Insights and Ideas Garnered from Marine Metabolites for Development of Dual-Function Acetylcholinesterase and Amyloid-β Aggregation Inhibitors

Abstract
:
1. Introduction to Acetylcholinesterase Structure and Function
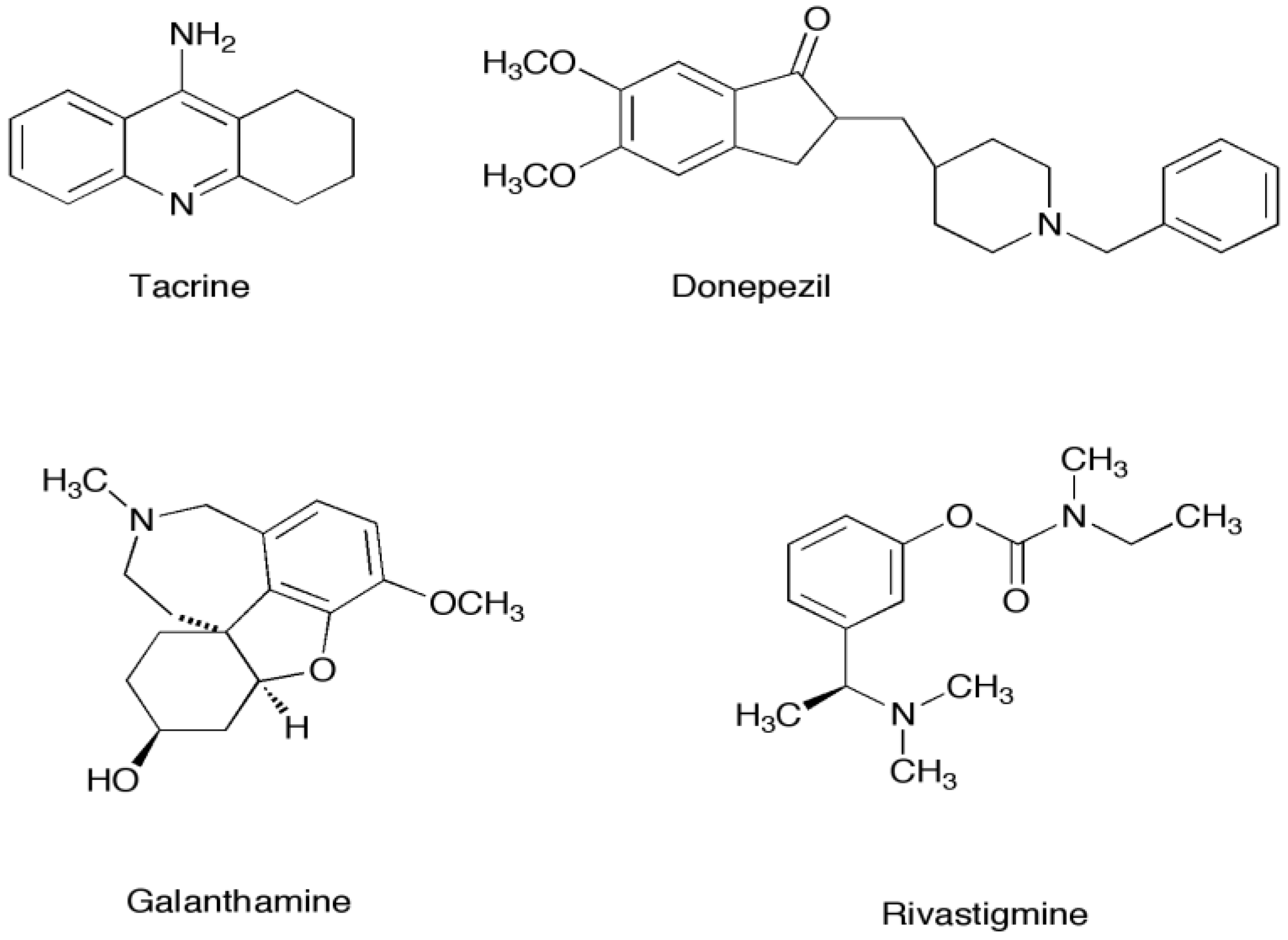
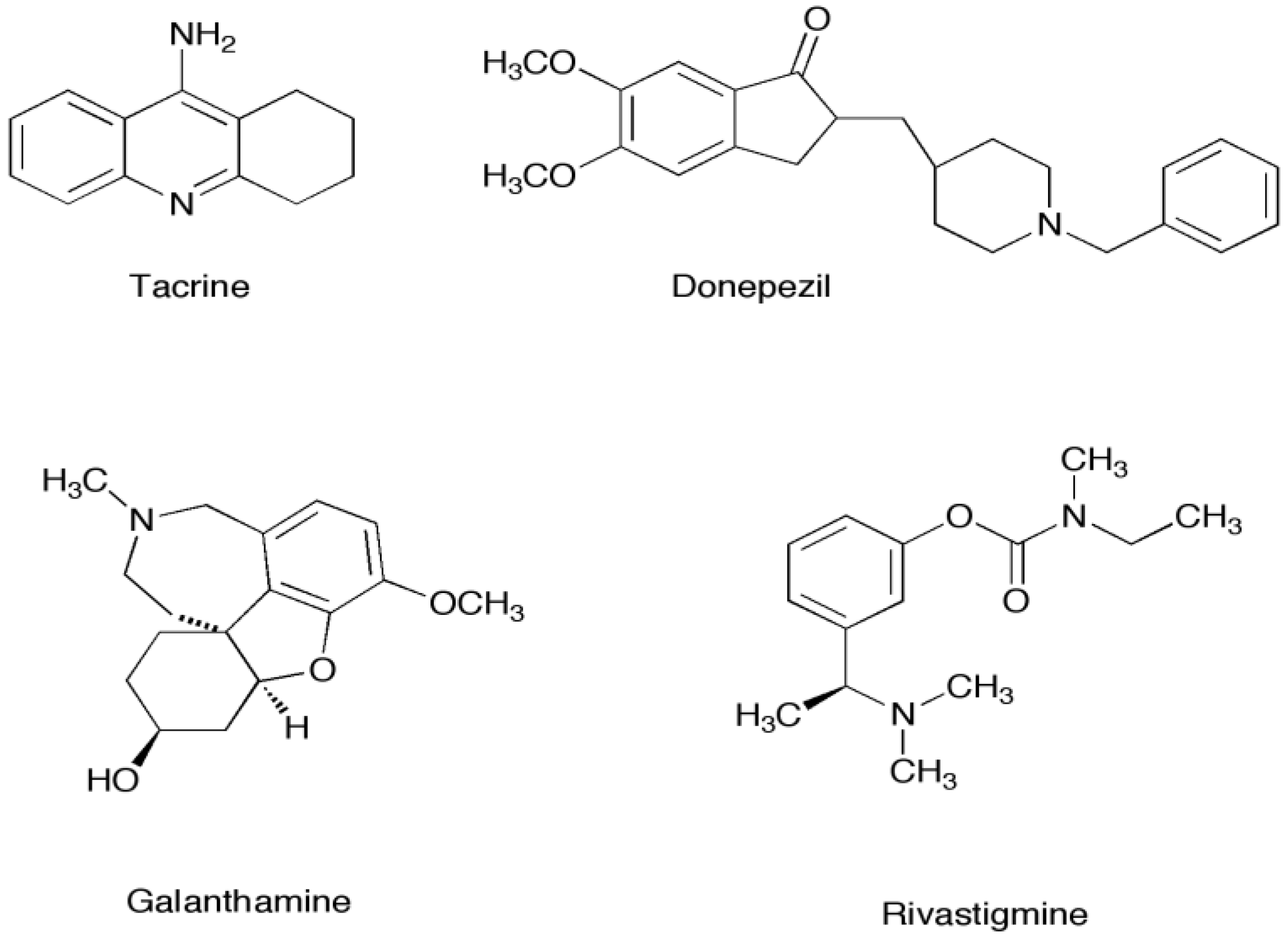
1.1. Marine Metabolites as Acetylcholinesterase Inhibitors
2. Results and Discussion
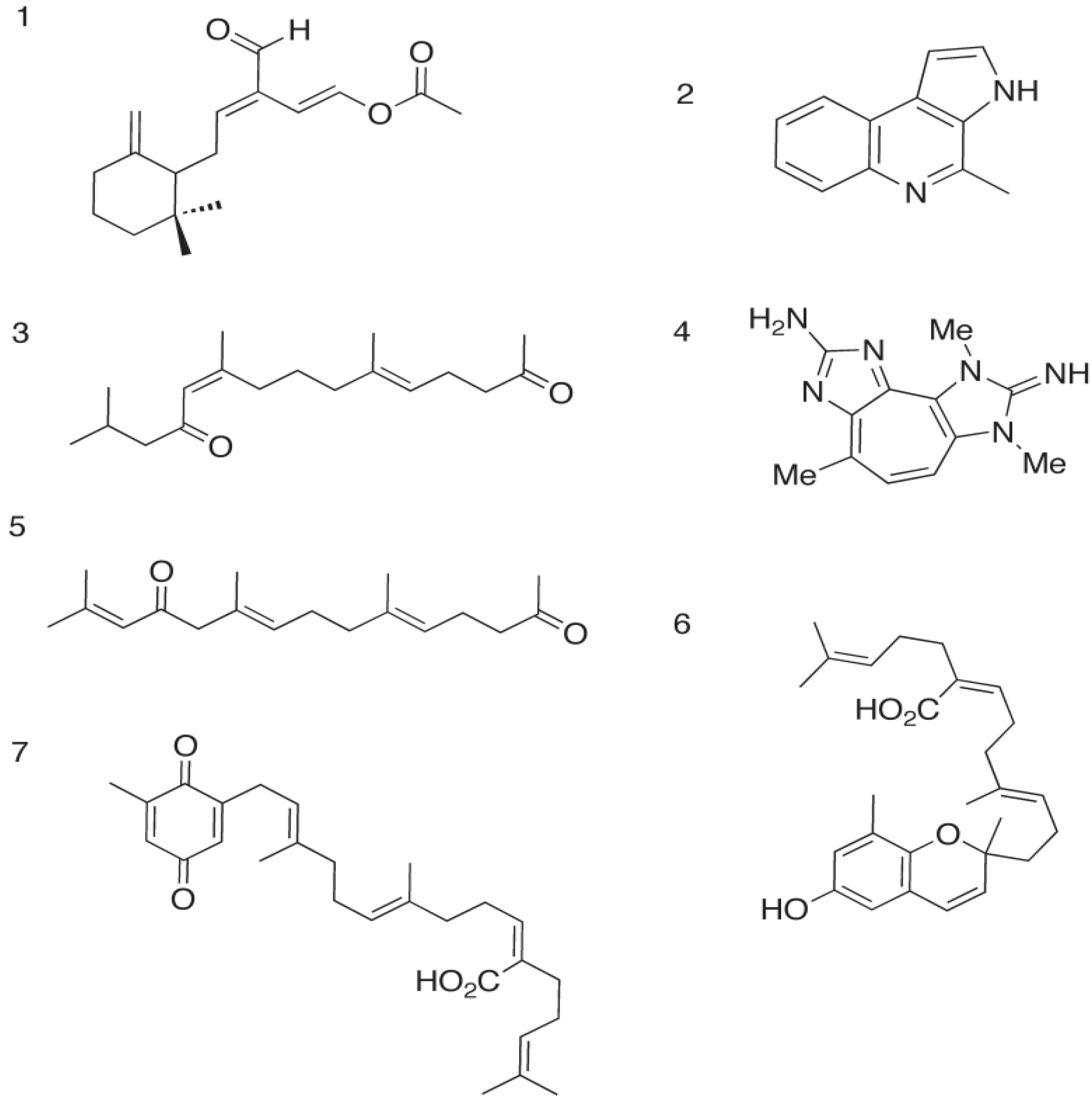
2.1. The Opistobranch Mollusk and its Metabolite, Onchidal: A Sesquiterpene Acetate
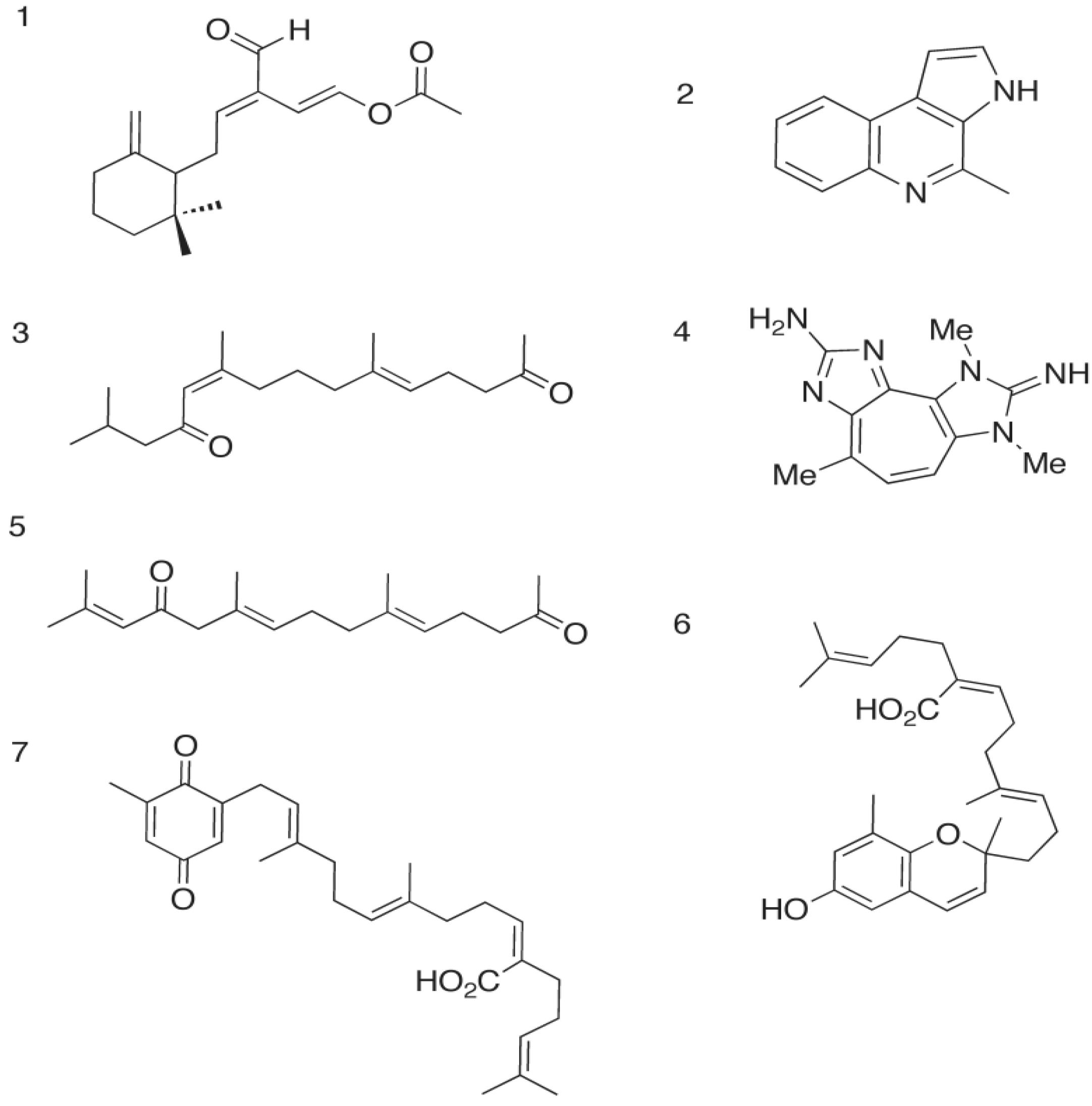
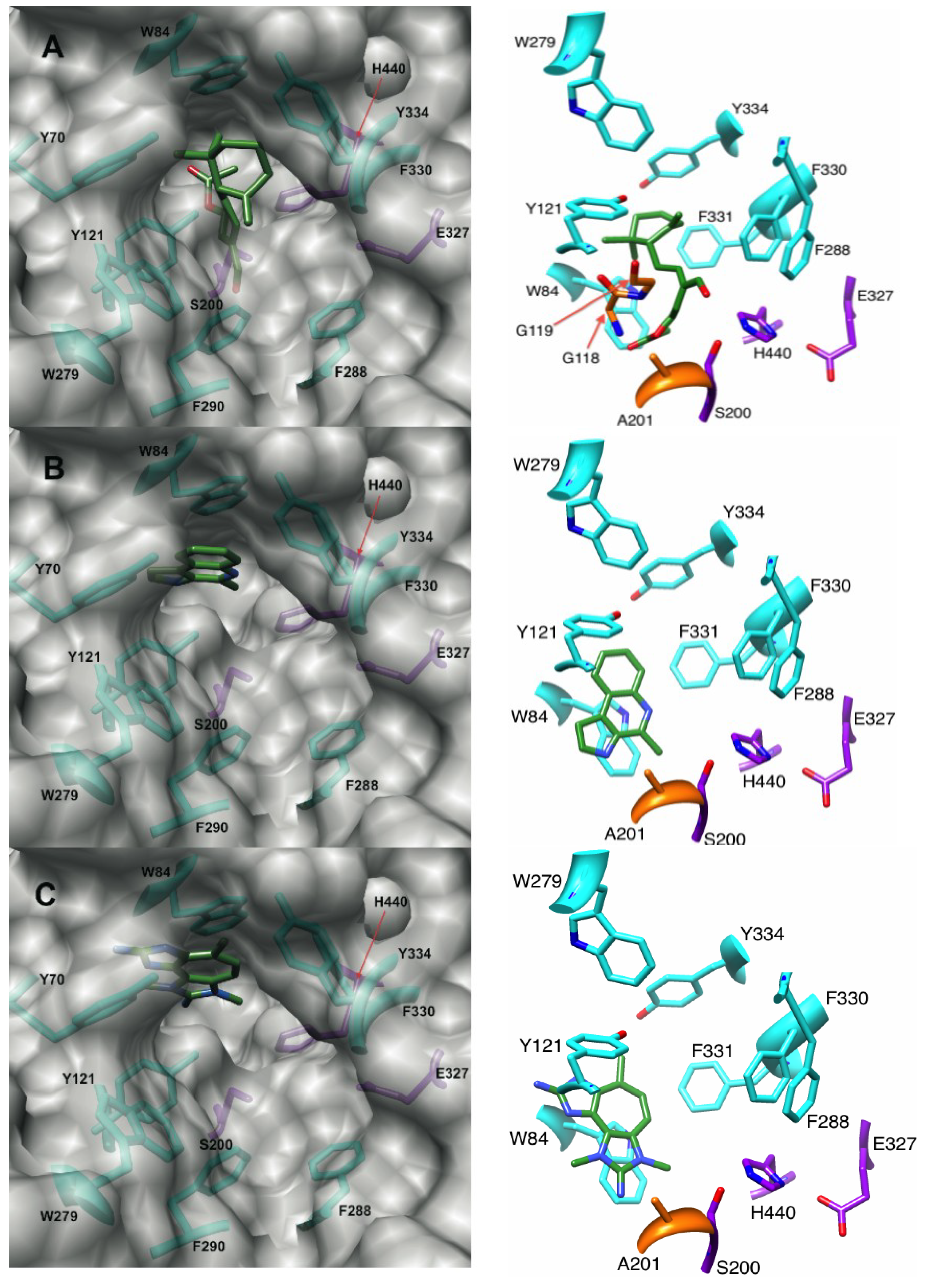
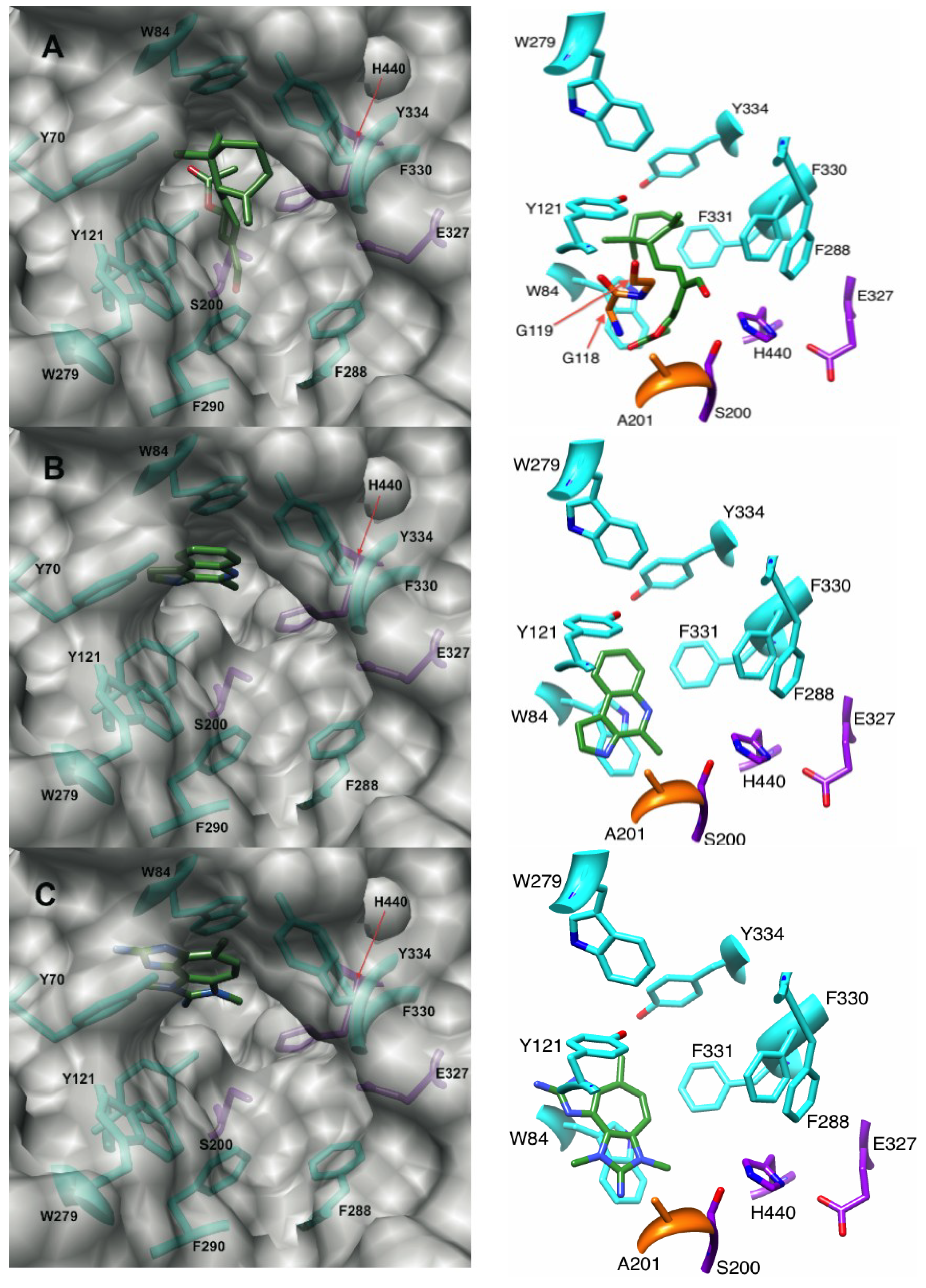
2.1.1. Docking of Onchidal into AChE
2.2. The Gliding Bacteria Rapidithrix thailandica and Its Pyrrole Metabolites
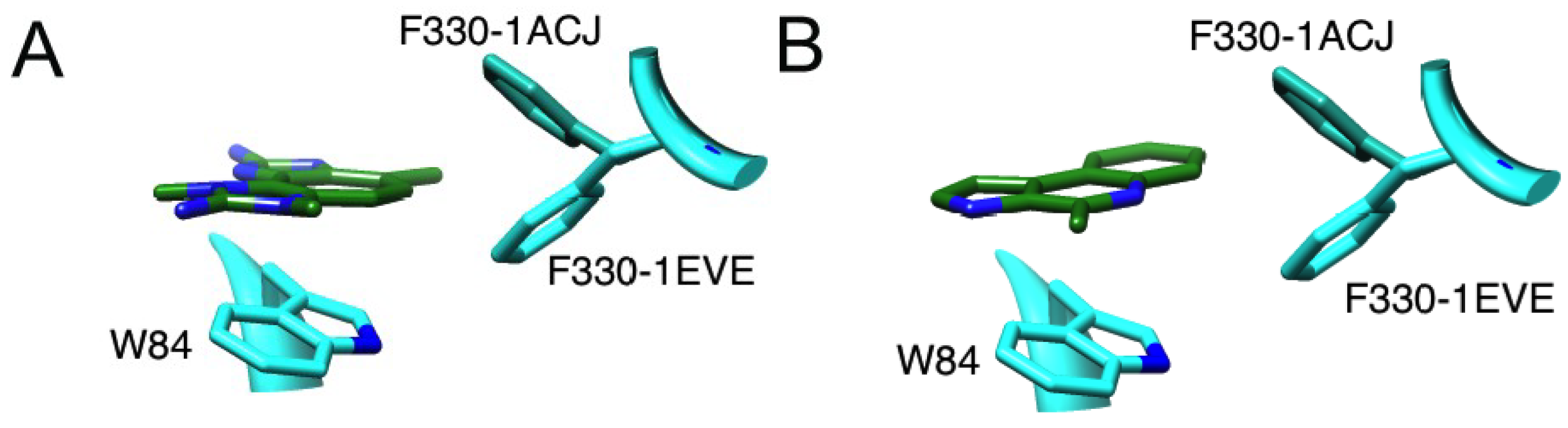
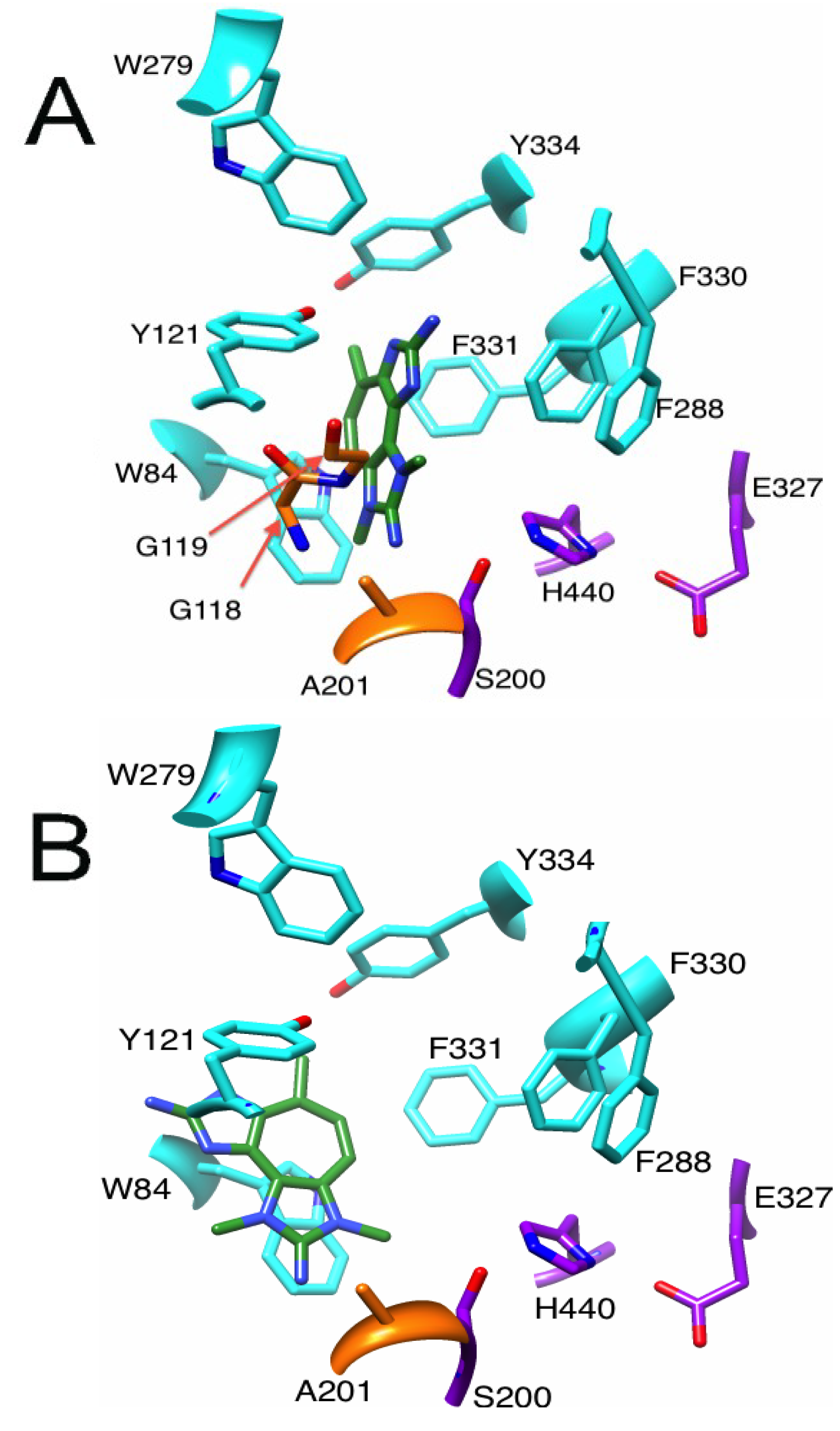
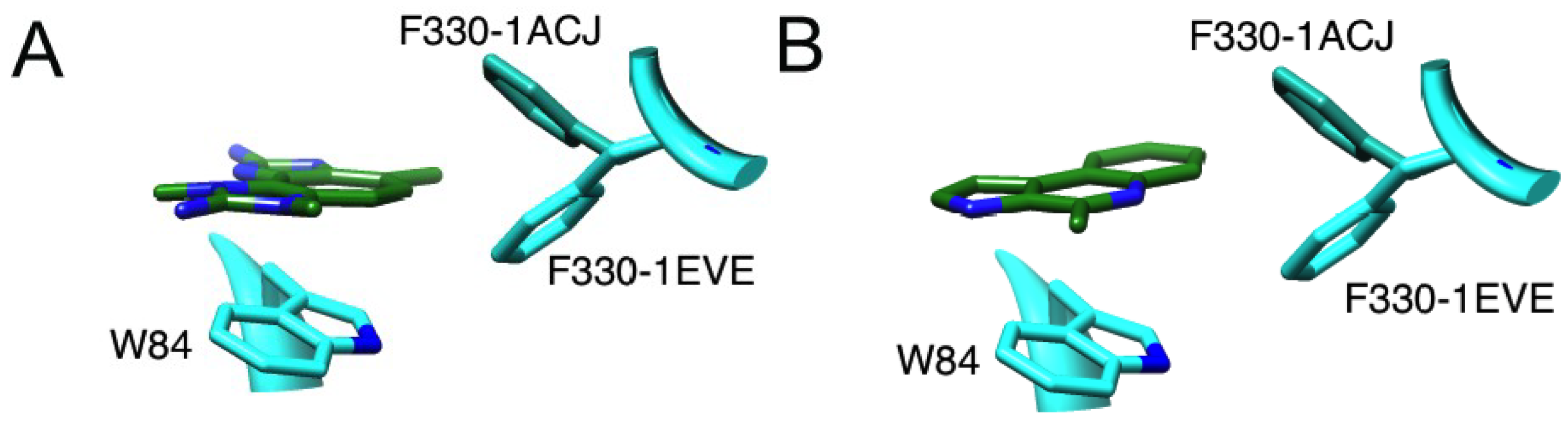
2.2.1. Docking of Marinoquinoline to AChE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | |||||
|---|---|---|---|---|---|
| Ligand | 1ACL | 1ACJ | 1DX6 | 1DX6 w/H2O | 1EVE |
| All Scores in kJ/mol | |||||
| Donepezil | −59.79 | −10.86 | −53.65 | −61.18 | −56.74 |
| Tacrine | −35.28 | −33.83 | −33.91 | −34.09 | −34.36 |
| Galanthamine | −35.28 | −35.77 | −36.47 | −38.76 | −36.38 |
| Acetylcholine | −31.78 | −34.06 | −31.99 | −31.25 | −31.61 |
| Onchidal | −47.51 | −44.28 | −48.55 | −47.37 | −46.14 |
| Marinoquinoline | −30.77 | −34.27 | −31.09 | −30.50 | −31.05 |
| Tetracyclopentazulene | −42.34 1 | −44.03 1 | −41.77 2 | −42.03 1 | −42.51 2 |
