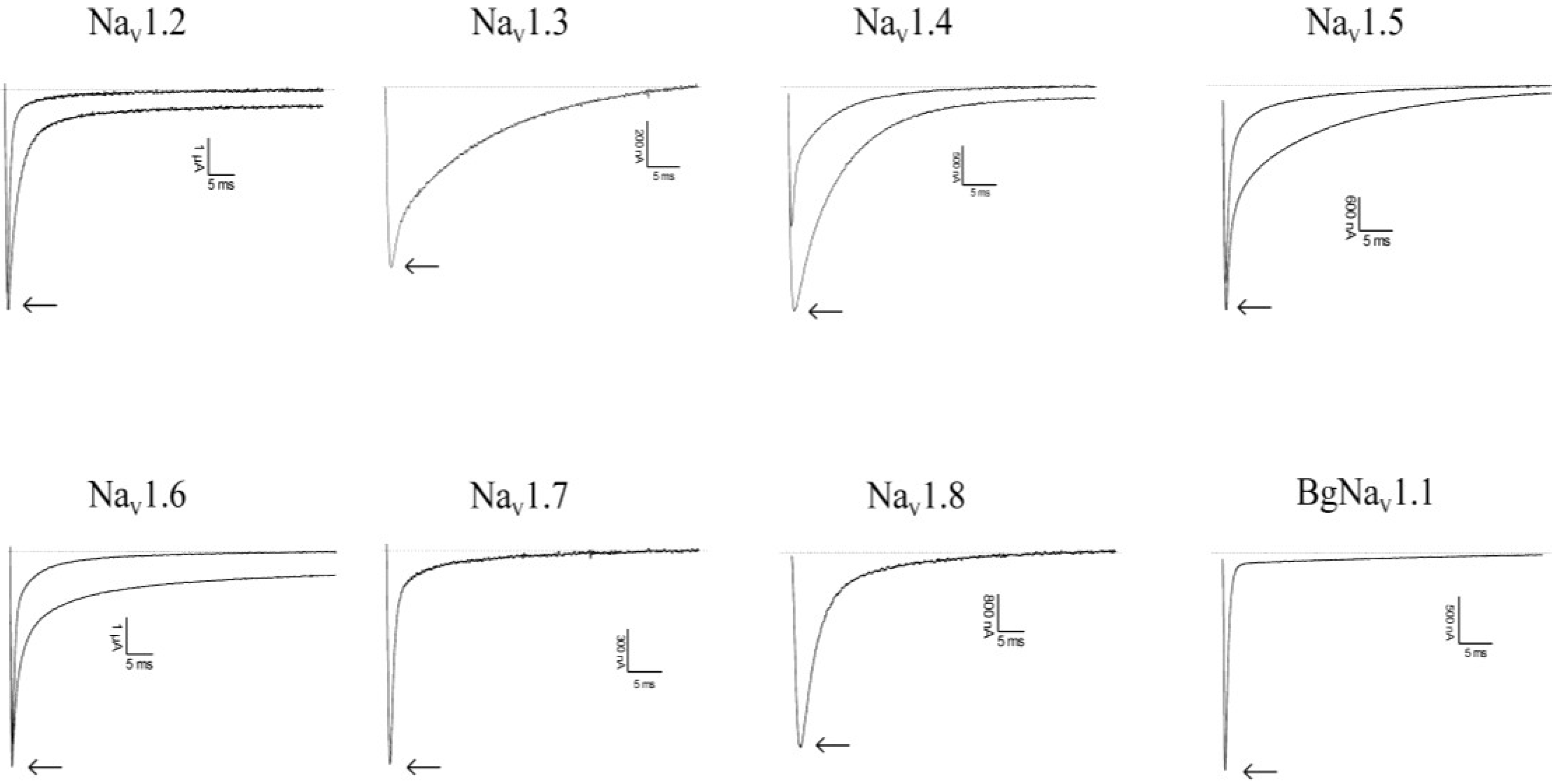
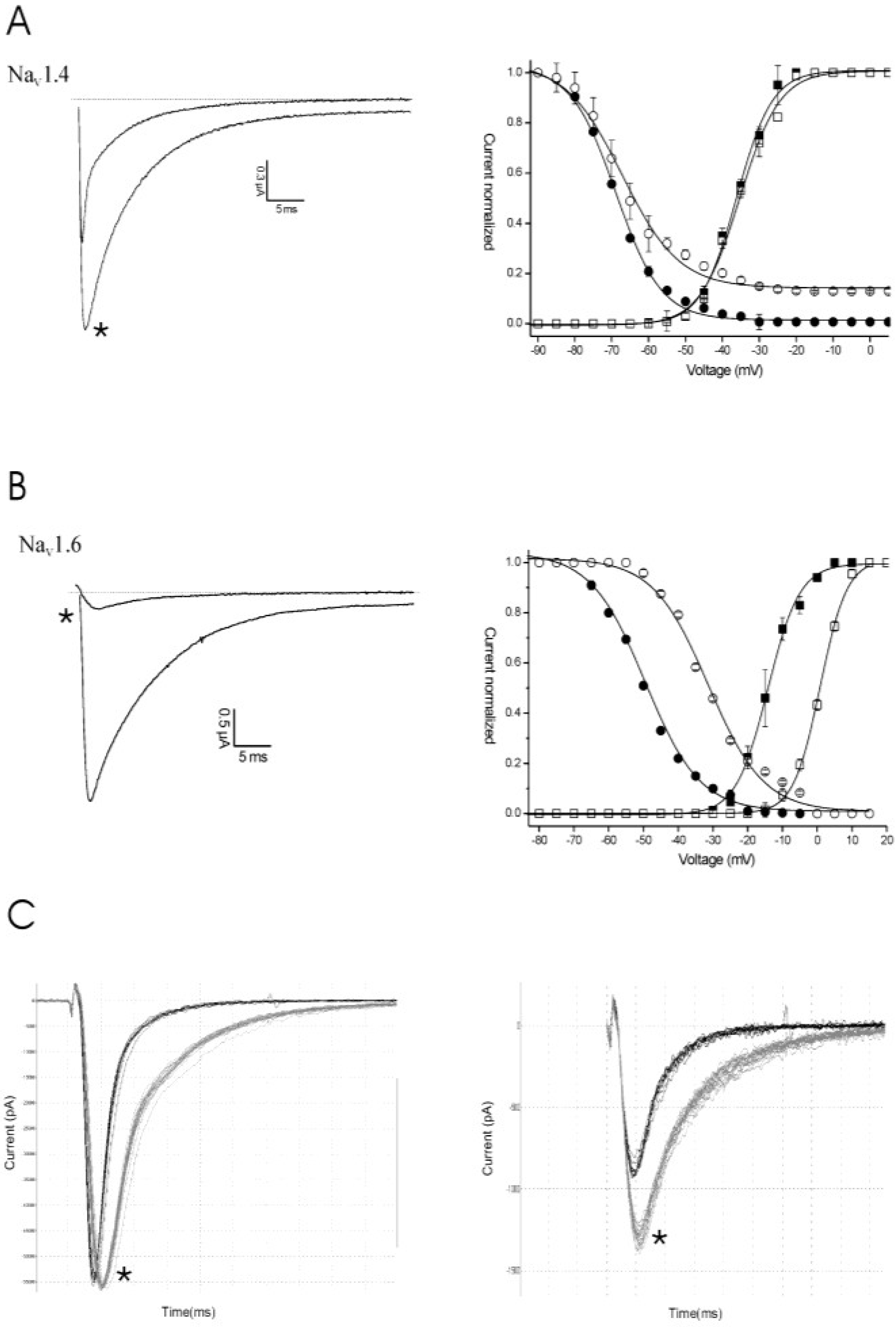
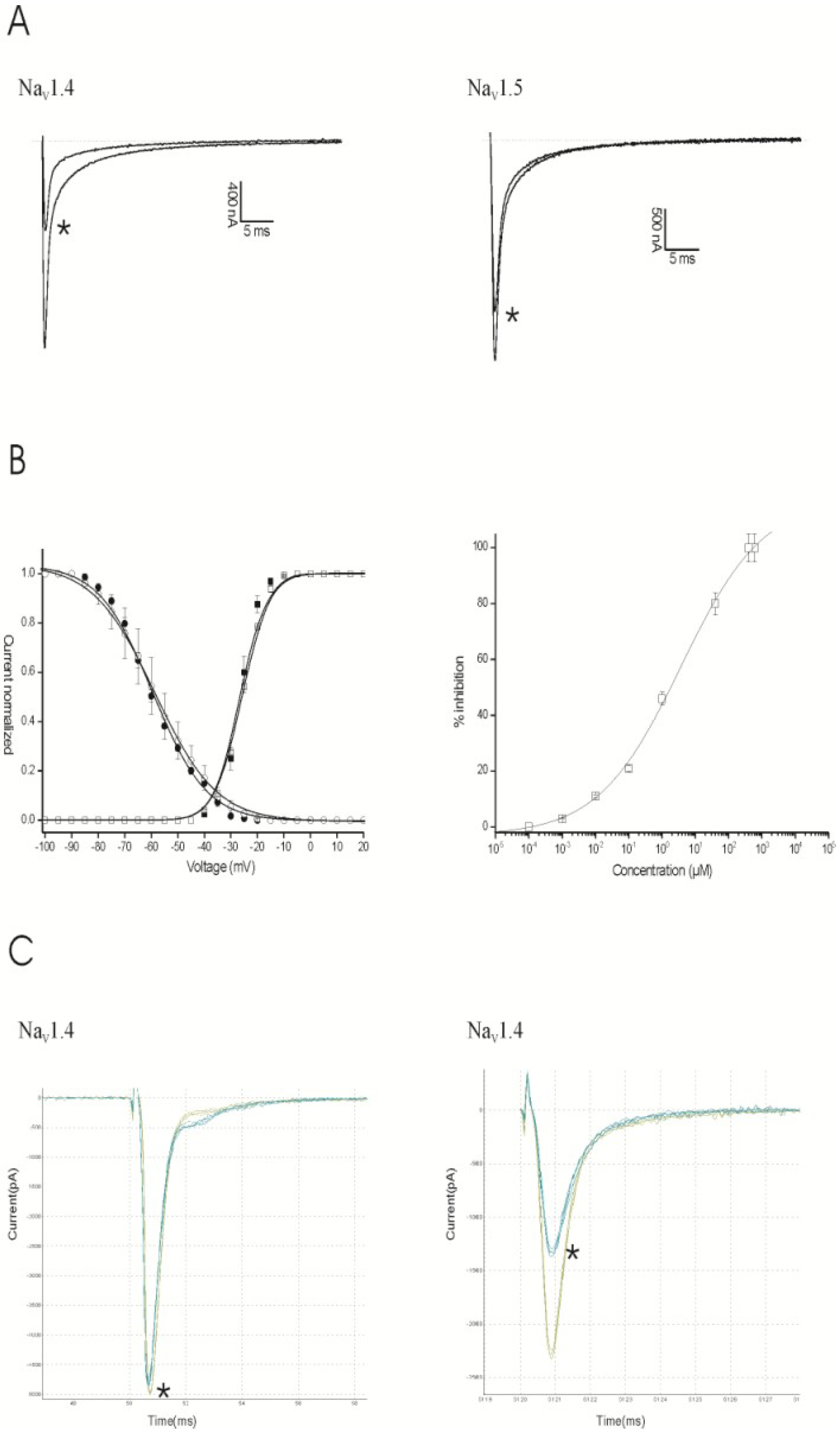
2.2. Discussion
3. Experimental Section
3.1. Compound Synthesis
3.2. Electrophysiology
3.3. Two-Electrode Voltage Clamp
3.3.1. Heterologous Expression
3.3.2. Electrophysiological Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Al-Sabi, A.; McArthur, J.; Ostroumov, V.; French, R.J. Marine toxins that target voltage-gated sodium channels. Mar. Drugs 2006, 4, 157–192. [Google Scholar] [CrossRef]
- Morales, J.J.; Rodriguez, A.D. The structure of Clathrodin, a novel alkaloid isolated from the Caribbean Sea sponge Agelas-Clathrodes. J. Nat. Prod. 1991, 54, 629–631. [Google Scholar] [CrossRef]
- Forenza, S.; Minale, L.; Riccio, R. New bromo-pyrrole derivatives from sponge Agelas-Oroides. J. Chem. Soc. Chem. Commun. 1971, 1129–1130. [Google Scholar] [CrossRef]
- Kobayashi, J.; Ohizumi, Y.; Nakamura, H.; Hirata, Y. A novel antagonist of serotonergic receptors, Hymenidin, isolated from the Okinawan marine sponge Hymeniacidon sp. Experientia 1986, 42, 1176–1177. [Google Scholar] [CrossRef]
- Rentas, A.L.R.; Rosa, R.; Rodriguez, A.D.; Demotta, G.E. Effect of alkaloid toxins from tropical marine sponges on membrane sodium currents. Toxicon 1995, 33, 491–497. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef]
