Spirobisnaphthalenes from the Mangrove-Derived Fungus Rhytidhysteron sp. AS21B

Abstract
:1. Introduction
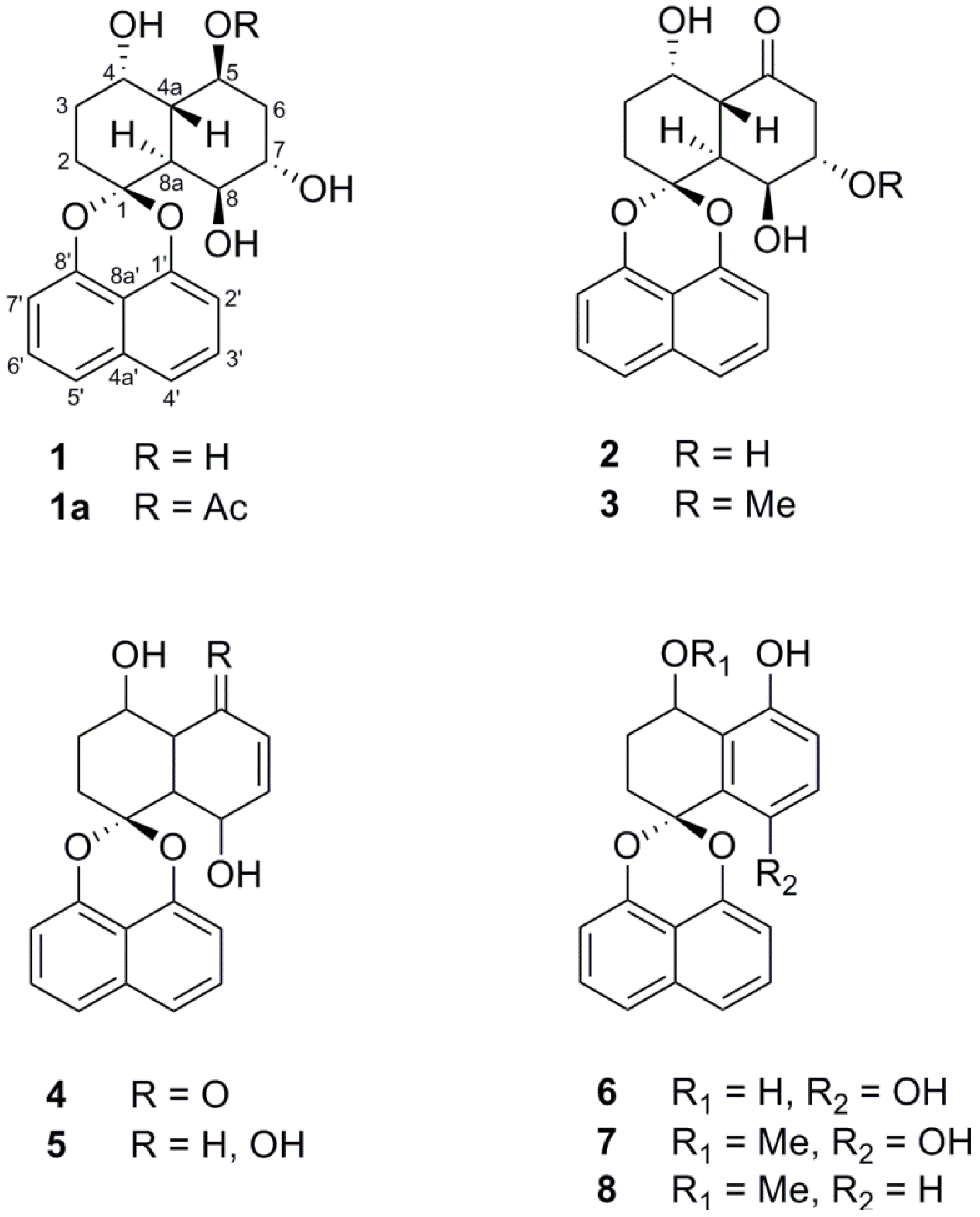
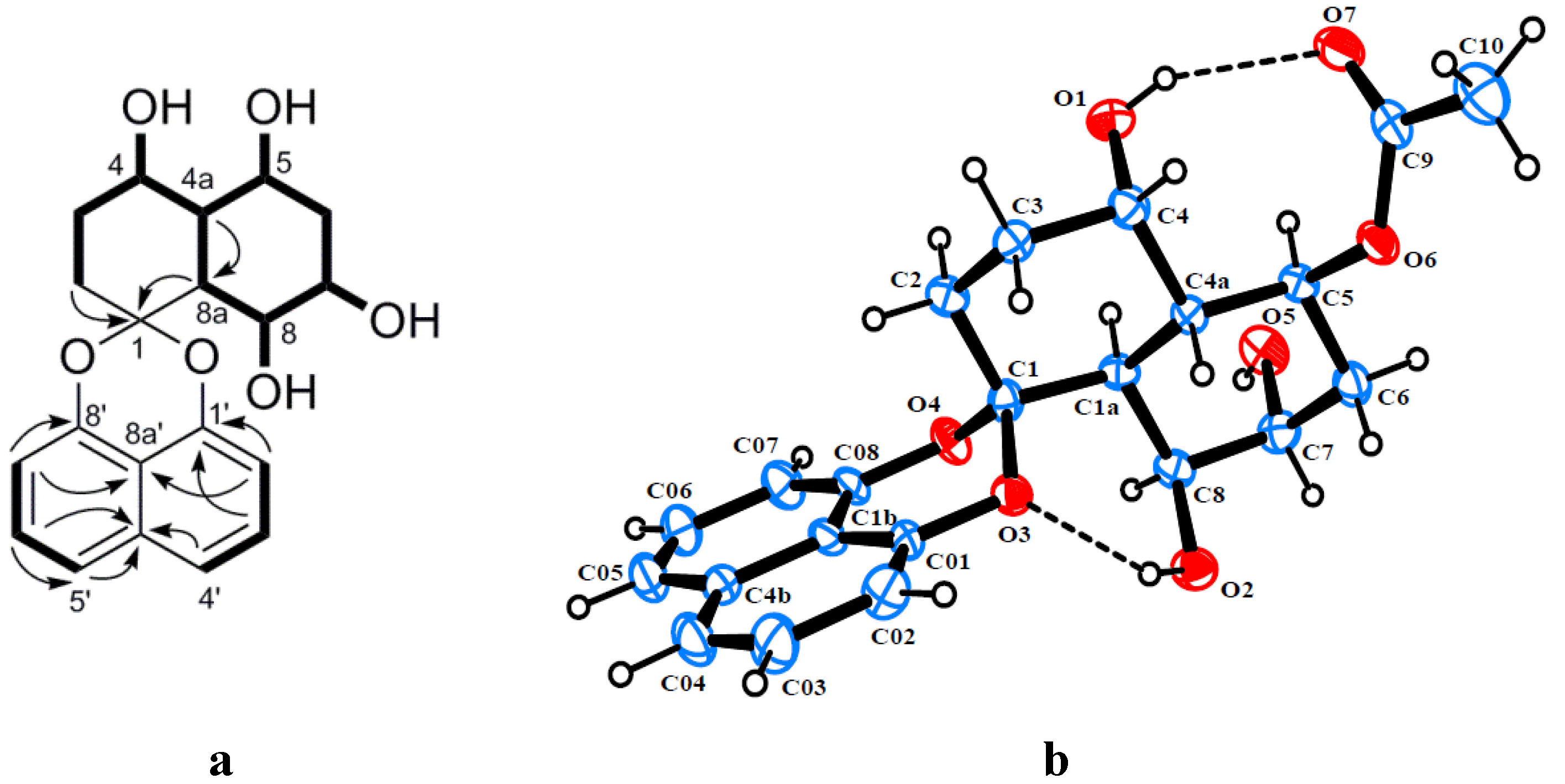
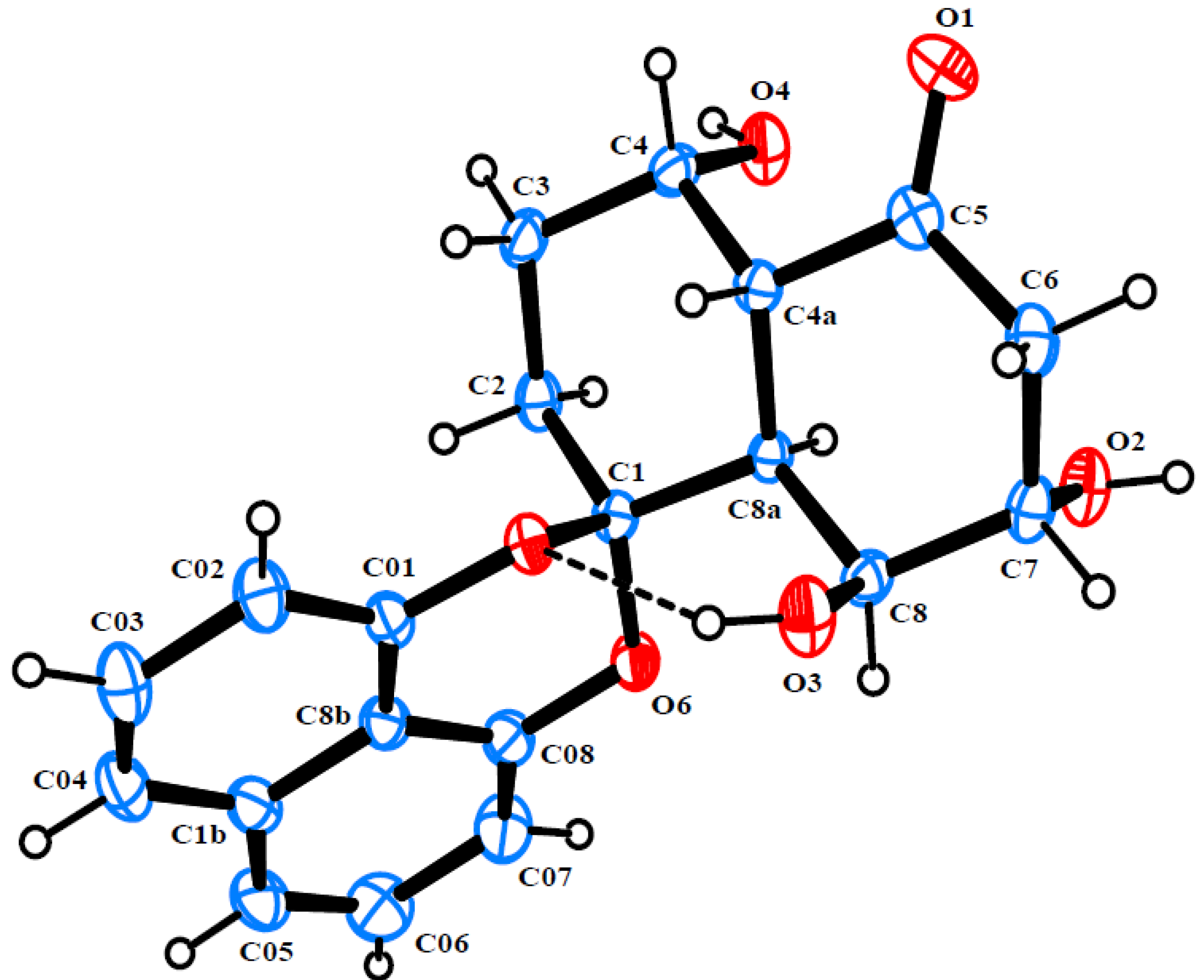
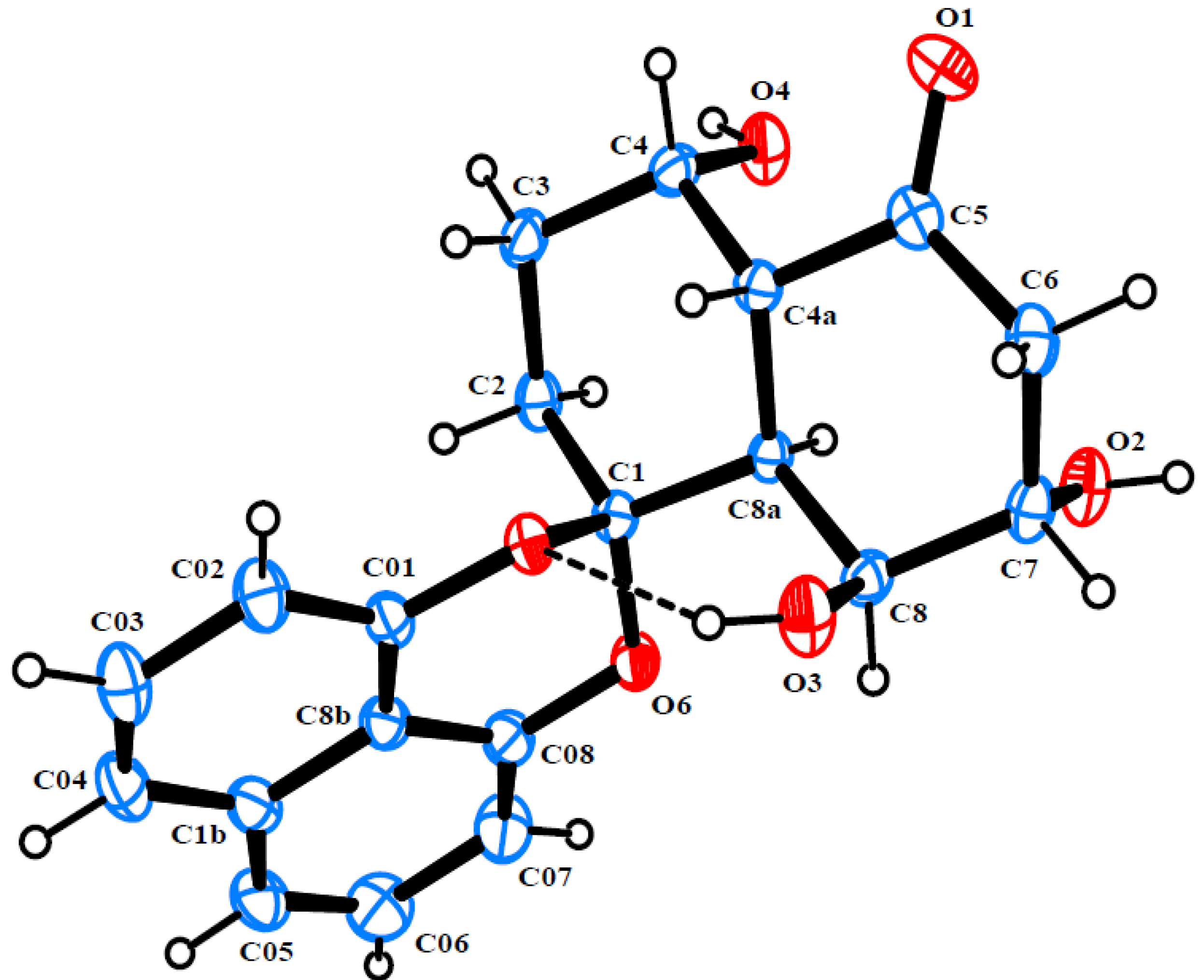
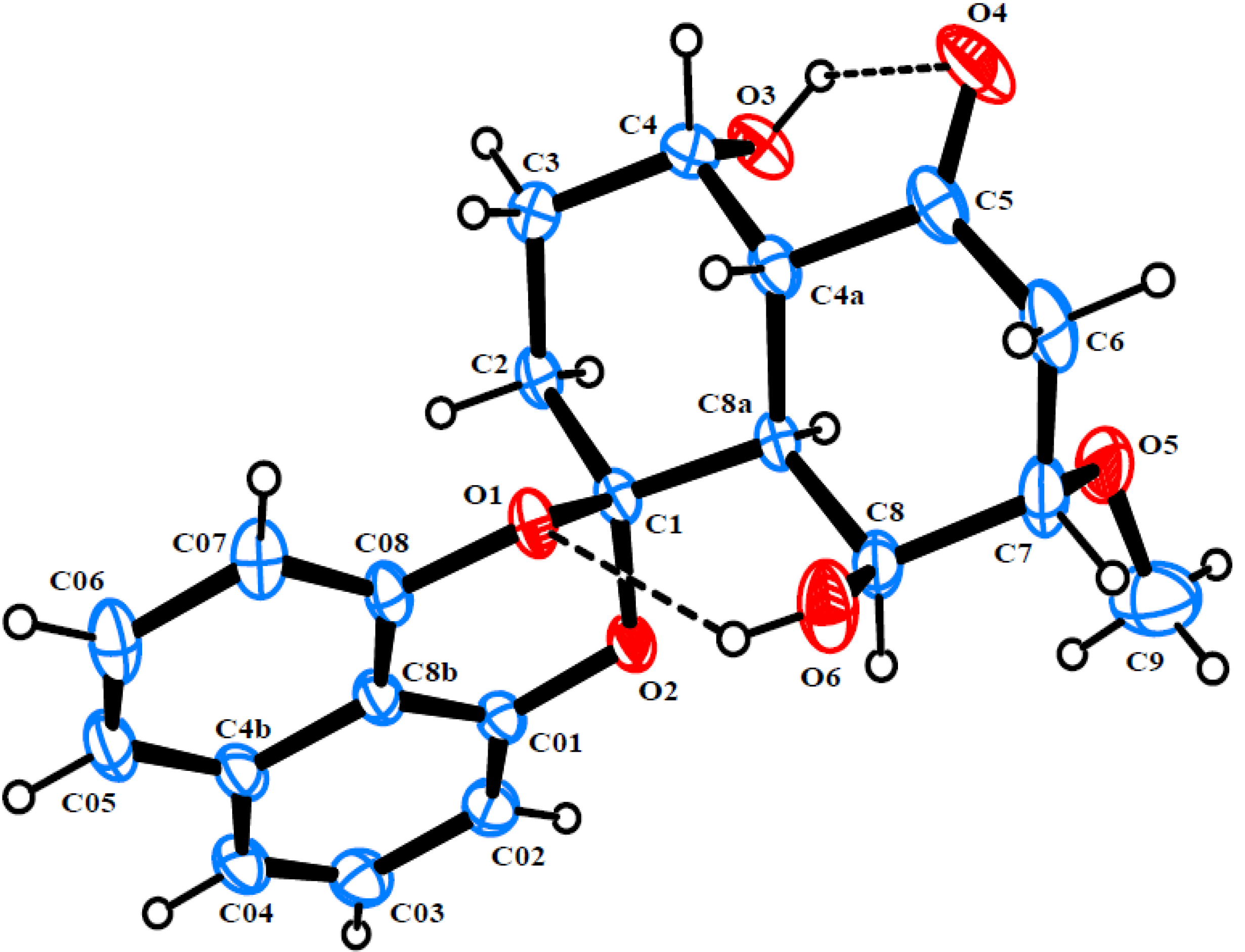
2. Results and Discussion

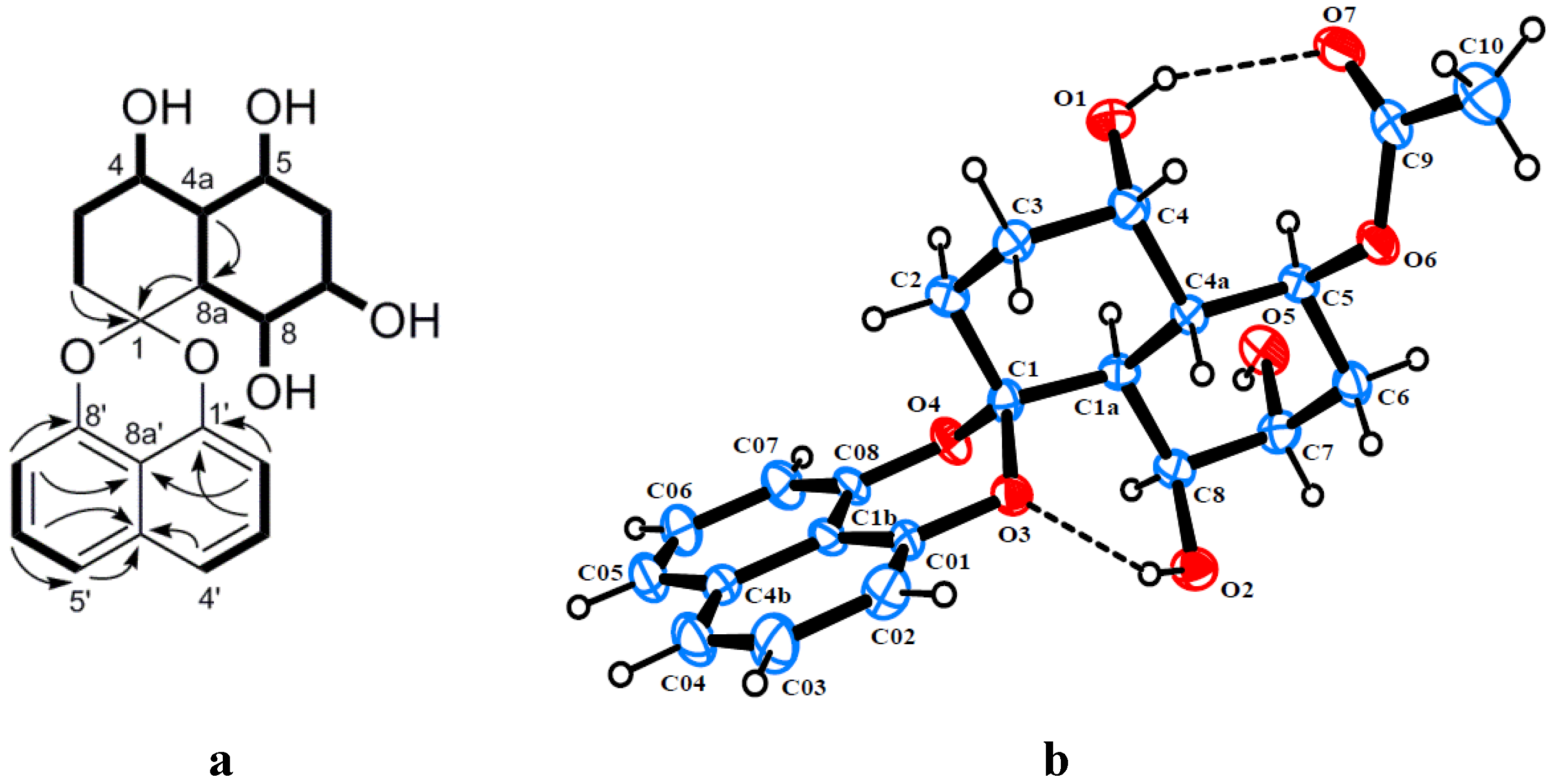

{kind=link}
{kind=link}
{kind=link}
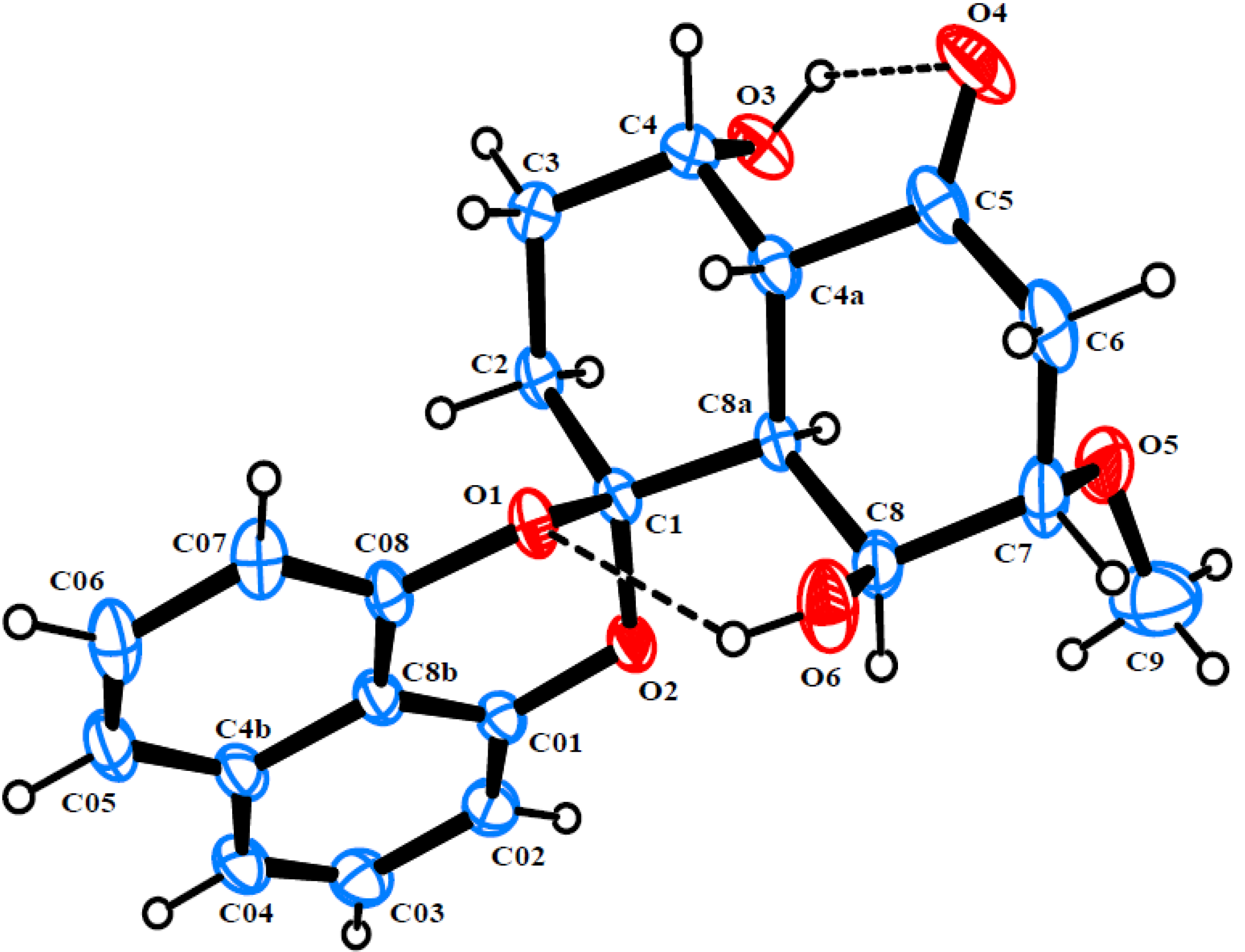{kind=link}
| No. | 1 a | 2 b | 3 b | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | - | 104.4 | - | 104.4 | - | 104.4 |
| 2 | 1.73, m | 25.6 | 1.95, m | 25.6 | 1.94, m | 25.6 |
| 3 | 1.53, m | 28.2 | 1.75, m 1.66, m | 26.5 | 1.72, m 1.62, m | 26.6 |
| 4 | 4.16, br s | 61.2 | 4.54, br s | 62.9 | 4.52, br s | 62.9 |
| 4a | 1.97, ddd (12.8, 10.0, 2.4) | 42.9 | 3.31, d (13.6) | 49.0 | 3.27, dd (13.2, 1.6) | 48.9 |
| 5 | 3.70, br s | 68.8 | - | 212.2 | - | 211.8 |