| Sargaquinoic acid | −67.92 | ND | −63.14 | −69.81 | −67.15 |
| Sargachromenol | −62.98 | −61.45 | −62.82 | −67.67 | −65.16 |
| Monooxofarnesylacetone | −59.00 | −55.34 | −57.81 | −50.22 | −57.36 |
| Dihydromonooxofarnesylacetone | −56.00 | −55.38 | −55.94 | −52.83 | −55.51 |
2.3. The Parazoanthus axinellae (O. Schmidt), Zoanthid Corals, and the Tetrazacyclopentazulene Natural Products
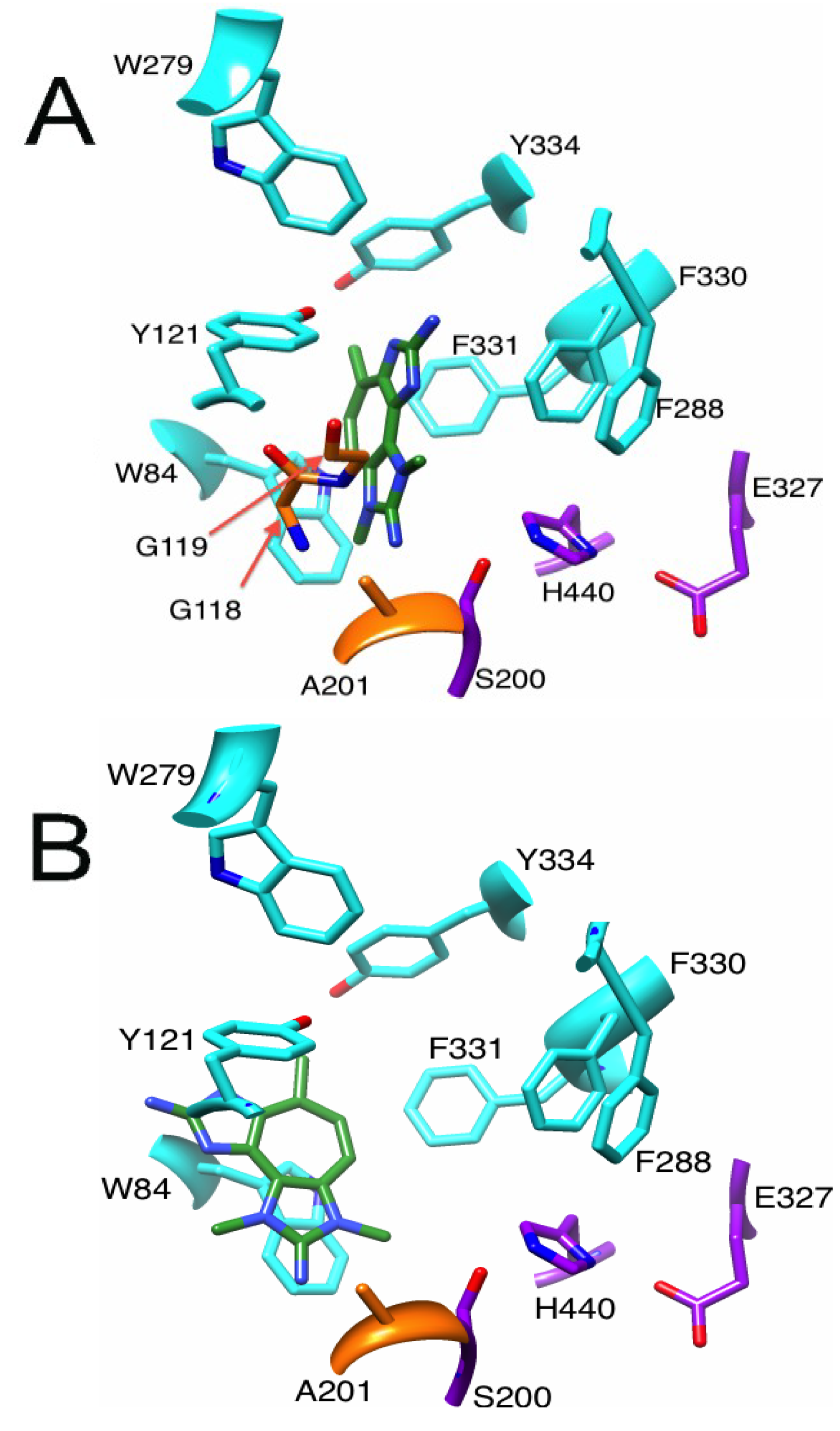
2.3.1. Binding of a Tetrazacyclopentazulene (PZT) Compound to AChE
2.4. The Brown Alga Sargassum Sagamianum and the Plastoquinones and Farnesylacetones Metabolites
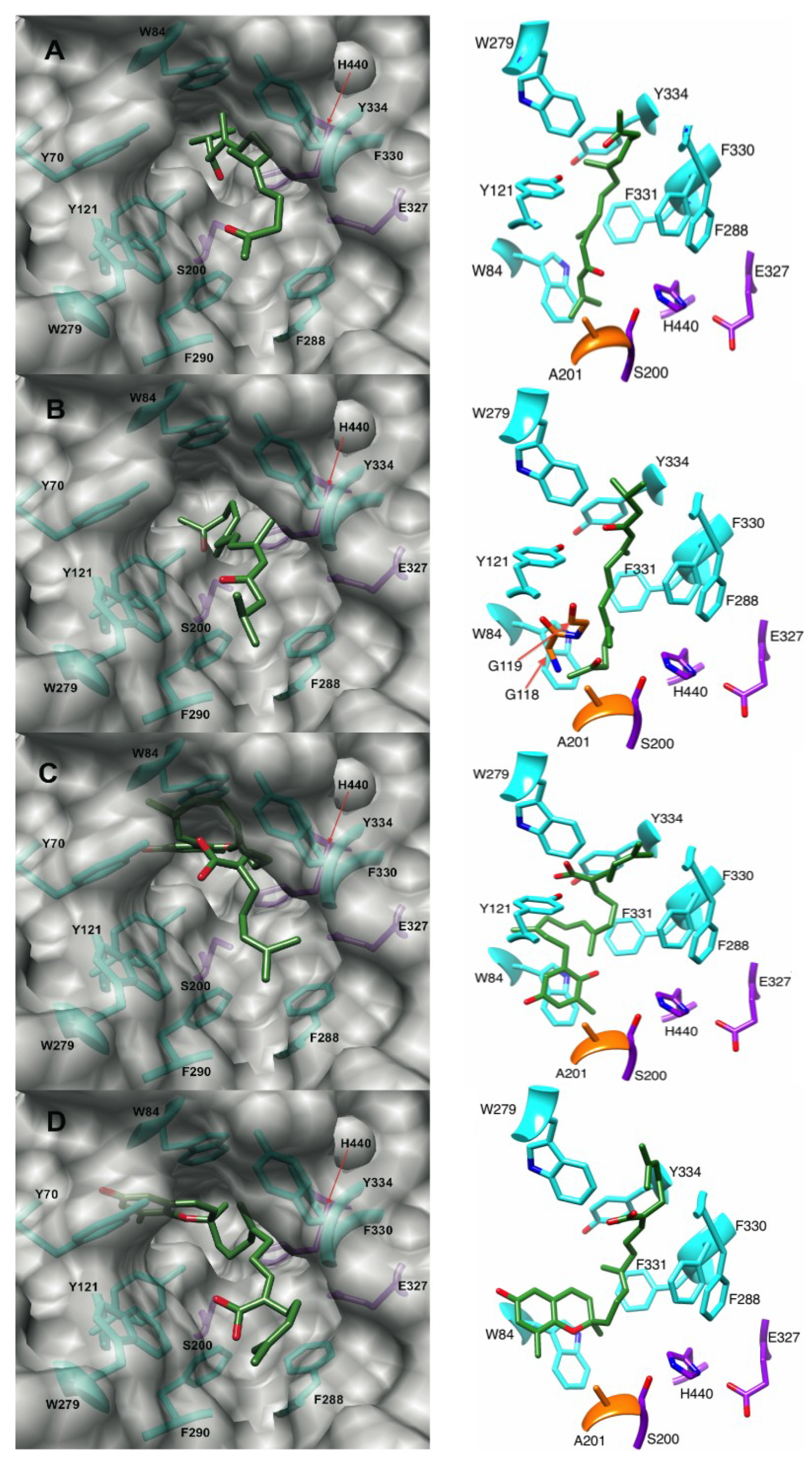
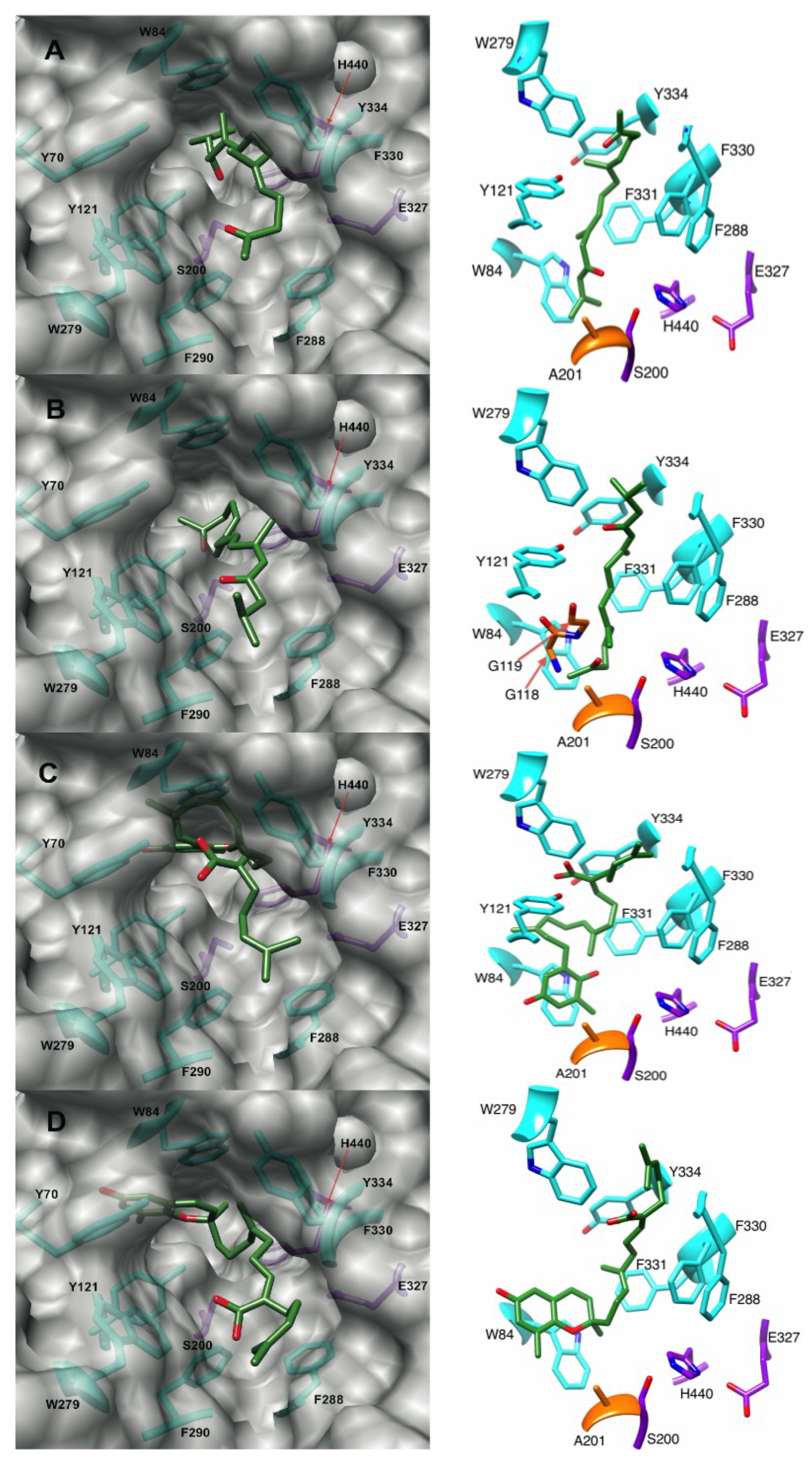
2.4.1. Docking of the Plastoquinones and Farnesylacetones into AChE
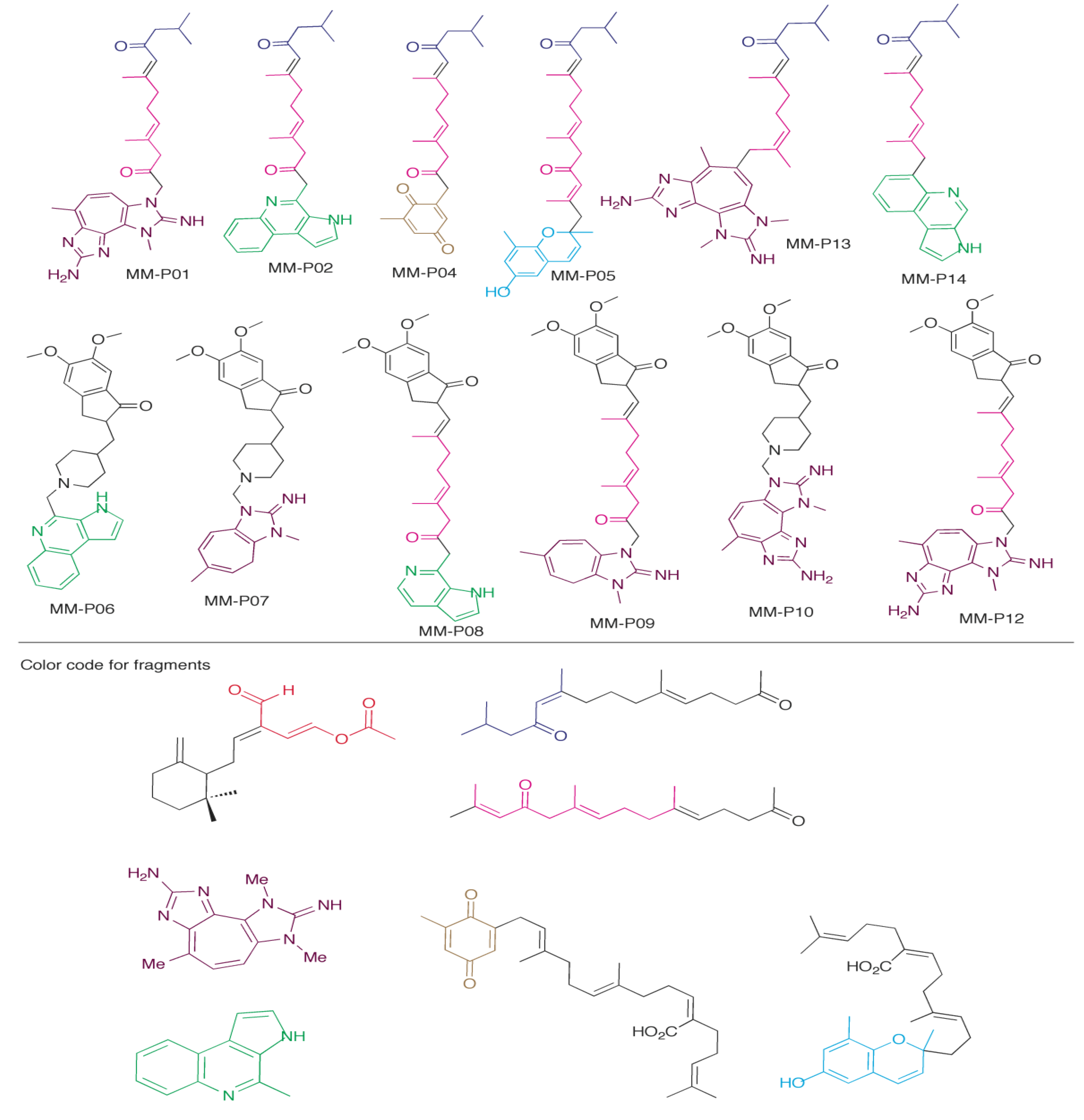
2.5. Design of New Dual AChE and Amyloid-β Aggregation Inhibitors
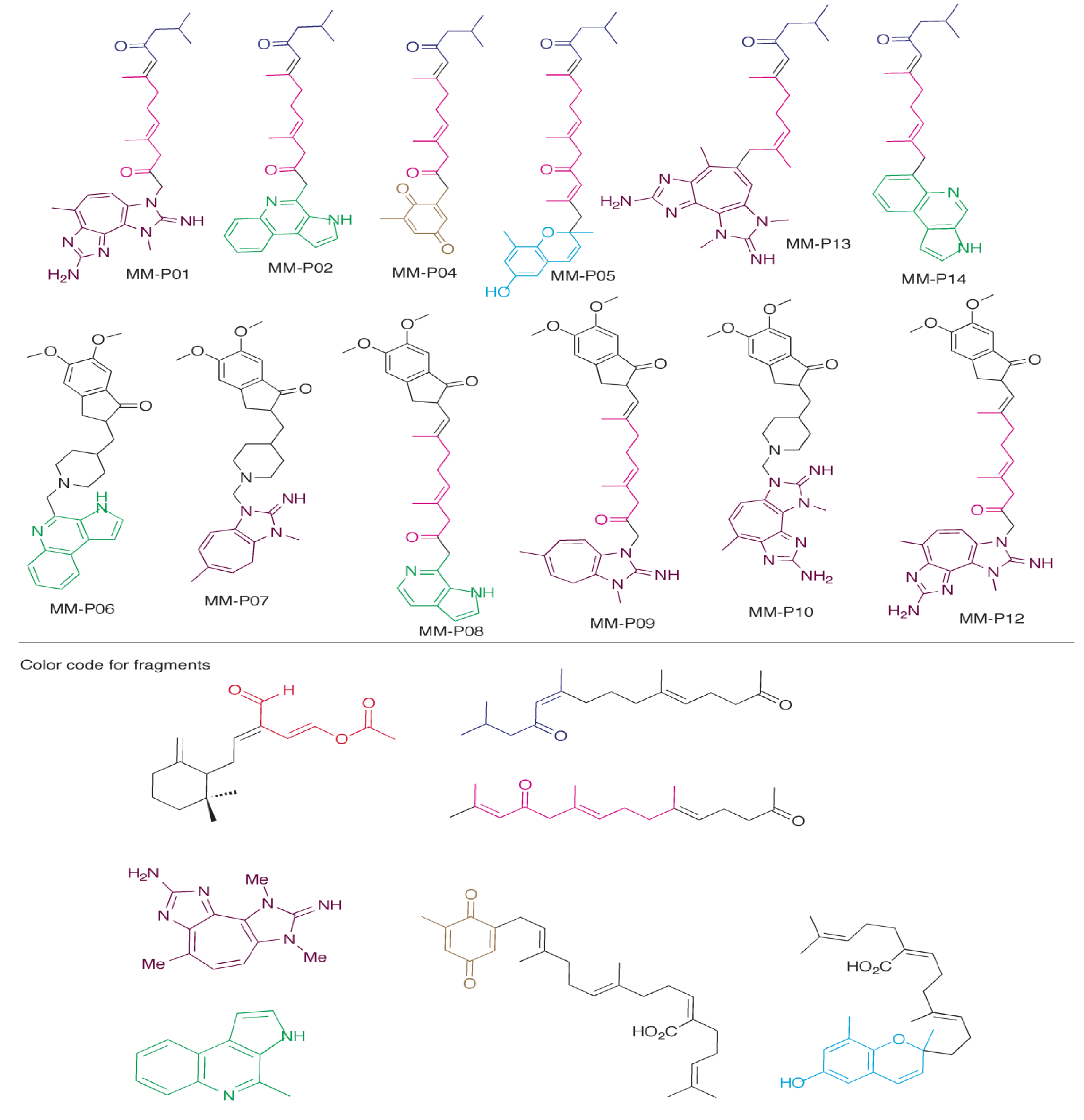
| Compound | Best Score (kJ/mol) | Receptor | Interacts with PAS | MM Scaffolds Used to Create Compound (Figure 2) | CLogP |
|---|---|---|---|---|---|
| MM-P01 | −72.93 | 1EVE | Yes | 3, 4, 5 | 4.60 |
| MM-P02 | −63.02 | 1EVE | No | 2, 3, 5 | 5.23 |
| MM-P04 | −62.62 | 1EVE | Yes | 3, 5, 7 | 3.81 |
| MM-P05 | −71.95 | 1EVE | Yes | 3, 5, 6 | 6.62 |
| MM-P06 | −62.00 | 1DX6 | Yes | 2, and donepezil | 5.01 |
| MM-P07 | −58.26 | 1DX6 | Yes | 4 and donepezil | 4.68 |
| MM-P08 | −65.76 | 1EVE | Yes | 2, 5, and donepezil | 4.78 |
| MM-P09 | −72.19 | 1EVE | Yes | 4, 5, and donepezil | 5.98 |
| MM-P10 | −64.77 | 1DX6 | Yes | 4 and donepezil | 4.24 |
| MM-P12 | −73.43 | 1EVE | No | 4, 5, and donepezil | 5.54 |
