Cytotoxic Polyketides from the Deep-Sea-Derived Fungus Engyodontium album DFFSCS021

Abstract
:1. Introduction
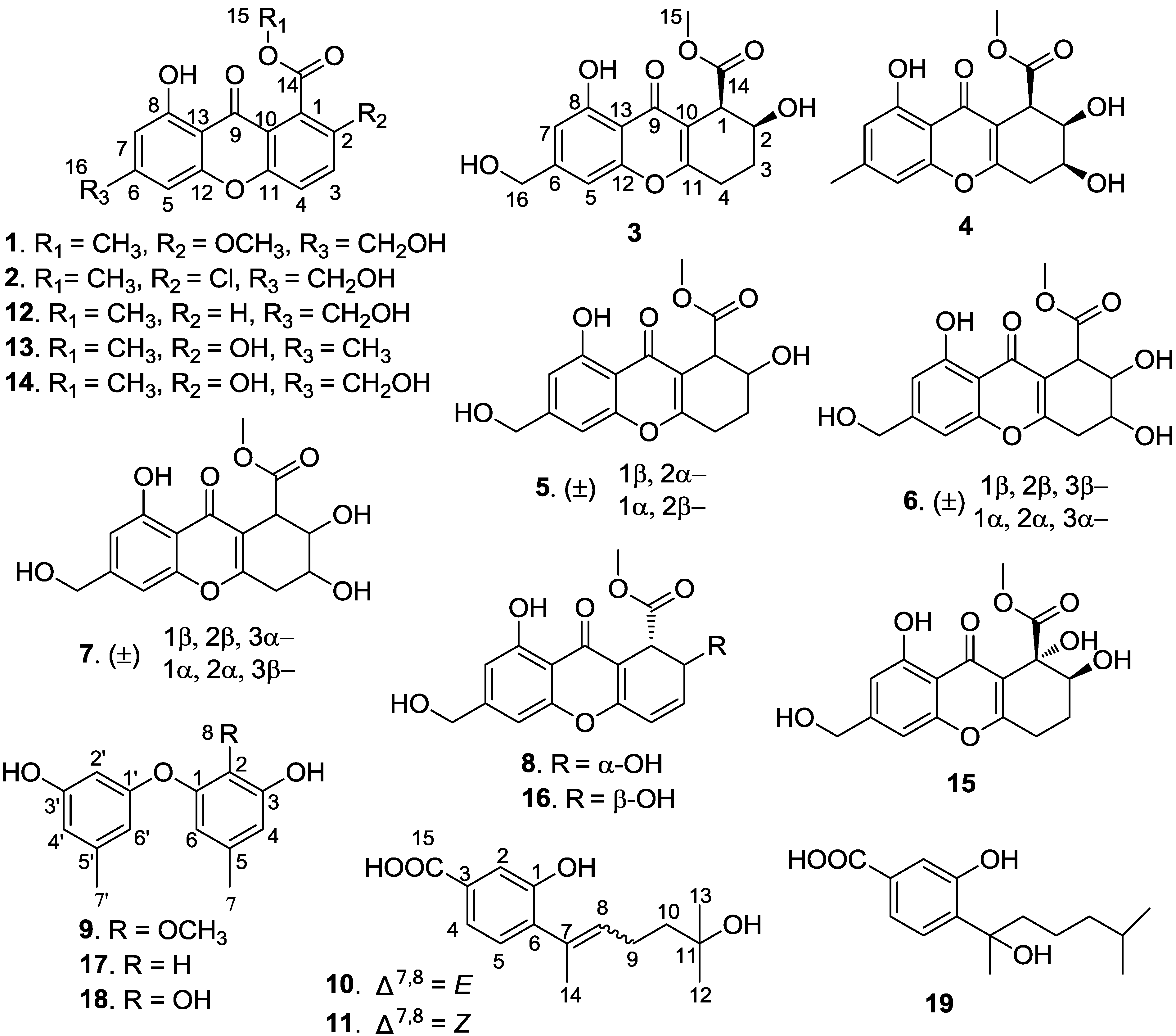
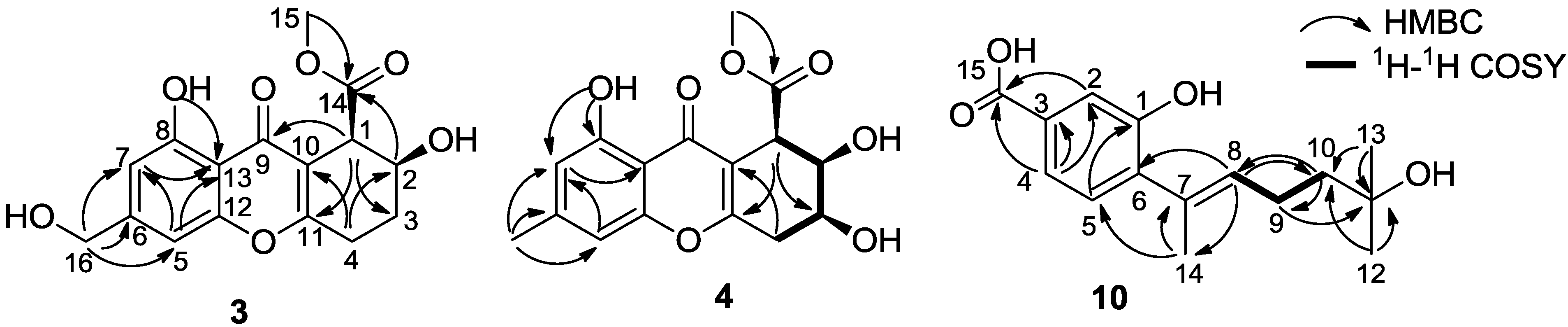
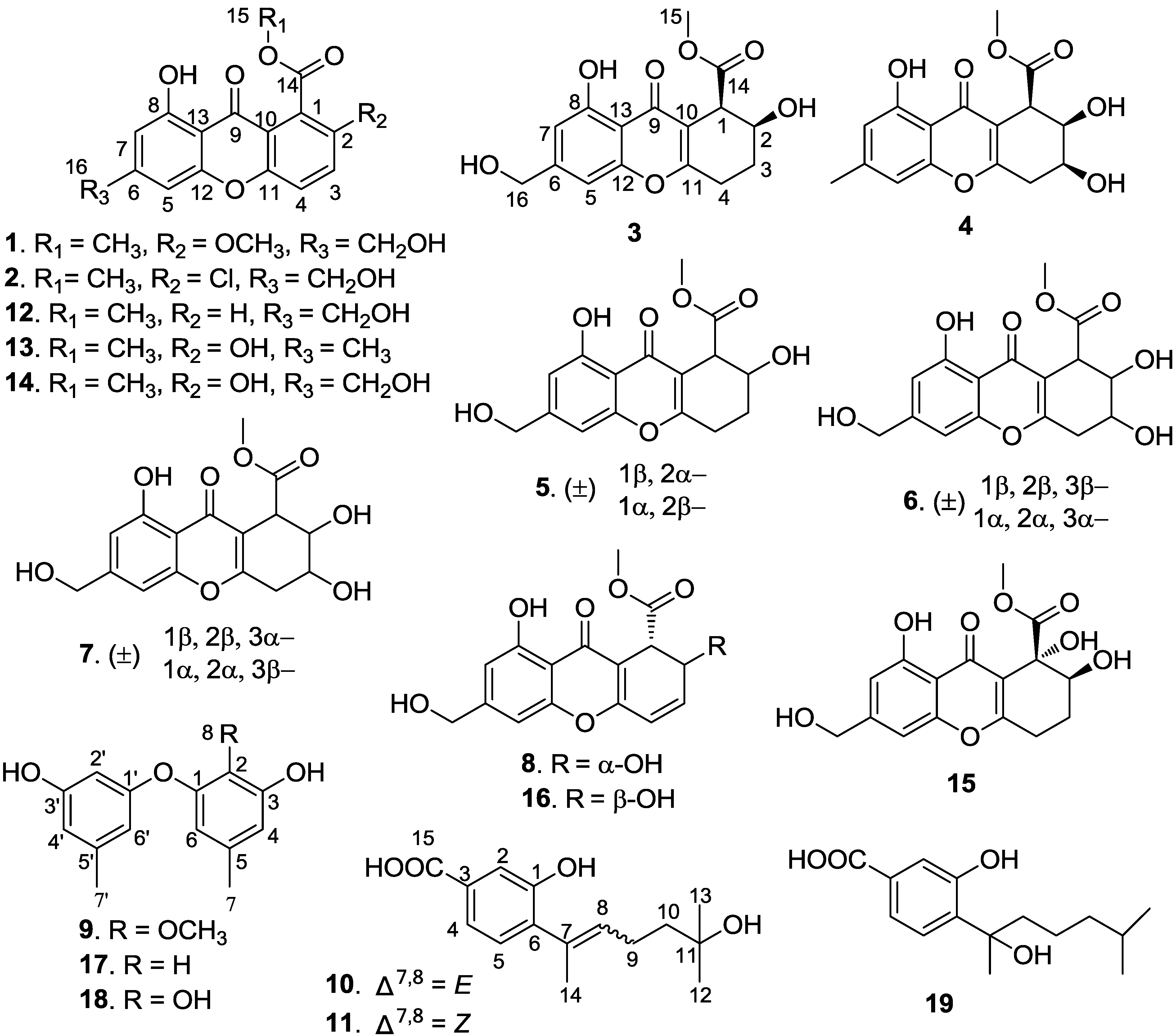
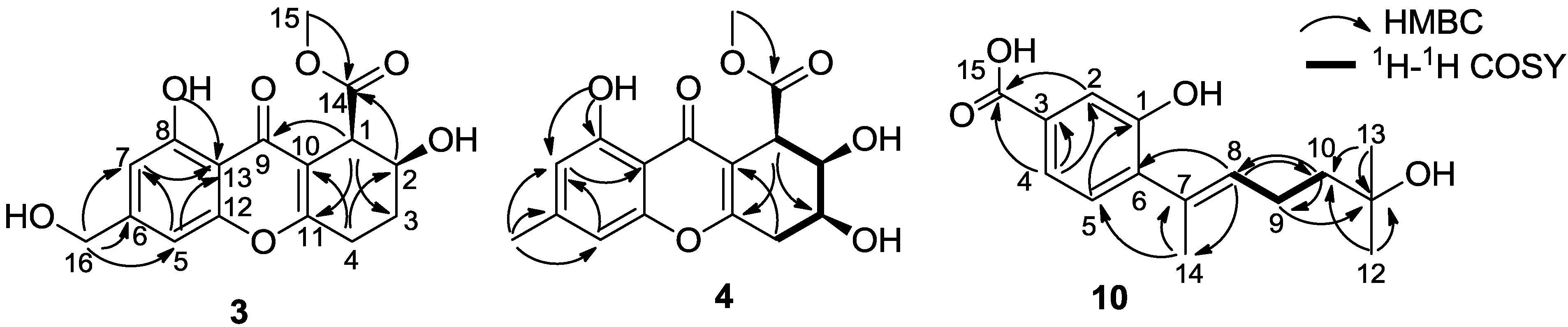
2. Results and Discussion

| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| δH (J) | δH (J) | δH (J) | δH (J) | δH (J) | δH (J) | δH (J) | δH (J) | |
| 1 | 3.84, d (6.0) | 3.78, d (5.5) | 3.62, d (3.5) | 3.60, d (4.5) | 3.63, d (6.0) | 4.03, d (10.0) | ||
| 2 | 4.11, ddd (3.5, 6.0, 9.5) | 3.99, br t (5.5) | 4.14, ddd (2.5, 3.5, 6.0) | 4.15, br t (4.5) | 3.99, dd (1.5, 6.0) | 5.01, br d (10.0) | ||
| 3 | 7.79, d (9.0) | 8.06, d (9.0) | 1.84, m 2.01, m | 4.04, ddd (4.5, 5.0, 5.5) | 1.80, m 1.90, m | 3.96, ddd (3.5, 4.5, 4.5) | 3.92, ddd (1.5, 4.5, 6.5) | 6.49, d (2.0) |
| 4 | 7.82, d (8.5) | 7.84, d (9.0) | 2.73–2.89, m | 3.10, dd (4.0, 18.5) | 2.73–2.87, m | 3.03, dd (4.5, 18.5) | 3.07, dd (4.5, 18.5) | 6.36, d (2.0) |
| 2.60, dd (5.0, 18.5) | 2.70, dd (3.5, 18.5) | 2.79, dd (6.5, 18.5) | ||||||
| 5 | 7.02, s | 7.05, s | 6.96, s | 6.89, s | 6.98, s | 6.98, s | 6.98, s | 6.98, s |
| 7 | 6.77, s | 6.81, s | 6.73, s | 6.65, s | 6.74, s | 6.74, s | 6.74, s | 6.75, s |
| 15 | 3.87, s | 3.95, s | 3.60, s | 3.60, s | 3.64, s | 3.65, s | 3.65, s | 3.58, s |
| 16 | 4.60, s | 4.62, d (4.5) | 4.56, s | 2.37, s | 4.58, s | 4.56, s | 4.56, s | 4.56, s |
| 2-OH | 5.49, brs | 5.90, brs | ||||||
| 8-OH | 12.11, s | 11.88, brs | 12.29, s | 12.29, s | 12.41, brs | 12.25, brs | 12.39, s | |
