Induced Marine Fungus Chondrostereum sp. as a Means of Producing New Sesquiterpenoids Chondrosterins I and J by Using Glycerol as the Carbon Source


Abstract
:1. Introduction

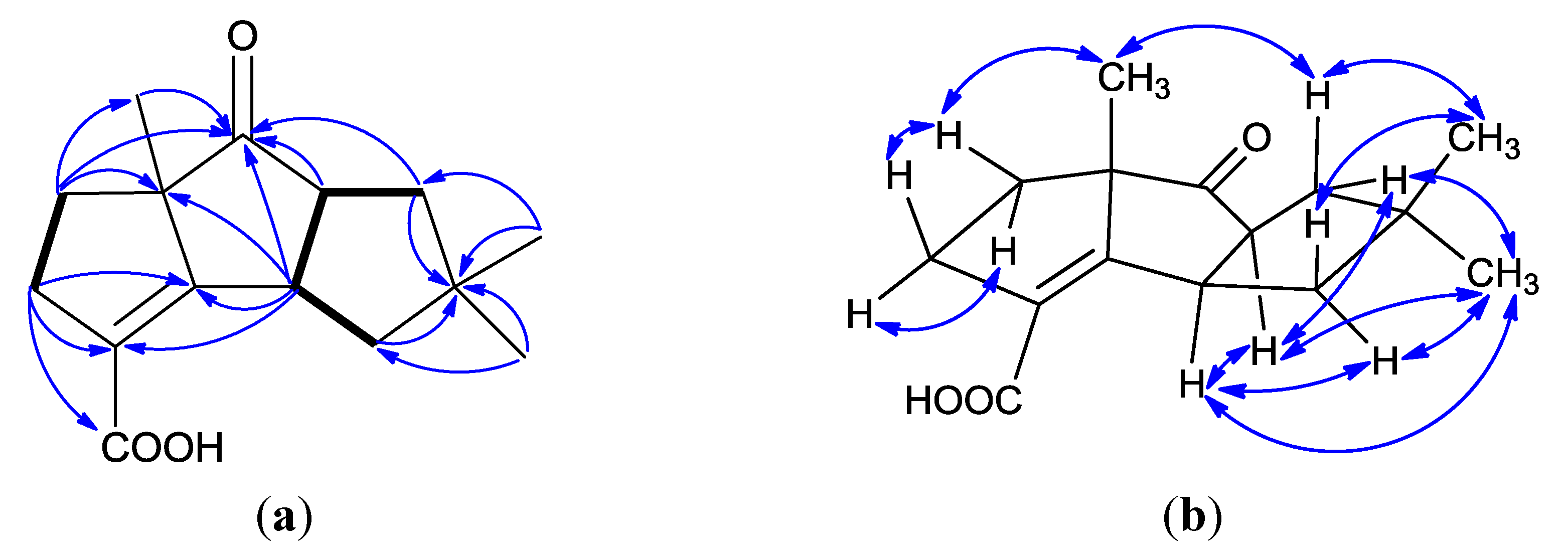
2. Results and Discussion