- Andavan, G.S.B.; Lemmens-Gruber, R. Voltage-gated sodium channels: Mutations, channelopathies and targets. Curr. Med. Chem. 2011, 18, 377–397. [Google Scholar]
- Nardi, A.; Damann, N.; Hertrampf, T.; Kless, A. Advances in targeting voltage-gated sodium channels with small molecules. Chemmedchem 2012, 7, 1712–1740. [Google Scholar] [CrossRef]
- Vimont, A.; Travert, A.; Binet, C.; Pichon, C.; Mialane, P.; Secheresse, F.; Lavalley, J.C. Relationship between infrared spectra and stoichiometry of pyridine-H3PW12O40 salts using a new TGA-infrared coupling. J. Catal. 2006, 241, 221–224. [Google Scholar] [CrossRef]
- McCormack, K.; Santos, S.; Chapman, M.L.; Krafte, D.S.; Marron, B.E.; West, C.W.; Krambis, M.J.; Antonio, B.M.; Zellmer, S.G.; Printzenhoff, D.; et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2013, 110, E2724–E2732. [Google Scholar] [CrossRef]
- Chi, V.; Pennington, M.W.; Norton, R.S.; Tarcha, E.J.; Londono, L.M.; Sims-Fahey, B.; Upadhyay, S.K.; Lakey, J.T.; Iadonato, S.; Wulff, H.; et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012, 59, 529–546. [Google Scholar] [CrossRef]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Expert Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef]