| 16-OH | 5.58, t (4.5) | 5.50, brs | 5.50, brs | |||||
| 8-OCH3 | 3.88, s |
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| δC | δC | δC | δC | δC | δC | δC | δC | |
| 1 | 120.3, C | 131.0, C | 43.5, CH | 42.3, CH | 46.2, CH | 43.5, CH | 44.6, CH | 41.7, CH |
| 2 | 154.4, C | 125.5, C | 65.1, CH | 68.1, CH | 65.4, CH | 66.3, CH | 65.8, CH | 65.9, CH |
| 3 | 121.3, CH | 136.4, CH | 26.1, CH2 | 66.1, CH | 26.1,CH2 | 69.8, CH | 69.9, CH | 145.0, CH |
| 4 | 120.8, CH | 121.3, CH | 25.7, CH2 | 32.8, CH2 | 23.8, CH2 | 32.9, CH2 | 33.4, CH2 | 119.7, CH |
| 5 | 103.9, CH | 104.1, CH | 103.8, CH | 107.2, CH | 103.9, CH | 103.9, CH | 103.9, CH | 104.1, CH |
| 6 | 155.5, C | 154.8, C | 155.5, C | 147.2, C | 152.3, C | 152.1, C | 152.3, C | 152.2, C |
| 7 | 107.2, CH | 107.8, CH | 107.7, CH | 111.4, CH | 107.8, CH | 107.9, CH | 107.9, CH | 108.3, C |
| 8 | 160.2, C | 160.4, C | 159.3, C | 159.2, C | 159.3, C | 159.4, C | 159.3, C | 159.3, C |
| 9 | 180.2, C | 179.2, C | 181.1, C | 180.7, C | 181.2, C | 180.1, C | 180.8, C | 180.0, C |
| 10 | 117.1, C | 118.0, C | 113.9, C | 113.1, C | 113.1, C | 112.7, C | 113.0, C | 111.0, C |
| 11 | 152.0, C | 154.3, C | 166.9, C | 164.1, C | 166.7, C | 164.3, C | 164.8, C | 160.4, C |
| 12 | 156.2, C | 155.3, C | 152.2, C | 155.6, C | 155.5, C | 155.7, C | 155.7, C | 154.9, C |
| 13 | 107.0, C | 106.9, C | 107.9, C | 107.2, C | 107.9, C | 107.8, C | 107.9, C | 108.3, C |
| 14 | 166.2, C | 165.7, C | 171.4, C | 170.0, C | 172.0, C | 170.7, C | 172.3, C | 169.7, C |