| MM-P13 | −68.90 | 1DX6 | No | 3, 4, 5 | 4.93 |
| MM-P14 | −60.78 | 1EVE | No | 2, 3, 5 | 6.24 |
3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J. The alpha/beta hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef]
- Silverman, R.B. The Organic Chemistry of Enzyme Catalyzed Reactions, revised ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Quinn, D.M. Acetylcholinesterase—Enzyme structure, reaction dynamics, and virtual transition-states. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo Californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef]
- Shafferman, A.; Velan, B.; Ordentlich, A.; Kronman, C.; Grosfeld, H.; Leitner, M.; Flashner, Y.; Cohen, S.; Barak, D.; Ariel, N. Substrate inhibition of acetylcholinesterase: Residues affecting signal transduction from the surface to the catalytic center. EMBO J. 1992, 11, 3561–3568. [Google Scholar]
- Lahiri, D.K.; Farlow, M.R.; Sambamurti, K.; Greig, N.H.; Giacobini, E.; Schneider, L.S. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer’s disease. Curr. Drug Targ. 2003, 4, 97–112. [Google Scholar] [CrossRef]
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M. Principles of Neural Science, 3rd ed.; Appleton & Lange: Norwalk, CT, USA, 1991; pp. 975–983. [Google Scholar]
- Fisher, A. Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: Perspectives and challenges in treatment of Alzheimer’s disease. J. Neurochem. 2011, 1, 22–33. [Google Scholar]