- De la Vega, R.C.R.; Possani, L.D. Overview of scorpion toxins specific for Na+ channels and related peptides: Biodiversity, structure-function relationships and evolution. Toxicon 2005, 46, 831–844. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Sodium channel activators: Model of binding inside the pore and a possible mechanism of action. FEBS Lett. 2005, 579, 4207–4212. [Google Scholar] [CrossRef]
- Du, Y.; Garden, D.P.; Wang, L.; Zhorov, B.S.; Dong, K. Identification of new batrachotoxin-sensing residues in segment IIIS6 of the sodium channel. J. Biol. Chem. 2011, 286, 13151–13160. [Google Scholar]
- Moczydlowski, E.G. The molecular mystique of tetrodotoxin. Toxicon 2013, 63, 165–183. [Google Scholar] [CrossRef]
- Schroif-Gregoire, C.; Travert, N.; Zaparucha, A.; Al-Mourabit, A. Direct access to marine pyrrole-2-aminoimidazoles, oroidin, and derivatives, via new acyl-1,2-dihydropyridin intermediates. Org. Lett. 2006, 8, 2961–2964. [Google Scholar]
- Olofson, A.; Yakushijin, K.; Horne, D.A. Synthesis of mauritiamine. J. Org. Chem. 1997, 62, 7918–7919. [Google Scholar] [CrossRef]
- Žula, A.; Kikelj, D.; Ilaš, J.J. 2-Aminoimidazoles in medicinal chemistry. Mini Rev. Med. Chem. 2013, 13, 1921–1943. [Google Scholar] [CrossRef]