| 15 | 52.4, CH3 | 52.9, CH3 | 51.5, CH3 | 51.5, CH3 | 52.0, CH3 | 51.5, CH3 | 51.9, CH3 | 51.5, CH3 |
| 16 | 62.3, CH2 | 62.2, CH2 | 62.1, CH2 | 21.7, CH3 | 62.2, CH2 | 62.2, CH2 | 62.1, CH2 | 62.1, CH2 |
| 2-OCH3 | 58.6, OCH3 |

{kind=link}
{kind=link}
| Compound | JH-1,H-2 | JH-2,H-3 | JH-3,H-4 | Orientations of H-1, H-2, H-3 |
|---|---|---|---|---|
| 3 | 6.0 | 3.5, 9.5 | H-1: equatorial, H-2: axial | |
| 4 | 5.5 | 5.5 | 4.5, 5.0 | H-1: equatorial, H-2: axial, H-3: equatorial |
| 5 | 3.5 | 2.5, 6.0 | H-1: equatorial, H-2: equatorial | |
| 6 | 4.5 | 4.5 | 3.5, 4.5 | H-1: equatorial, H-2: axial, H-3: equatorial |
| 7 | 6.0 | 1.5 | 4.5, 6.5 | H-1: axial, H-2: equatorial, H-3: equatorial |
| Compound | Zone of Inhibition (mm) a | MIC (μg/mL) | Cytotoxicity (IC50 μM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| E. coli | B. subtilis | E. coli | B. subtilis | U937 | Hela | MCF-7 | HepG2 | Huh7 | |
| 2 | - b | - | - | - | 55.5 | 96.1 | 172.3 | 73.8 | >300 |
| 3 | - | - | - | - | 218.4 | >300 | >300 | >300 | >300 |
| 4 | - | - | - | - | 208.6 | >300 | >300 | >300 | >300 |
| 5 | - | - | - | - | 15.9 | 205.2 | >300 | >300 | >300 |
| 6 | - | - | - | - | 192.7 | >300 | >300 | >300 | >300 |
| 7 | - | - | - | - | 287.2 | >300 | >300 | >300 | >300 |
| 8 | 13.8 | 16.5 | 64.0 | 32.0 | 4.9 | 24.8 | 38.5 | 60.5 | 53.3 |
| 10 | - | 10.0 | - | 256.0 | c NT | NT | NT | NT | NT |
| 12 | - | - | - | - | 75.6 | >300 | >300 | >300 | >300 |
| 14 | - | - | - | - | 127.0 | >300 | >300 | >300 | >300 |