| 6 | 1.75, m | 35.7 | 3.13, br s 2.47, dd (14.4, 2.8) | 44.2 | 3.05, m 2.60, dd (14.4, 2.0) | 41.2 |
| 7 | 4.21, br s | 66.9 | 4.43, t (3.2) | 71.3 | 3.89, m | 80.1 |
| 8 | 3.93, m | 63.3 | 4.77, d (3.2) | 67.4 | 4.88, br d (3.6) | 65.1 |
| 8a | 2.44, dd (12.8, 1.6) | 38.6 | 3.17, br s | 41.5 | 3.03, m | 41.9 |
| 1' | - | 146.5 | - | 147.3 | - | - |
| 2' | 6.96, d (7.2) | 109.4 | 6.94, d (7.2) | 109.8 | 6.96, d (7.6) | 109.8 |
| 3' | 7.45, t (7.6) | 127.6 | 7.43, t (8.0) | 127.8 | 7.44, t (7.6) | 127.7 |
| 4' | 7.50, d (8.0) | 119.7 | 7.53, d (8.4) | 121.5 | 7.48, d (8.4) | 120.5 |
| 4a' | - | 133.6 | - | 134.2 | - | 134.2 |
| 5' | 7.52, d (8.0) | 120.1 | 7.49, d (8.4) | 120.5 | 7.53, d (8.4) | 121.4 |
| 6' | 7.45, t (7.6) | 127.5 | 7.43, t (8.0) | 127.1 | 7.42, t (7.6) | 127.1 |
| 7' | 6.94, d (7.2) | 108.8 | 6.95, d (7.2) | 109.6 | 6.93, d (7.6) | 109.6 |
| 8' | - | 147.6 | - | 145.9 | - | 145.9 |