- Colombres, M.; Sagal, J.P.; Inestrosa, N.C. An overview of the current and novel drugs for Alzheimer’s disease with particular reference to anti-cholinesterase compounds. Curr. Pharm. Des. 2004, 10, 3121–3130. [Google Scholar] [CrossRef]
- Farlow, M.R. Pharmacokinetic profiles of current therapies for Alzheimer’s disease: Implications for switching to galantamine. Clin. Ther. 2001, 23, A13–A24. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar]
- Inestrosa, N.C.; Alvarez, A.; Perez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Tennis, M.K.; Brown, L.B.; Gomez-Isla, T.; Hayden, D.L.; Schoenfeld, D.A.; Walsh, K.L.; Corwin, C.; Daffner, K.R.; Friedman, P.; et al. Donepezil therapy in clinical practice: A randomized crossover study. Arch. Neurol. 2000, 57, 94–99. [Google Scholar] [CrossRef]
- Gupta, S.; Fallarero, A.; Jarvinen, P.; Karlsson, D.; Johnson, M.S.; Vuorela, P.M.; Mohan, C.G. Discovery of dual binding site acetylcholinesterase inhibitors identified by pharmacophore modeling and sequential virtual screening techniques. Bioorg. Med. Chem. Lett. 2011, 21, 1105–1112. [Google Scholar] [CrossRef]
- Castro, A.; Martinez, A. Peripheral and dual binding site acetylcholinesterase inhibitors: Implications in treatment of Alzheimer’s disease. Mini Rev. Med. Chem. 2001, 1, 267–272. [Google Scholar]
- Mohamed, T.; Zhao, X.; Habib, L.K.; Yang, J.; Rao, P.P. Design, synthesis and structure-activity relationship (SAR) studies of 2,4-disubstituted pyrimidine derivatives: Dual activity as cholinesterase and Abeta-aggregation inhibitors. Bioorg. Med. Chem. 2011, 19, 2269–2281. [Google Scholar] [CrossRef]