| 15 | 11.0 | 14.4 | 64.0 | 64.0 | NT | NT | NT | NT | NT |
| 16 | 15.8 | 17.5 | 64.0 | 64.0 | 8.8 | 60.0 | 102.2 | 52.7 | 133.3 |
| 19 | 11.4 | 13.6 | 64.0 | 128.0 | NT | NT | NT | NT | NT |
| d Dox | 0.06 | 0.8 | 23.1 | 3.3 | 1.2 | ||||
| d PG | 31.8 | 43.3 | 2.0 | 2.0 | |||||
3. Experimental Section
3.1. General Experimental Procedure
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Purification
3.5. Cytotoxicity
3.6. Antibacterial Activities
3.7. Barnacle Balanus amphitrite Larval Settlement Bioassays
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268. [Google Scholar] [CrossRef] [PubMed]
- Skropeta, D. Deep-sea natural products. Nat. Prod. Rep. 2008, 25, 1131–1166. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, X.Y.; Tu, Z.C.; Xu, X.Y.; Qi, S.H. Alkaloids from the deep-sea-derived fungus Aspergillus westerdijkiae DFFSCS013. J. Nat. Prod. 2013, 76, 983–987. [Google Scholar] [CrossRef]
- Hamasaki, T.; Sato, Y.; Hatsuda, Y. Structure of sydowinin A, sydowinin B, and sydowinol, metabolites from Aspergillus sydowi. Agric. Biol. Chem. 1975, 39, 1345–2341. [Google Scholar]
- Healy, P.C.; Hocking, A.; Tran-Dinh, N.; Pitt, J.I.; Shivas, R.G.; Mitchell, J.K.; Kotiw, M.; Davis, R.A. Xanthones from a microfungus of the genus Xylaria. Phytochemistry 2003, 65, 2373–2378. [Google Scholar] [CrossRef]
- Little, A.; Porco, J.J. Total syntheses of graphisin A and sydowinin B. Org. Lett. 2012, 14, 2862–2865. [Google Scholar] [CrossRef] [PubMed]