| 8a' | - | 113.3 | - | 113.8 | - | 113.9 |
| 4-OH | 3.70, br s | - | - | - | - | - |
| 5-OH | 4.81, d (2.8) | - | - | - | - | - |
| 7-OH | 3.85, d (2.4) | - | - | - | - | - |
| 8-OH | 4.21, br s | - | 3.77, s | - | 3.71, s | - |
| 7-OMe | - | - | - | - | 3.43, s | 56.9 |
| Compound | IC50 (μM) | |
|---|---|---|
| MCF-7 | CaSki | |
| 2 | − a | 22.81 ± 1.33 |
| 3 | 17.30 ± 2.11 | 24.44 ± 0.22 |
| 4 | 20.10 ± 1.52 | 25.59 ± 1.70 |
| 5 | 14.47 ± 0.51 | 21.95 ± 2.56 |
| Doxorubicin | 0.06 ± 0.01 | 0.20 ± 0.02 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material and Fermentation
3.3. Extraction and Isolation
3.4. Cytotoxicity Assay
4. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Jiao, P.; Swenson, D.C.; Gloer, J.B.; Campbell, J.; Shearer, C.A. Decaspirones A‒E, bioactive spirodioxynaphthalenes from the freshwater aquatic fungus Decaisnella thyridioides. J. Nat. Prod. 2006, 69, 1667–1671. [Google Scholar] [CrossRef]