- Munoz-Ruiz, P.; Rubio, L.; Garcia-Palomero, E.; Dorronsoro, I.; del Monte-Millan, M.; Valenzuela, R.; Usan, P.; de Austria, C.; Bartolini, M.; Andrisano, V.; Bidon-Chanal, A.; et al. Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: New disease-modifying agents for Alzheimer’s disease. J. Med. Chem. 2005, 48, 7223–7233. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Mayer, A.M.; Rodriguez, A.D.; Berlinck, R.G.; Hamann, M.T. Marine pharmacology in 2003–4: Marine compounds with anthelmintic antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiplatelet, antiprotozoal, antituberculosis, and antiviral activities; affecting the cardiovascular, immune and nervous systems, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. 2007, 145, 553–581. [Google Scholar]
- Ireland, C.; Faulkner, D.J. The defensive secretion of the opisthobranch mollusc Onchidella binneyi. Bioorg. Chem. 1978, 7, 125–131. [Google Scholar]
- Abramson, S.N.; Radic, Z.; Manker, D.; Faulkner, D.J.; Taylor, P. Onchidal: A naturally occurring irreversible inhibitor of acetylcholinesterase with a novel mechanism of action. Mol. Pharmacol. 1989, 36, 349–354. [Google Scholar]
- Kanjana-Opas, A.; Panphon, S.; Fun, H.K. Chantrapromma S 4-methyl-3H-pyrrolo[2,3-c]quinoline. Acta Crystallogr. Sect. E 2006, 62, O2728–O2730. [Google Scholar] [CrossRef]
- Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K. Acetylcholinesterase-inhibiting activity of pyrrole derivatives from a novel marine gliding bacterium, Rapidithrix thailandica. Mar. Drugs 2008, 6, 578–586. [Google Scholar] [CrossRef]
