A Chemoinformatics Approach to the Discovery of Lead-Like Molecules from Marine and Microbial Sources En Route to Antitumor and Antibiotic Drugs



Abstract
:1. Introduction
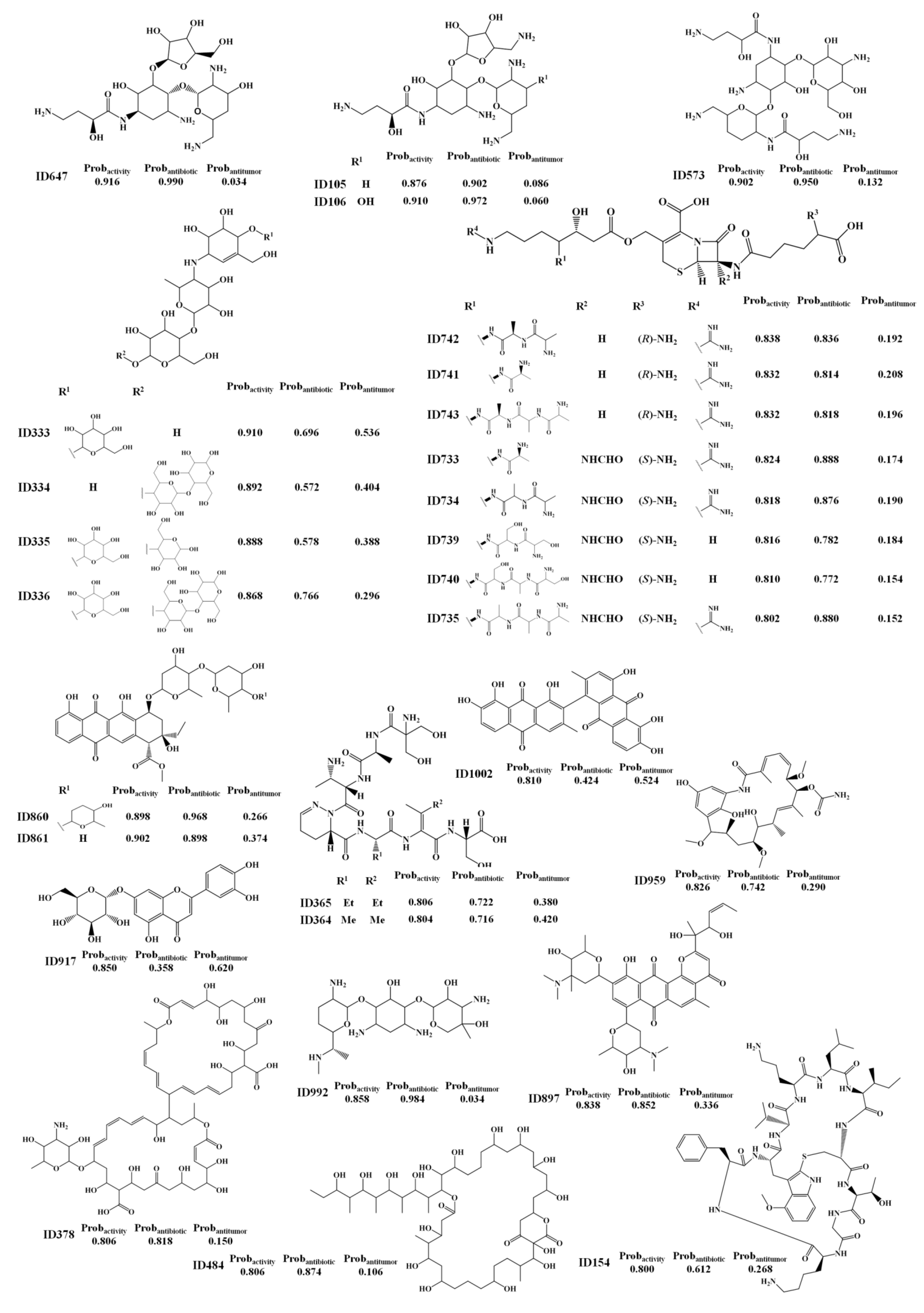
2. Results and Discussion
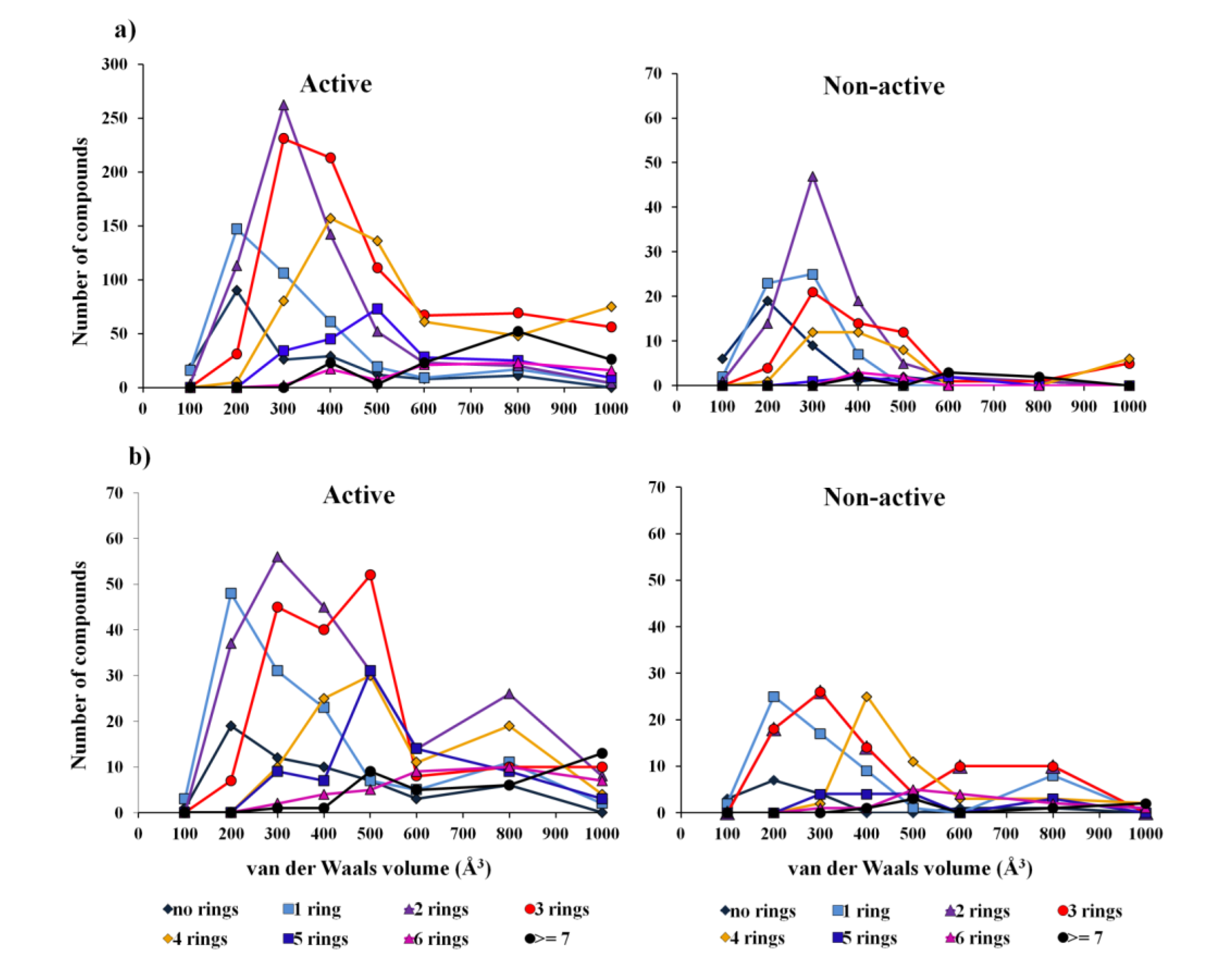
2.1. Establishment of Quantitative Structure–Activity Relationship (QSAR) Classification Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
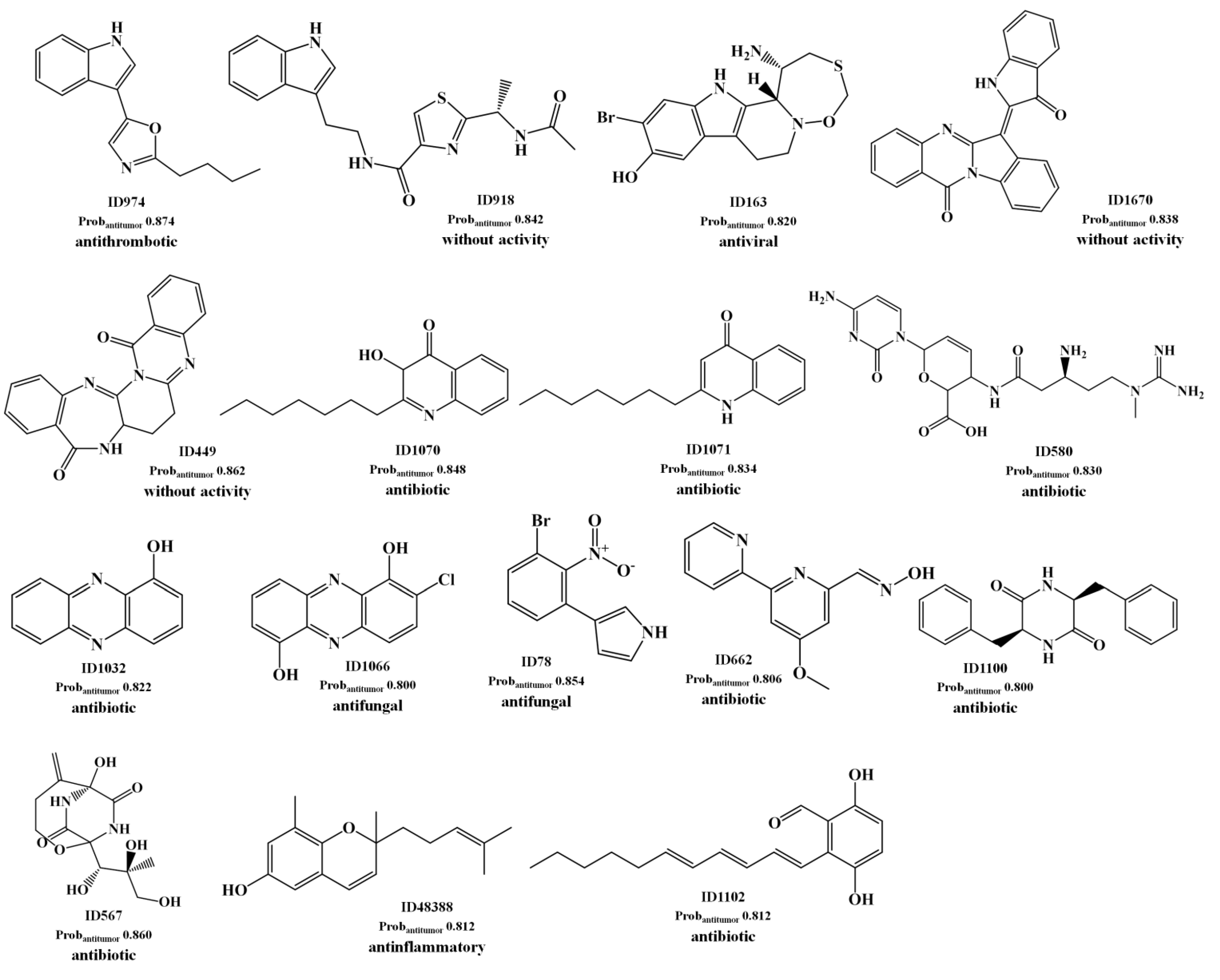{kind=link}
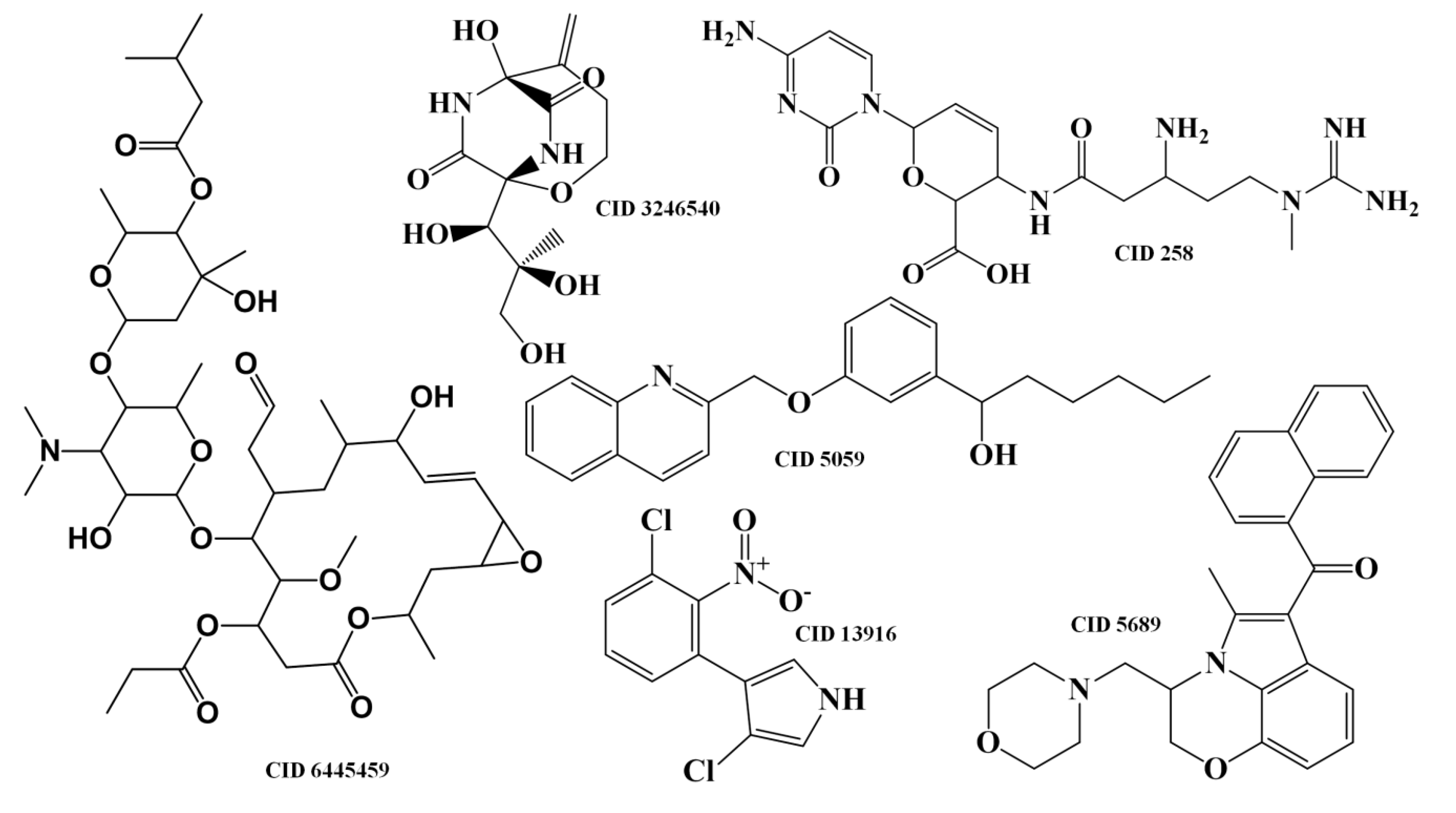{kind=link}
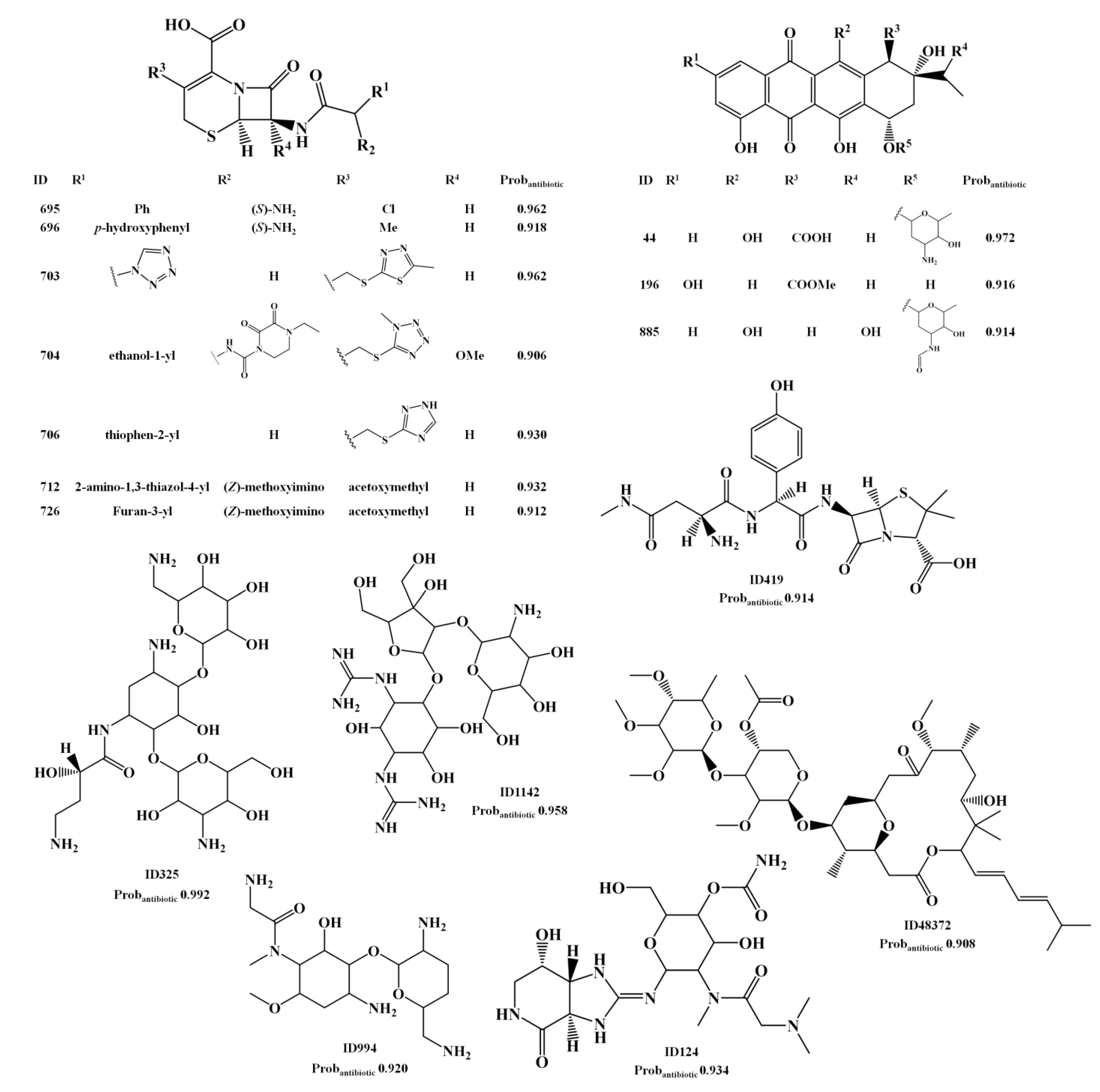{kind=link}
{kind=link}
| ML | SVM a | RF b | CT | |
|---|---|---|---|---|
| Training set/Test Set I/Test Set II | ||||