- Hu, H.; Guo, H.; Li, E.; Zhou, Y.; Che, Y. Decaspirones F‒I, bioactive secondary metabolites from the saprophytic fungus Helicoma viridis. J. Nat. Prod. 2006, 69, 1672–1675. [Google Scholar] [CrossRef]
- Macías-Rubalcava, M.L.; Hernández-Bautista, B.E.; Jiménez-Estrada, M.; González, M.C.; Glenn, A.E.; Hanlin, R.T.; Hernández-Ortega, S.; Saucedo-García, A.; Muria-González, J.M.; Anaya, A.L. Naphthaquinone spiroketal with allelochemical activity from the newly discovered endophytic fungus Edenia gomezpompae. Phytochemistry 2008, 69, 1185–1196. [Google Scholar]
- Matínez-Luis, S.; Della-Togna, G.; Coley, P.D.; Kursar, T.A.; Gerwick, W.H.; Cubilla-Rios, L. Antileishmanial constituents of the Panamanian endophytic fungus Edenia sp. J. Nat. Prod. 2008, 71, 2011–2014. [Google Scholar]
- Van der Sar, S.A.; Blunt, J.W.; Munro, M.H.G. Spiro-Mamakone A, a unique relative of the spironaphthalene class of compounds. Org. Lett. 2006, 8, 2059–2061. [Google Scholar]