- Turk, T.; Macek, P.; Suput, D. Inhibition of acetylcholinesterase by a pseudozoanthoxanthin-like compound isolated from the zoanthid Parazoanthus axinellae (O. Schmidt). Toxicon 1995, 33, 133–142. [Google Scholar] [CrossRef]
- Sepcic, K.; Mancini, I.; Vidic, I.; Franssanito, R.; Pietra, F.; Macek, P.; Turk, T. Antibacterial and anticholinesterase activities of aplysamine-4, a bromotyrosine-derived metabolite of a Red Sea marine sponge. J. Nat. Toxins 2001, 10, 181–191. [Google Scholar]
- Goud, T.V.; Srinivasulu, M.; Reddy, V.L.; Reddy, A.V.; Rao, T.P.; Kumar, D.S.; Murty, U.S.; Venkateswarlu, Y. Two new bromotyrosine-derived metabolites from the sponge Psammaplysilla purpurea. Chem. Pharm. Bull. 2003, 51, 990–993. [Google Scholar]
- Choi, B.W.; Ryu, G.; Park, S.H.; Kim, E.S.; Shin, J.; Roh, S.S.; Shin, H.C.; Lee, B.H. Anticholinesterase activity of plastoquinones from Sargassum sagamianum: Lead compounds for Alzheimer’s disease therapy. Phytother. Res. 2007, 21, 423–426. [Google Scholar] [CrossRef]
- Ryu, G.; Park, S.H.; Kim, E.S.; Choi, B.W.; Ryu, S.Y.; Lee, B.H. Cholinesterase inhibitory activity of two farnesylacetone derivatives from the brown alga Sargassum sagamianum. Arch. Pharm. Res. 2003, 26, 796–799. [Google Scholar] [CrossRef]
- Turk, T.; Frangez, R.; Sepcic, K. Mechanisms of toxicity of 3-alkylpyridinium polymers from marine sponge Reniera sarai. Mar. Drugs 2007, 5, 157–167. [Google Scholar] [CrossRef]