| Model | Class Size | Correct Predictions SensitivitycSpecificitydG-meane | Correct Predictions SensitivitycSpecificitydG-meane | Correct Predictions SensitivitycSpecificitydG-meane |
| Overall | YES f 1666/834/879 NO g 138/68/313 | YES 1295/647/671 NO 73/37/80 0.78/0.78/0.76 0.53/0.54/0.26 0.64/0.65/0.44 | YES 1152/591/660 NO 87/53/101 0.69/0.71/0.75 0.63/0.78/0.32 0.66/0.74/0.49 | YES 1657/821/864 NO 24/3/3 0.99/0.98/0.98 0.17/0.04/0.01 0.42/0.21/0.10 |
| Antitumor | YES 918/456/208 NO 886/446/984 | YES 744/377/79 NO 648/337/650 0.81/0.83/0.38 0.73/0.76/0.66 0.77/0.79/0.50 | YES 792/388/86 NO 689/357/686 0.86/0.85/0.41 0.78/0.80/0.70 0.82/0.82/0.54 | YES 775/369/84 NO 617/295/643 0.84/0.81/0.40 0.70/0.66/0.65 0.77/0.73/0.51 |
| Antibiotic | YES 654/338/625 NO 1150/564/567 | YES 501/262/419 NO 1061/516/337 0.77/0.78/0.67 0.92/0.91/0.59 0.84/0.84/0.63 | YES 548/271/433 NO 1090/531/376 0.84/0.80/0.69 0.95/0.94/0.66 0.89/0.87/0.68 | YES 521/249/391 NO 1094/516/396 0.80/0.74/0.63 0.95/0.91/0.70 0.87/0.82/0.66 |
2.1.1. Overall Biological Activity Model
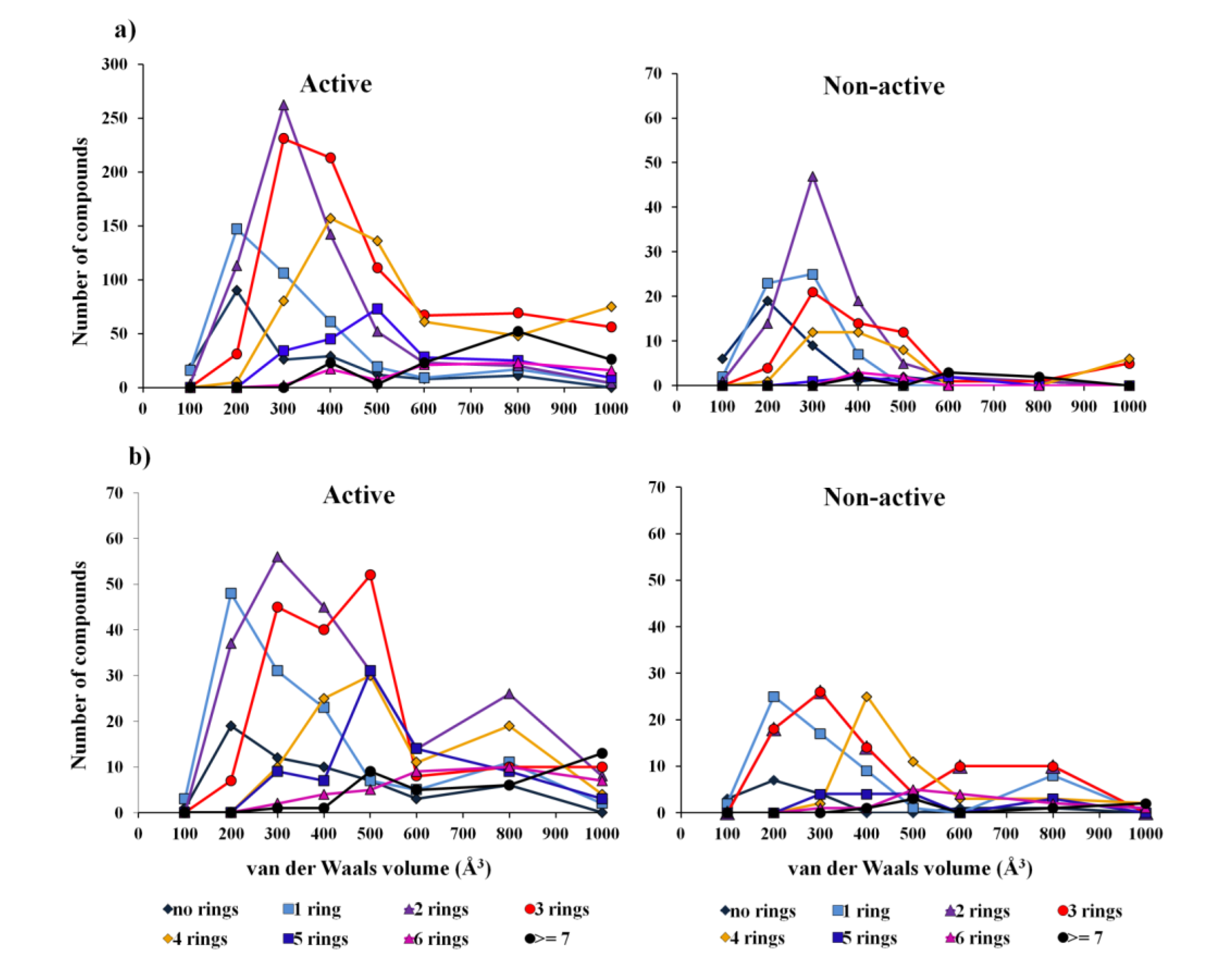
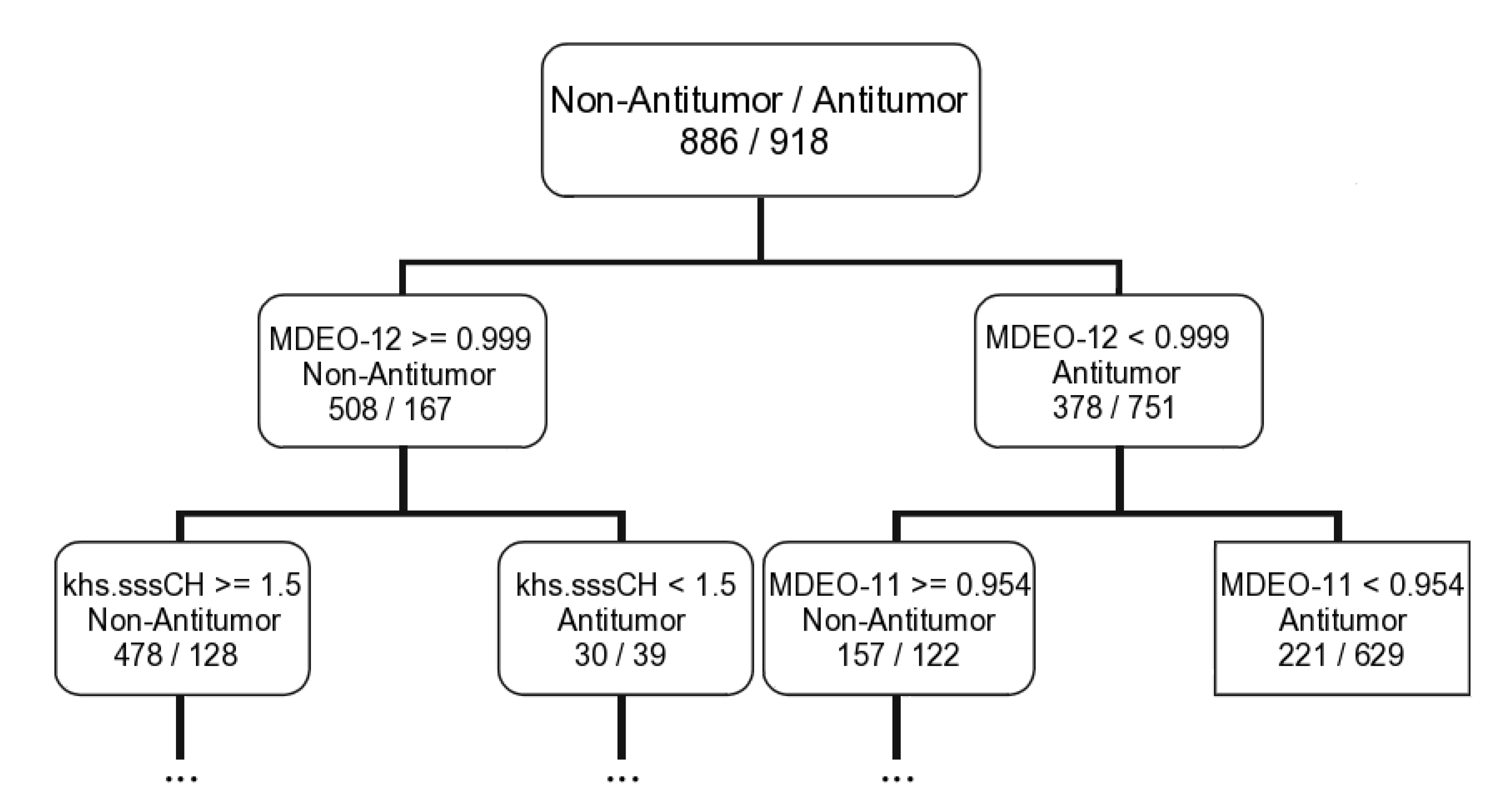
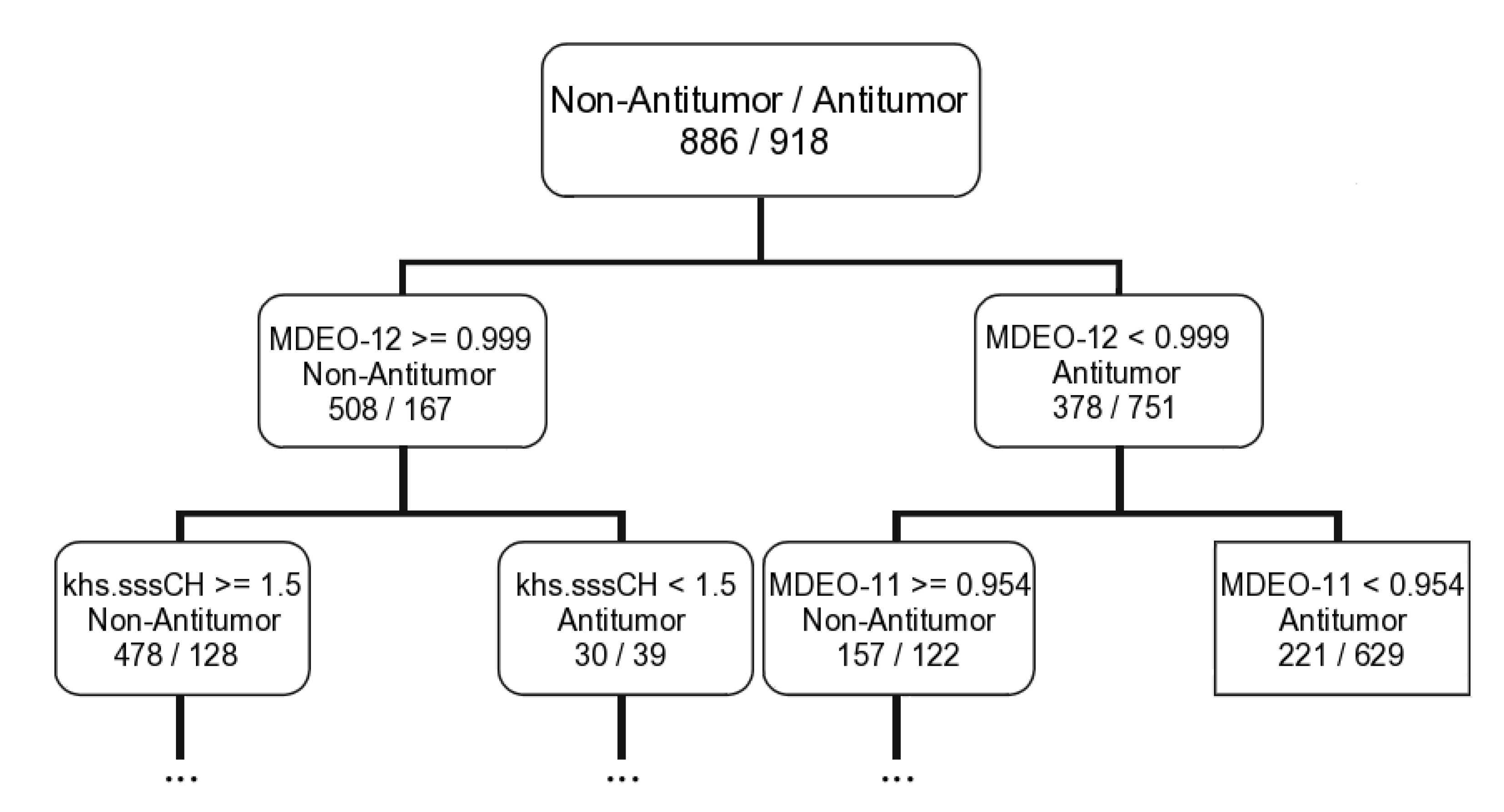
2.1.2. Antitumor Activity Model

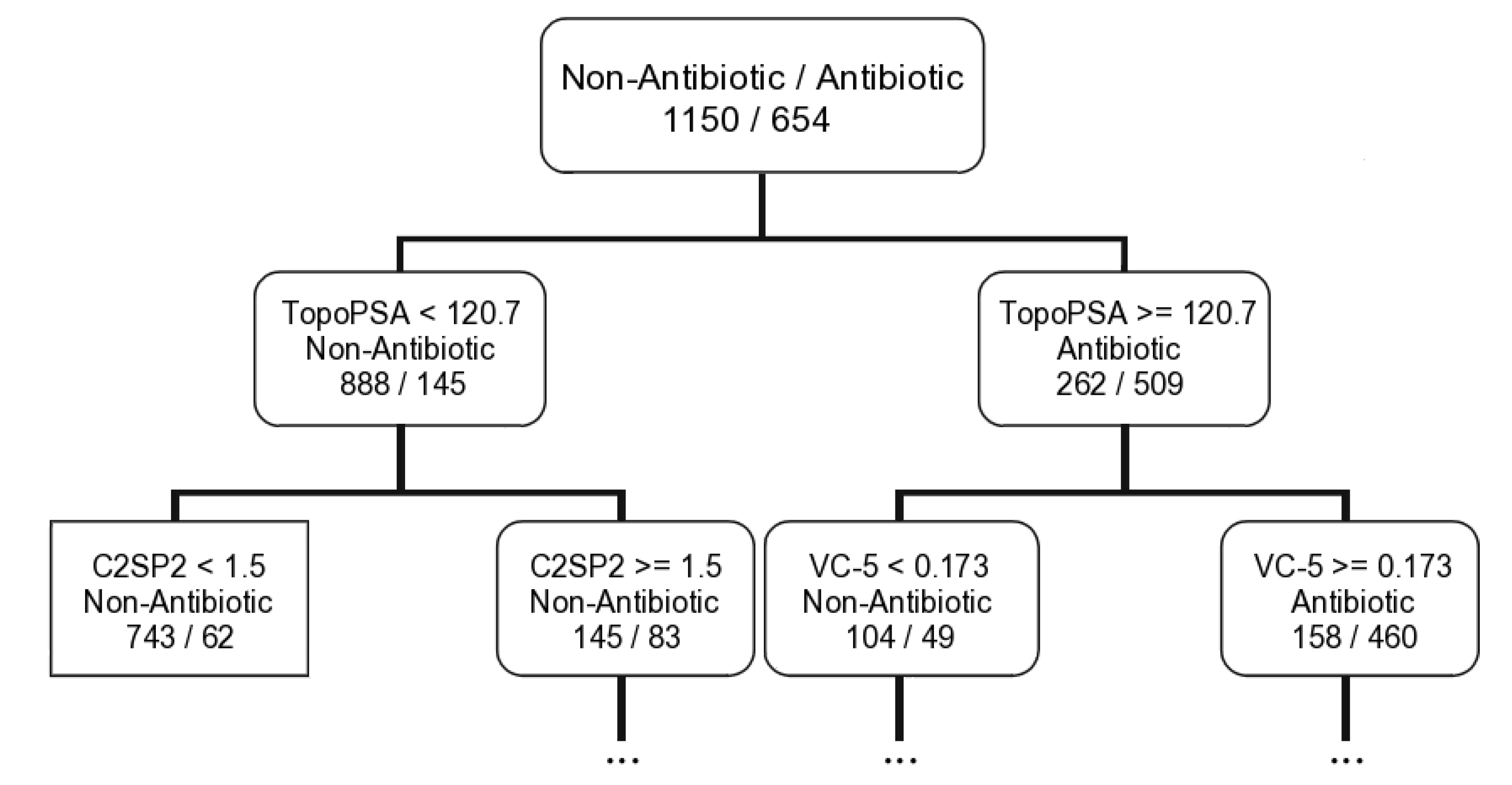
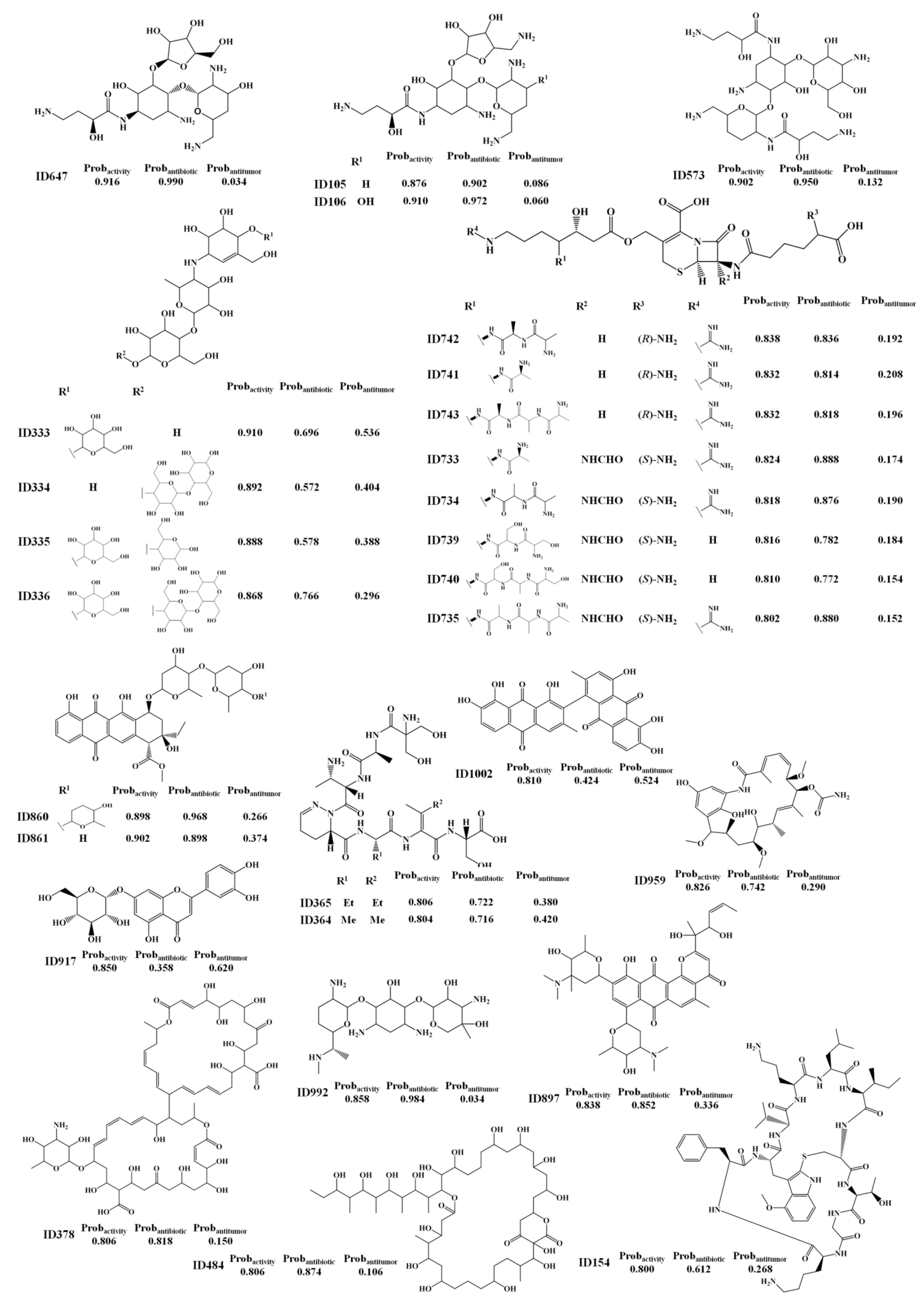
2.1.3. Antibiotic Activity Model
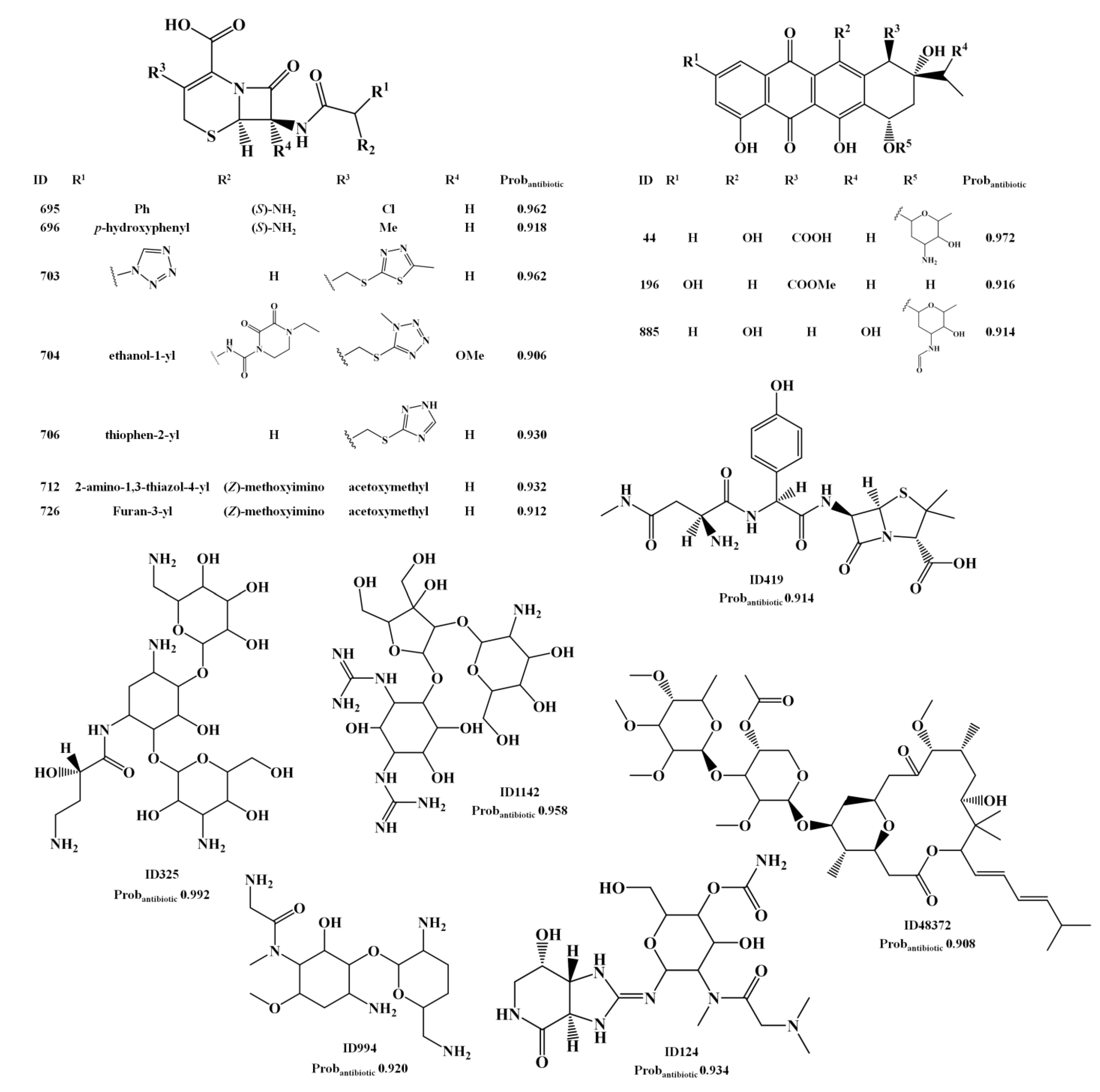