- Chen, X.; Shi, Q.; Lin, G.; Guo, S.; Yang, J. Spirobisnaphthalene analogues from the endophytic fungus Preussia sp. J. Nat. Prod. 2009, 72, 1712–1715. [Google Scholar]
- Cai, Y.-S.; Guo, Y.-W.; Krohn, K. Structure, bioactivities, biosynthetic relationships and chemical synthesis of the spirodioxynaphthalenes. Nat. Prod. Rep. 2010, 27, 1840–1870. [Google Scholar]
- Zhang, H.W.; Song, Y.C.; Tan, R.X. Biology and chemistry of endophytes. Nat. Prod. Res. 2006, 23, 753–771. [Google Scholar]
- Gunatilaka, A.A.L. Natural products from plant-associated microorganisms: Distribution, structural diversity, bioactivity and implications of their occurrence. J. Nat. Prod. 2006, 69, 509–526. [Google Scholar] [CrossRef]
- Strobel, G.; Diasy, B.; Castillo, U.; Harper, J. Natural products form endophytic microorganisms. J. Nat. Prod. 2004, 67, 257–268. [Google Scholar]
- Strobel, G.A. Endophytes as sources of bioactive products. Microbes Infect. 2003, 5, 535–544. [Google Scholar]
- Debbab, A.; Aly, A.H.; Proksch, P. Mangrove derived fungal endophytes—A chemical and biological perception. Fungal Divers. 2013, 61, 1–27. [Google Scholar] [CrossRef]
- Sakemi, S.; Inagaki, T.; Kaneda, K.; Hirai, H.; Iwata, E.; Sakakibara, T.; Yamauchi, Y.; Norcia, M.; Wondrack, L.M.; Sutcliffe, J.A.; Kojima, N. CJ-12,371 and CJ-12,372, two novel DNA gyrase inhibitors, fermentation, isolation, structural elucidation and biological activities. J. Antibiot. 1995, 48, 134–142. [Google Scholar]
- Wipf, P.; Lynch, S.M.; Birmingham, A.; Tamayo, G.; Jiménez, A.; Campos, N.; Powis, G. Natural product based inhibitors of the thioredoxin-thioredoxin reductase system. Org. Biomol. Chem. 2004, 2, 1651–1658. [Google Scholar]