- Sepcic, K.; Marcel, V.; Klaebe, A.; Turk, T.; Suput, D.; Fournier, D. Inhibition of acetylcholinesterase by an alkylpyridinium polymer from the marine sponge, Reniera sarai. Biochim. Biophys. Acta 1998, 1387, 217–225. [Google Scholar] [CrossRef]
- Sepcic, K. Bioactive alkylpyridinium compounds from marine sponges. J. Toxicol. Toxin Rev. 2000, 19, 139–160. [Google Scholar] [CrossRef]
- Hostettmann, K.; Borloz, A.; Urbain, A.; Marston, A. Natural product inhibitors of acetylcholinesterase. Curr. Org. Chem. 2006, 10, 825–847. [Google Scholar] [CrossRef]
- Robin, G.; Chappell, K.; Stoermer, M.J.; Hu, S.H.; Young, P.R.; Fairlie, D.P.; Martin, J.L. Structure of West Nile virus NS3 protease: Ligand stabilization of the catalytic conformation. J. Mol. Biol. 2009, 385, 1568–1577. [Google Scholar] [CrossRef]
- Zhu, L.; George, S.; Schmidt, M.F.; Al-Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011, 92, 204–212. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Kryger, G.; Lewis, T.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with (−)-galanthamine at 2.3 angstrom resolution. FEBS Lett. 1999, 463, 321–326. [Google Scholar]
- Srisukchayakul, P.; Suwanachart, C.; Sangnoi, Y.; Kanjana-Opas, A.; Hosoya, S.; Yokota, A.; Arunpairojana, V. Rapidithrix thailandica gen. nov., sp. nov., a marine gliding bacterium isolated from samples collected from the Andaman sea, along the southern coastline of Thailand. Int. J. Syst. Evol. Microbiol. 2007, 57, 2275–2279. [Google Scholar] [CrossRef]