2.2. Analysis of Molecular Descriptors Identified as Relevant for Modeling Overall Biological Activity, Antitumor and Antibiotic Activities
| Model | CDK Descriptors | ||
|---|---|---|---|
| Overall biological activity | SVM a | 20D: ALogp2; BCUTc-1l; BCUTp-1h; PPSA-2; FPSA-3; TPSA; RHSA; Wlambda2.unity; ATSc3; ATSc4; C3SP2; SCH-5; SP-6; VP-7; khs.ssCH2; khs.dsCH; khs.sssCH; khs.sNH2; MDEC-33; TopoPSA | |
| RF | MeanDecreaseAccuracy b | Weta1.unity; FMF; BCUTw-1l; HybRatio; PNSA-2; ATSm1; FPSA-1; bpol; BCUTp-1l; TPSA | |
| CT | 7D: SP-6; BCUTc-1h; Wnu1.unity; Weta1.unity; SC-5; ATSc4; PPSA-3 | ||
| Antitumor activity | SVM a | 43D: ALogp2; AMR; BCUTw-1h; BCUTp-1l; PNSA-3; FPSA-3; FNSA-2; WNSA-3; TPSA; naAromAtom; nAromBond; ATSc2; ATSc3; ATSc4; ATSc5; ATSm5; bpol; C1SP2; C2SP2; SCH-4; VCH-4; VCH-5; VCH-7; VC-6; SPC-4; FMF; HybRatio; khs.dsCH; khs.aaCH; khs.sssCH; khs.tsC; khs.sNH2; khs.dO; khs.ssO; khs.sF; LOBMIN; MDEC-12; MDEC-13; MDEC-22; MDEO-11; MDEO-12; MDEO-22; TopoPSA | |
| RF | MeanDecreaseAccuracy b | MDEO-12; XlogP; khs.sssCH; ATSc5; FMF; MDEC-33; TopoPSA; MDEO-11; VC-5; MDEC-22 | |
| CT | 9D: MDEO-12; Khs.sssCH; MDEO-11; VC-6; ALogp2; SCH-7; BCUTc-1h; C2SP2; BCUTp-1l | ||
| Antibiotic activity | SVM a | 38D: ALogP; BCUTw-1h; BCUTp-1l; DPSA-3; FPSA-3; FNSA-2; RPCG; RNCS; TPSA; Wnu1.unity; nAromBond; ATSc1; ATSc5; ATSm5; nBase; C2SP2; C3SP3; SCH-4; SCH-5; VCH-4; VCH-7; VPC-5; khs.sssCH; khs.tsC; khs.dssC; khs.ssO; khs.sF; nAtomLC; MDEC-13; MDEC-22; MDEC-24; MDEC-33; MDEO-11; MDEO-12; MDEO-22; MOMI-XZ; TopoPSA; XLogP | |
| RF | MeanDecreaseAccuracy b | MDEO-12; MDEC-22; C2SP2; khs.dsCH; khs.sssCH; khs.dssC; VCH-5; MDEC-33; TopoPSA; XlogP | |
| CT | 16D: TopoPSA; C2SP2; VC-5; MDEC-22; XlogP; BCUTp-1h; VP-0; SCH-7; DPSA-1; Khs.dssC; Khs.ssCH2; Khs.sssCH; Khs.sssN; MDEO-12; THSA; VC-4 | ||

3. Material and Methods
3.1. Data Sets
3.2. Molecular Descriptors
3.3. Selection of Descriptors and Optimization of QSAR Classification Methods
3.4. ML Techniques
4. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- MarinLit. Available online: http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml (accessed on 9 January 2014).