- Ragot, J.P.; Steeneck, C.; Alcaraz, M.-L.; Taylor, R.J.K. The synthesis of 1,8-dihydroxynaphthalene-derived natural products: Palmarymycin CP1, Palmarymycin CP2, Palmarymycin CP11, CJ-12,371, deoxypreussomerin A and novel analogues. J. Chem. Soc. Perkin Trans. 1 1999, 1073–1082. [Google Scholar] [CrossRef]
- Ohishi, H.; Chiba, N.; Mikawa, T.; Sakaki, T.; Miyaji, S.; Sezaki, M. Novel Antibiotic MK3018 substance— Useful as antimicrobial agent. Jpn. Pat. 01294686, 28 November 1989. [Google Scholar]
- Chokpaiboon, S.; Sommit, D.; Teerawatananond, T.; Muangsin, N.; Bunyapaiboonsri, T.; Pudhom, K. Cytotoxic nor-chamigrane and chamigrane endoperoxides from a basidiomycetous fungus. J. Nat. Prod. 2010, 73, 1005–1007. [Google Scholar]
- Sheldrick, G.M. SHELXS-97, Program for Crystal Structure Solution, University of Göttingen: Göttingen, Germany, 1997.
- Sheldrick, G.M. SHELXS-97, Program for Crystal Structure Refinement, University of Göttingen: Göttingen, Germany, 1997.
- Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 24 February 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pudhom, K.; Teerawatananond, T.; Chookpaiboon, S. Spirobisnaphthalenes from the Mangrove-Derived Fungus Rhytidhysteron sp. AS21B. Mar. Drugs 2014, 12, 1271-1280. https://doi.org/10.3390/md12031271
Pudhom K, Teerawatananond T, Chookpaiboon S. Spirobisnaphthalenes from the Mangrove-Derived Fungus Rhytidhysteron sp. AS21B. Marine Drugs. 2014; 12(3):1271-1280. https://doi.org/10.3390/md12031271
Chicago/Turabian StylePudhom, Khanitha, Thapong Teerawatananond, and Supichar Chookpaiboon. 2014. "Spirobisnaphthalenes from the Mangrove-Derived Fungus Rhytidhysteron sp. AS21B" Marine Drugs 12, no. 3: 1271-1280. https://doi.org/10.3390/md12031271
APA StylePudhom, K., Teerawatananond, T., & Chookpaiboon, S. (2014). Spirobisnaphthalenes from the Mangrove-Derived Fungus Rhytidhysteron sp. AS21B. Marine Drugs, 12(3), 1271-1280. https://doi.org/10.3390/md12031271