- Trisuwan, K.; Rukachaisirikul, V.; Kaewpet, M.; Phongpaichit, S.; Hutadilok-Towatana, N.; Preedanon, S.; Sakayaroj, J.; Rukachaisirikul, V. Sesquiterpene and xanthone derivatives from the sea fan-derived fungus Aspergillus sydowii PSU-F154. J. Nat. Prod. 2011, 74, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Park, I.Y.; Park, Y.J.; Lee, J.H.; Hong, Y.S.; Lee, J.J. A novel dihydroxanthenone, AGI-B4 with inhibition of VEGF-induced endothelial cell growth. J. Antibiot 2002, 55, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.H.; Bijay, K.S.; Iwona, T.S.; Jeffrey, H.B. Synthesis and herbicidal activity of cyperin. J. Agric. Food Chem. 1995, 43, 804–808. [Google Scholar]
- Bunyapaiboonsri, T.; Yoiprommarat, S.; Intereya, K.; Kocharin, K. New diphenyl ethers from the insect pathogenic fungus Cordyceps sp. BCC 1861. Chem. Pharm. Bull. 2007, 55, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Takashi, H.; Kouzou, N.; Yuichi, H. Two new metabolites, sydonic acid and hydroxysydonic acid, from Aspergillus sydowi. Agric. Biol. Chem. 1978, 42, 37–40. [Google Scholar] [CrossRef]
- Ratnayake, R.; Lace, E.; Tennant, S.; Gill, J.H.; Capon, R.J. Kibdelones: Novel anticancer polyketides from a rare Australian actinomycete. Chem. Eur. J. 2007, 13, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Lange, G.L.; Lee, M. 13C NMR determination of the configuration of methyl substituted double bonds in medium- and large-ring terpenoids. Mag. Res. Chem. 1986, 24, 656–658. [Google Scholar] [CrossRef]