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef]
- Koch, M.A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proc. Natl. Acad. Sci. USA 2005, 102, 17272–17277. [Google Scholar] [CrossRef]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 46, 68–74. [Google Scholar]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar]
- Liu, B.; Zhou, J. SARS-CoV protease inhibitors design using virtual screening method from natural products libraries. J. Comput. Chem. 2005, 26, 484–490. [Google Scholar] [CrossRef]
- Wright, A.D.; de Nys, R.; Angerhofer, C.K.; Pezzuto, J.M.; Gurrath, M. Biological activities and 3D QSAR studies of a series of Delisea pulchra (cf. fimbriata) derived natural products. J. Nat. Prod. 2006, 69, 1180–1187. [Google Scholar] [CrossRef]
- Hussain, A.; Melville, J.L.; Hirst, J.D. Molecular docking and QSAR of aplyronine A and analogues: Potent inhibitors of actin. J. Comput. Aided Mol. Des. 2010, 24, 1–15. [Google Scholar] [CrossRef]
- Hassan, H.M.; Elnagar, A.Y.; Khanfar, M.A.; Sallam, A.A.; Mohammed, R.; Shaala, L.A.; Youssef, D.T.A.; Hifnawy, M.S.; El Sayed, K.A. Design of semisynthetic analogues and 3D-QSAR study of eunicellin-based diterpenoids as prostate cancer migration and invasion Inhibitors. Eur. J. Med. Chem. 2011, 46, 1122–1130. [Google Scholar] [CrossRef]
- Lang, G.; Mayhudin, N.A.; Mitova, M.I.; Sun, L.; van der Sar, S.; Blunt, J.W.; Cole, A.L.J.; Ellis, G.; Laatsch, H.; Munro, M.H.G. Evolving trends in the dereplication of natural product extracts: New methodology for rapid, small-scale investigation of natural product extracts. J. Nat. Prod. 2008, 71, 1595–1599. [Google Scholar] [CrossRef]
- AntiMarin Database; Blunt, J.W.; Munro, M.H.G.; Laatsch, H. (Eds.) University of Canterbury: Christchurch, New Zealand; University of Göttingen: Göttingen, Germany, 2007.
- AntiBase Home Page. Available online: http://wwwuser.gwdg.de/~hlaatsc/antibase.htm (accessed on 9 January 2014).
- Bérdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Woo, P.W.K.; Haskell, T.H. Deoxy derivatives of butirosin A and 5′-amino-5′-deoxybutirosin A, aminoglycoside antibiotics resistant to bacterial 3′-phosphorylative enzymatic inactivation. Synthesis and NMR studies. J. Antibiot. 1982, 35, 692–702. [Google Scholar] [CrossRef]
- Mingeot-Leclercq, M.-P.; Glupczynski, Y.; Tulkens, P.M. Aminoglycosides: Activity and resistance. Antimicrob. Agents Chemother. 1999, 43, 727–737. [Google Scholar]
- Matsukawa, Y.; Marui, N.; Sakai, T.; Satomi, Y.; Yoshida, M.; Matsumoto, K.; Nishino, H.; Aoike, A. Genistein arrests cell cycle progression at G2-M. Cancer Res. 1993, 53, 1328–1331. [Google Scholar]
- Ryu, S.; Choi, S.U.; Lee, C.O.; Lee, S.H.; Ahn, J.W.; Zee, O.P. Antitumor triterpenes from medicinal plants. Arch. Pharm. Res. 1994, 17, 42–44. [Google Scholar] [CrossRef]
- Le Bail, J.C.; Varnat, F.; Nicolas, J.C.; Habrioux, G. Estrogenic and antiproliferative activities on MCF-7 human breast cancer cells by flavonoids. Cancer Lett. 1998, 130, 209–216. [Google Scholar] [CrossRef]
- Post, J.F.M.; Varma, R.S. Growth inhibitory effects of bioflavonoids and related compounds on human leukemic CEM-C1 and CEM-C7 cells. Cancer Lett. 1992, 67, 207–213. [Google Scholar] [CrossRef]
- Gálvez, M.; Martín-Cordero, C.; López-Lázaro, M.; Cortés, F.; Ayuso, M.J. Cytotoxic effect of Plantago spp. on cancer cell lines. J. Ethnopharmacol. 2003, 88, 125–130. [Google Scholar] [CrossRef]
- Matthies, L.; Laatsch, H. Cortinarins in Cortinarius speciosissimus? A critical revision. Experientia 1991, 47, 634–640. [Google Scholar] [CrossRef]
- Fath, M.A.; Ahmad, I.M.; Smith, C.J.; Spence, J.; Spitz, D.R. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin. Cancer Res. 2011, 17, 6206–6217. [Google Scholar] [CrossRef]
- Ivanova, V.; Kolarova, M.; Aleksieva, K.; Gräfe, U.; Dahse, H.-M.; Laatsch, H. Microbiaeratin, a new natural indole alkaloid from a microbispora aerata strain, isolated from Livingston Island, Antarctica. Prep. Biochem. Biotechnol. 2007, 37, 161–168. [Google Scholar] [CrossRef]
- Kruft, B.I.; Greer, A. Photosensitization reactions in vitro and in vivo. Photochem. Photobiol. 2011, 87, 1204–1213. [Google Scholar] [CrossRef]
- Olano, C.; Méndez, C.; Salas, J.A. Antitumor compounds from marine actinomycetes. Mar. Drugs 2009, 7, 210–248. [Google Scholar] [CrossRef]
- Yamagishi, H.; Matsumoto, K.; Iwasaki, K.; Miyazaki, T.; Yokoshima, S.; Tokuyama, H.; Fukuyama, T. Synthesis of eudistomin C and E: Improved preparation of the indole unit. Org. Lett. 2008, 10, 2369–2372. [Google Scholar] [CrossRef]
- Berkaew, P.; Soonthornchareonnon, N.; Salasawadee, K.; Chanthaket, R.; Isaka, M. Aurocitrin and related metabolites from the wood-decay fungus Hypocrea sp. J. Nat. Prod. 2008, 71, 902–904. [Google Scholar] [CrossRef]
- PubChem Bioassay Web Page. Cerulomycin—Compound Summary. Available online: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=5381230 (accessed on 12 November 2013).