- Hosoya, S.; Arunpairojana, V.; Suwannachart, C.; Kanjana-Opas, A.; Yokota, A. Aureispira marina gen. nov., sp. nov., a gliding, arachidonic acid-containing bacterium isolated from the southern coastline of Thailand. Int. J. Syst. Evol. Microbiol. 2006, 56, 2931–2935. [Google Scholar] [CrossRef]
- Leach, A.R.; Shoichet, B.K.; Peishoff, C.E. Prediction of protein-ligand interactions. Docking and scoring: Successes and gaps. J. Med. Chem. 2006, 49, 5851–5855. [Google Scholar] [CrossRef]
- Ewing, T.J.; Makino, S.; Skillman, A.G. Kuntz ID DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stoddard, S.V.; Hamann, M.T.; Wadkins, R.M. Insights and Ideas Garnered from Marine Metabolites for Development of Dual-Function Acetylcholinesterase and Amyloid-β Aggregation Inhibitors. Mar. Drugs 2014, 12, 2114-2131. https://doi.org/10.3390/md12042114
Stoddard SV, Hamann MT, Wadkins RM. Insights and Ideas Garnered from Marine Metabolites for Development of Dual-Function Acetylcholinesterase and Amyloid-β Aggregation Inhibitors. Marine Drugs. 2014; 12(4):2114-2131. https://doi.org/10.3390/md12042114
Chicago/Turabian StyleStoddard, Shana V., Mark T. Hamann, and Randy M. Wadkins. 2014. "Insights and Ideas Garnered from Marine Metabolites for Development of Dual-Function Acetylcholinesterase and Amyloid-β Aggregation Inhibitors" Marine Drugs 12, no. 4: 2114-2131. https://doi.org/10.3390/md12042114
APA StyleStoddard, S. V., Hamann, M. T., & Wadkins, R. M. (2014). Insights and Ideas Garnered from Marine Metabolites for Development of Dual-Function Acetylcholinesterase and Amyloid-β Aggregation Inhibitors. Marine Drugs, 12(4), 2114-2131. https://doi.org/10.3390/md12042114