- Tan, Q.W.; Ouyang, M.A.; Shen, S.; Li, W. Bioactive metabolites from a marine-derived strain of the fungus Neosartorya fischeri. Nat. Prod. Res. 2012, 26, 1402–1407. [Google Scholar] [CrossRef]
- Lai, X.; Cao, L.; Tan, H.; Fang, S.; Huang, Y.; Zhou, S. Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea. ISME J. 2007, 1, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Lorian, V. Antibiotics in Laboratory Medicine: The Disc Susceptibility Test; Williams & Wilkins: Baltimore, MD, USA, 1980; pp. 24–54. [Google Scholar]
- Brantner, A.; Grein, E. Antibacterial activity of plant extracts used externally in traditional medicine. J. Ethnopharmacol. 1994, 44, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.H.; Zhang, S.; Qian, P.Y.; Xiao, Z.H.; Li, M.Y. Ten new antifouling briarane diterpenoids from the South China Sea gorgonian Junceella juncea. Tetrahedron 2006, 62, 9123–9130. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Q.; Wang, J.; Zhang, X.; Nong, X.; Xu, X.; Qi, S. Cytotoxic Polyketides from the Deep-Sea-Derived Fungus Engyodontium album DFFSCS021. Mar. Drugs 2014, 12, 5902-5915. https://doi.org/10.3390/md12125902
Yao Q, Wang J, Zhang X, Nong X, Xu X, Qi S. Cytotoxic Polyketides from the Deep-Sea-Derived Fungus Engyodontium album DFFSCS021. Marine Drugs. 2014; 12(12):5902-5915. https://doi.org/10.3390/md12125902
Chicago/Turabian StyleYao, Qifeng, Jie Wang, Xiaoyong Zhang, Xuhua Nong, Xinya Xu, and Shuhua Qi. 2014. "Cytotoxic Polyketides from the Deep-Sea-Derived Fungus Engyodontium album DFFSCS021" Marine Drugs 12, no. 12: 5902-5915. https://doi.org/10.3390/md12125902