- PubChem Bioassay Web Page. Pyrrolnitrin—Compound Summary. Available online: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=13916 (accessed on 12 November 2013).
- Resch, M.; Steigel, A.; Chen, Z.-l.; Bauer, R. 5-Lipoxygenase and cyclooxygenase-1 inhibitory active compounds from atractylodes lancea. J. Nat. Prod. 1998, 61, 347–350. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Phcog. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Jao, C.-W.; Lin, W.-C.; Wu, Y.-T.; Wu, P.-L. Isolation, structure elucidation, and synthesis of cytotoxic tryptanthrin analogues from phaius mishmensis. J. Nat. Prod. 2008, 71, 1275–1279. [Google Scholar] [CrossRef]
- Scuderi, M.R.; Cantarella, G.; Scollo, M.; Lempereur, L.; Palumbo, M.; Saccani-Jotti, G.; Bernardini, R. The antimitogenic effect of the cannabinoid receptor agonist WIN55212–2 on human melanoma cells is mediated by the membrane lipid raft. Cancer Lett. 2011, 310, 240–249. [Google Scholar] [CrossRef]
- Harada, S.; Zukishi, T. Isolation and characterization of a nucleoside antibiotic, amipurimycin. J. Antibiot. 1977, 30, 11–16. [Google Scholar] [CrossRef]
- Kida, T.; Ishikawa, T.; Shibai, H. Isolation of two streptothricin-like antibiotics, Nos. 6241-A and B, as inhibitors of de novo starch synthesis and their herbicidal activity. Agric. Biol. Chem. 1985, 49, 1839–1844. [Google Scholar] [CrossRef]
- Kondo, S.; Ikeda, Y.; Ikeda, T.; Gomi, S.; Nagarawa, H.; Hamada, M.; Umezawa, H. 5′-Hydroxy-2′-N-demethyl-dihydrostreptomycin produced by a streptomyces. J. Antibiot. 1985, 38, 433–435. [Google Scholar] [CrossRef]
- Yotsu-Yamashita, M.; Haddock, R.L.; Yasumoto, T.J. Polycavernoside A: A novel glycosidic macrolide from the red alga Polycavernosa tsudai (Gracilaria edulis). J. Am. Chem. Soc. 1993, 115, 1147–1148. [Google Scholar] [CrossRef]
- Stanton, D.T.; Jurs, P.C. Development and use of charged partial surface area structural descriptors in computer assisted quantitative structure property relationship studies. Anal. Chem. 1990, 62, 2323–2329. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Li, S.; Fedorowicz, A.; Singh, H.; Soderholm, S.C. Application of the random forest method in studies of local lymph node assay based skin sensitization data. J. Chem. Inf. Model. 2005, 45, 952–964. [Google Scholar] [CrossRef]
- Pearlman, R.S.; Smith, K.M. Metric validation and the receptor relevant subspace concept. J. Chem. Inf. Comput. Sci. 1999, 39, 28–35. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; WILEY-VCH: Weinheim, Germany, 2009; Volumes 1 and 2. [Google Scholar]
- Pereira, F.; Latino, D.A.R.S.; Aires-de-Sousa, J. Estimation of mayr electrophilicity with a quantitative structure–property relationship approach using empirical and DFT descriptors. J. Org. Chem. 2011, 76, 9312–9319. [Google Scholar] [CrossRef]
- Dutta, D.; Guha, R.; Wild, D.; Chen, T. Ensemble feature selection: Consistent descriptor subsets for multiple QSAR models. J. Chem. Inf. Model. 2007, 47, 989–997. [Google Scholar] [CrossRef]
- Mattioni, B.E.; Jurs, P.C. Prediction of dihydrofolate reductase inhibition and selectivity using computational neural network and linear discriminant analysis. J. Mol. Graph. Model. 2003, 21, 391–419. [Google Scholar] [CrossRef]
- Rapposelli, S.; Coi, A.; Imbriani, M.; Bianucci, A.M. Development of classification models for identifying “true” p-glycoprotein (p-gp) inhibitors through inhibition, ATPase activation and monolayer efflux assays. Int. J. Mol. Sci. 2012, 13, 6924–6943. [Google Scholar] [CrossRef]
- Eitrich, T.; Kless, A.; Druska, C.; Meyer, W.; Grotendorst, J. Classification of highly unbalanced CYP450 data of drugs using cost sensitive machine learning techniques. J. Chem. Inf. Model. 2007, 47, 92–103. [Google Scholar] [CrossRef]
- Fernandes, J.; Gattass, C.R. Topological polar surface area defines substrate transport by multidrug resistance associated protein 1 (MRP1/ABCC1). J. Med. Chem. 2009, 52, 1214–1218. [Google Scholar] [CrossRef]
- PubChem Home Page. Available online: http://pubchem.ncbi.nlm.nih.gov (accessed on 12 November 2013).
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. PubChem’s bioAssay database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar] [CrossRef]
- CDK Descriptor Calculator, Version 1.3.2. Available online: http://cdk.sourceforge.net/ (accessed on 10 January 2013).
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E.L. Recent developments of the chemistry development kit (CDK)—An open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Weka. Available online: http://www.cs.waikato.ac.nz/ml/weka/ (accessed on January 2014).
- Hall, M.A.; Smith, L.A. Correlation-based feature selection for machine learning. In. In Proceedings of the Twelfth International FLAIRS Conference, Orlando, FL, USA, 1–5 May 1999; AAAI Press: Menlo Park, CA, USA, 1999; pp. 235–239. [Google Scholar]
- Aha, D.W.; Kibler, D.; Albert, M.K. Instance-based learning algorithms. Mach. Learn. 1991, 6, 37–66. [Google Scholar]
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees; Chapman & Hall/CRC: Boca Raton, FL, USA, 2000. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2004. Available online: http://www.r-project.org/ (accessed on 9 January 2014).
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- LIBSVM. Available online: http://www.csie.ntu.edu.tw/~cjlin/libsvm/ (accessed on 9 January 2014).
- El Manzalawy, Y.; Honavar, V. WLSVM: Integrating LibSVM into Weka Environment. 2005. Available online: http://www.cs.iastate.edu/~yasser/wlsvm/ (accessed on 9 January 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pereira, F.; Latino, D.A.R.S.; Gaudêncio, S.P. A Chemoinformatics Approach to the Discovery of Lead-Like Molecules from Marine and Microbial Sources En Route to Antitumor and Antibiotic Drugs. Mar. Drugs 2014, 12, 757-778. https://doi.org/10.3390/md12020757
Pereira F, Latino DARS, Gaudêncio SP. A Chemoinformatics Approach to the Discovery of Lead-Like Molecules from Marine and Microbial Sources En Route to Antitumor and Antibiotic Drugs. Marine Drugs. 2014; 12(2):757-778. https://doi.org/10.3390/md12020757
Chicago/Turabian StylePereira, Florbela, Diogo A. R. S. Latino, and Susana P. Gaudêncio. 2014. "A Chemoinformatics Approach to the Discovery of Lead-Like Molecules from Marine and Microbial Sources En Route to Antitumor and Antibiotic Drugs" Marine Drugs 12, no. 2: 757-778. https://doi.org/10.3390/md12020757
APA StylePereira, F., Latino, D. A. R. S., & Gaudêncio, S. P. (2014). A Chemoinformatics Approach to the Discovery of Lead-Like Molecules from Marine and Microbial Sources En Route to Antitumor and Antibiotic Drugs. Marine Drugs, 12(2), 757-778. https://doi.org/10.3390/md12020757