- Zidar, N.; Jakopin, Ž.; Madge, D.J.; Chan, F.; Tytgat, J.; Peigneur, S.; Sollner Dolenc, M.; Tomašić, T.; Ilaš, J.; Pweterlin Mašič, L.; et al. Ligand- and structure-based virtual screening for clathrodin-derived human voltage-gated sodium channel modulators. J. Chem. Inf. Model. 2013, 53, 3223–3232. [Google Scholar] [CrossRef]
- Liman, E.R.; Tytgat, J.; Hess, P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992, 9, 861–871. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peigneur, S.; Žula, A.; Zidar, N.; Chan-Porter, F.; Kirby, R.; Madge, D.; Ilaš, J.; Kikelj, D.; Tytgat, J. Action of Clathrodin and Analogues on Voltage-Gated Sodium Channels. Mar. Drugs 2014, 12, 2132-2143. https://doi.org/10.3390/md12042132
Peigneur S, Žula A, Zidar N, Chan-Porter F, Kirby R, Madge D, Ilaš J, Kikelj D, Tytgat J. Action of Clathrodin and Analogues on Voltage-Gated Sodium Channels. Marine Drugs. 2014; 12(4):2132-2143. https://doi.org/10.3390/md12042132
Chicago/Turabian StylePeigneur, Steve, Aleš Žula, Nace Zidar, Fiona Chan-Porter, Robert Kirby, David Madge, Janez Ilaš, Danijel Kikelj, and Jan Tytgat. 2014. "Action of Clathrodin and Analogues on Voltage-Gated Sodium Channels" Marine Drugs 12, no. 4: 2132-2143. https://doi.org/10.3390/md12042132