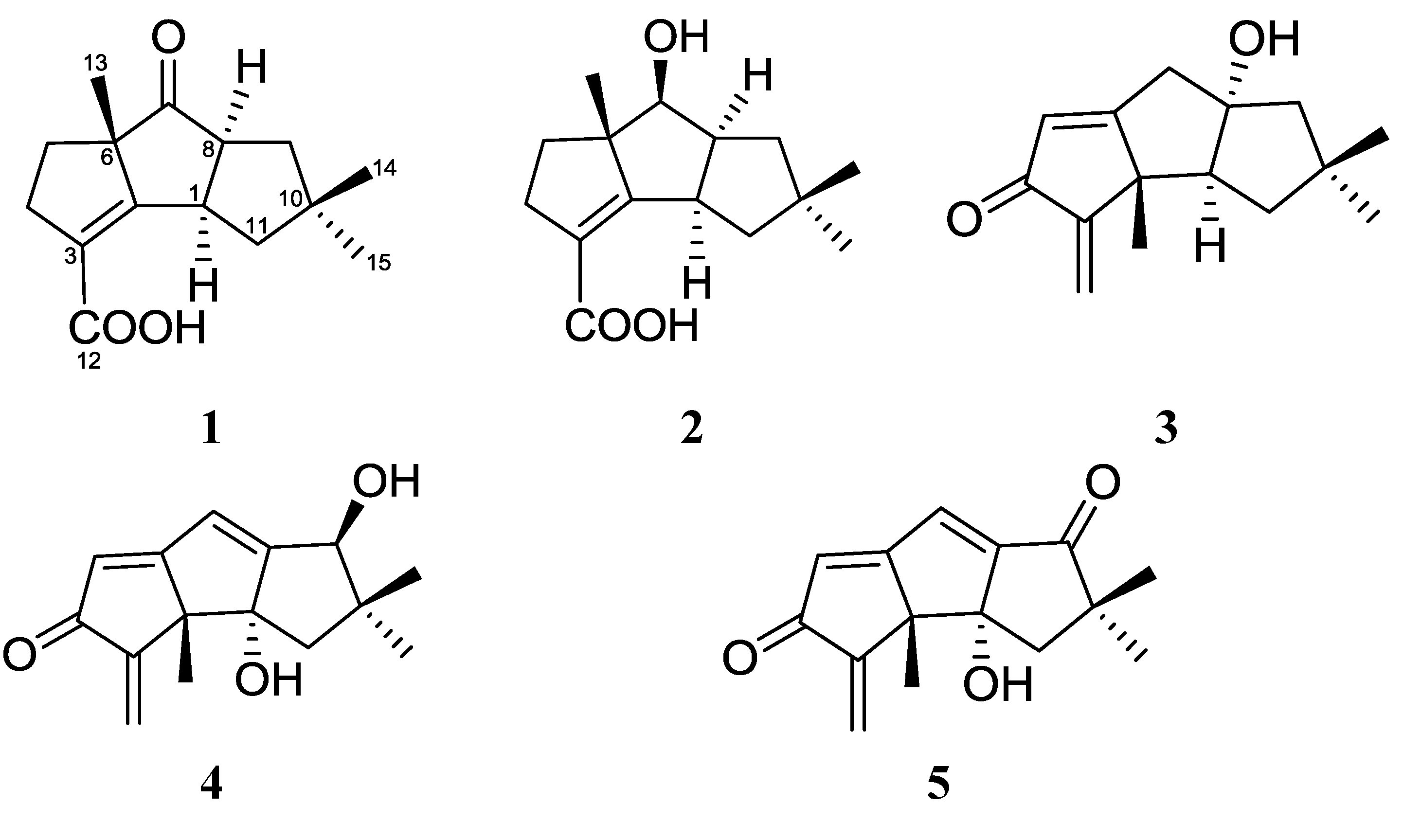{kind=link}
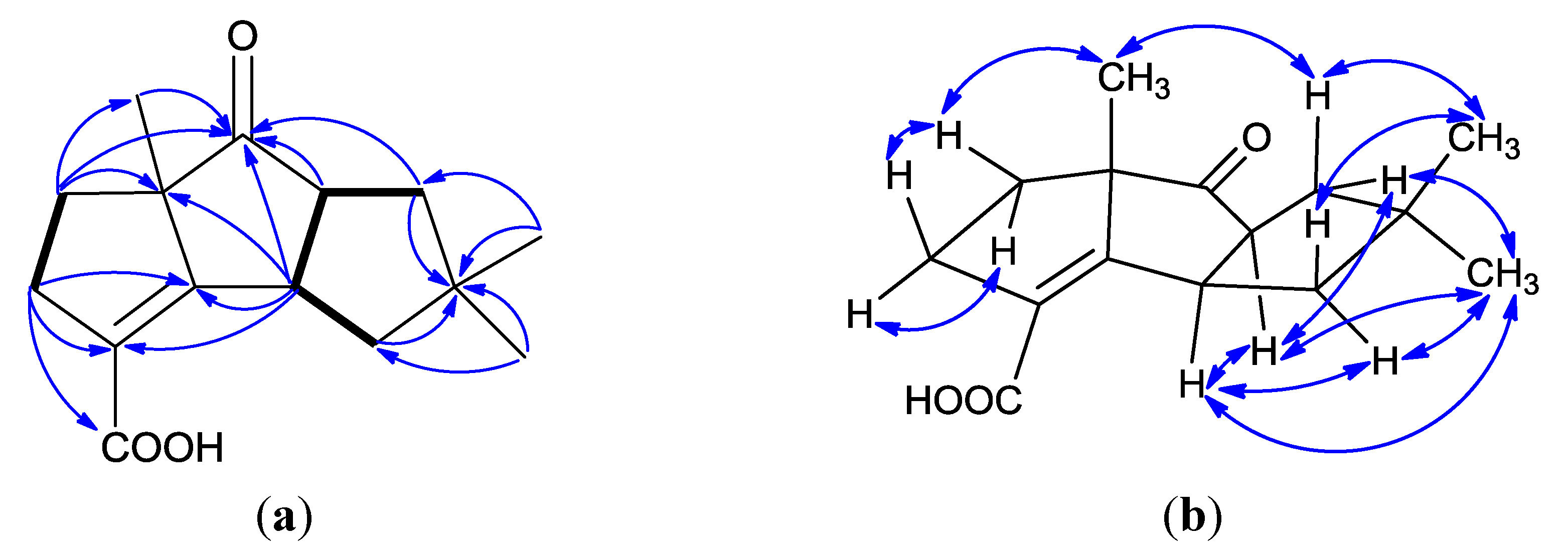{kind=link}
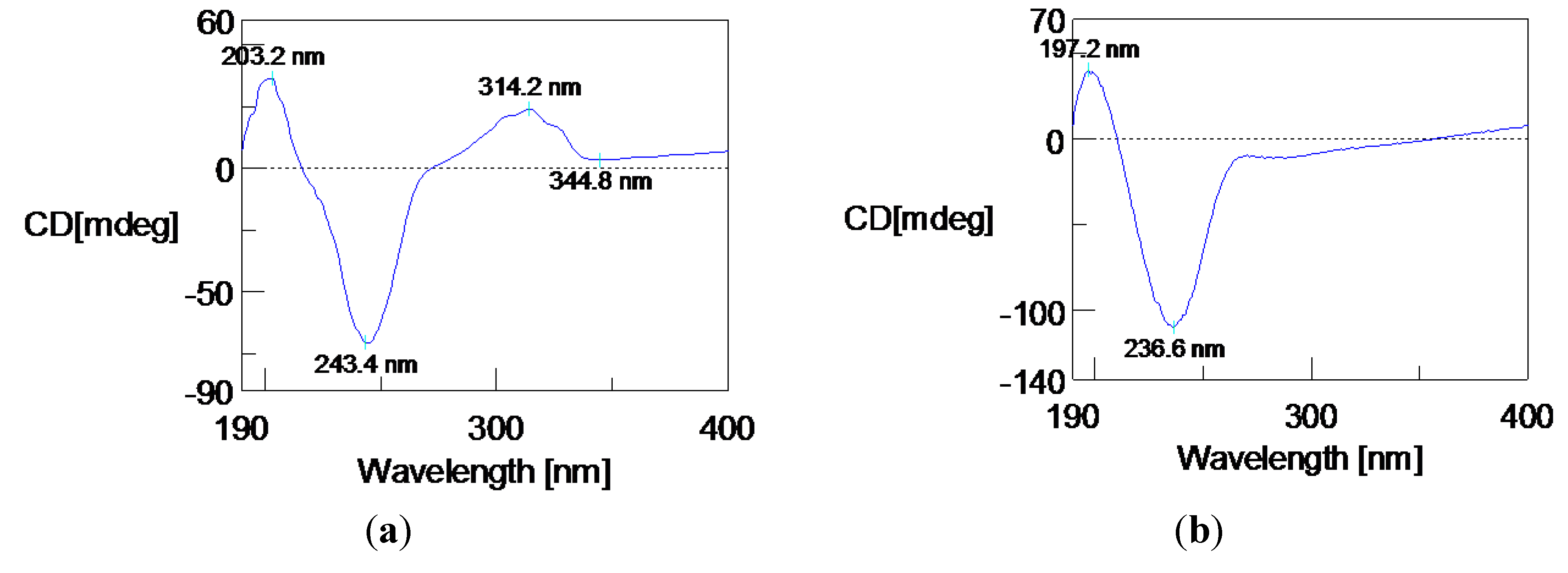{kind=link}
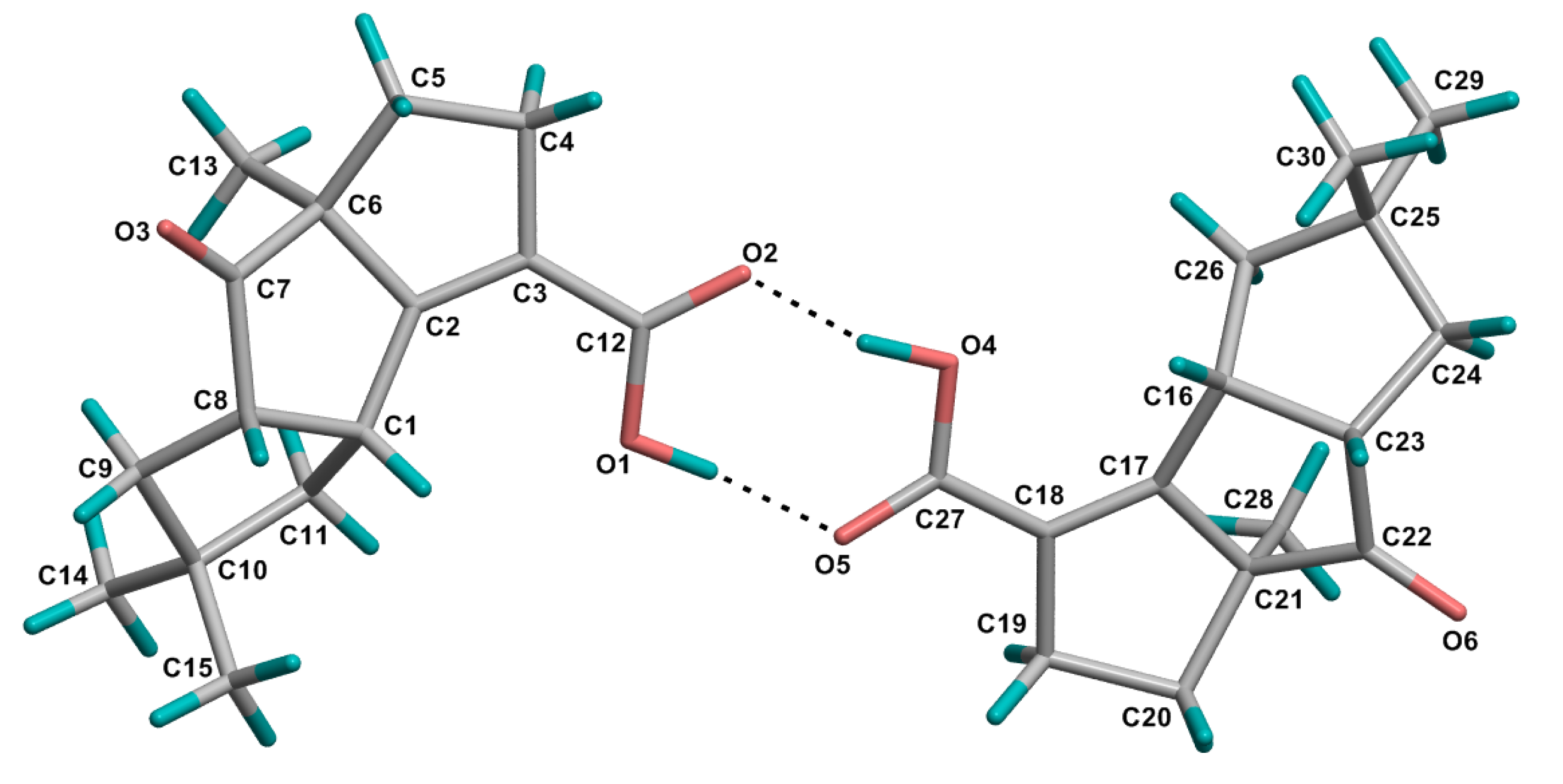{kind=link}
{kind=link}
| Position | δC, Type | δH, Mult., ( J) |
|---|---|---|
| 1 | 42.2, CH | 3.96, ddd (9.6, 9.6, 9.6) |
| 2 | 169.9, C | |
| 3 | 126.6, C | |
| 4 | 32.5, CH2 | α: 2.89, ddd (16.0, 10.4, 6.4); β: 2.72, dd (16.0, 8.8) |
| 5 | 35.6, CH2 | α: 1.99, m; β: 1.85, m |
| 6 | 63.8, C | |
| 7 | 220.1, C | |
| 8 | 58.2, CH | 3.22, ddd (9.6, 9.6, 9.6) |
| 9 | 46.1, CH2 | α: 2.03, m; β: 1.68, m |
| 10 | 43.5, C | |
| 11 | 46.1, CH2 | α: 2.03, m; β: 1.59, m |
| 12 | 170.1, C | |
| 13 | 23.8, CH3 | 1.44, s |
| 14 | 28.2, CH3 | 1.14, s |
| 15 | 26.5, CH3 | 1.00, s |
| 12-OH | 10.50, brs |

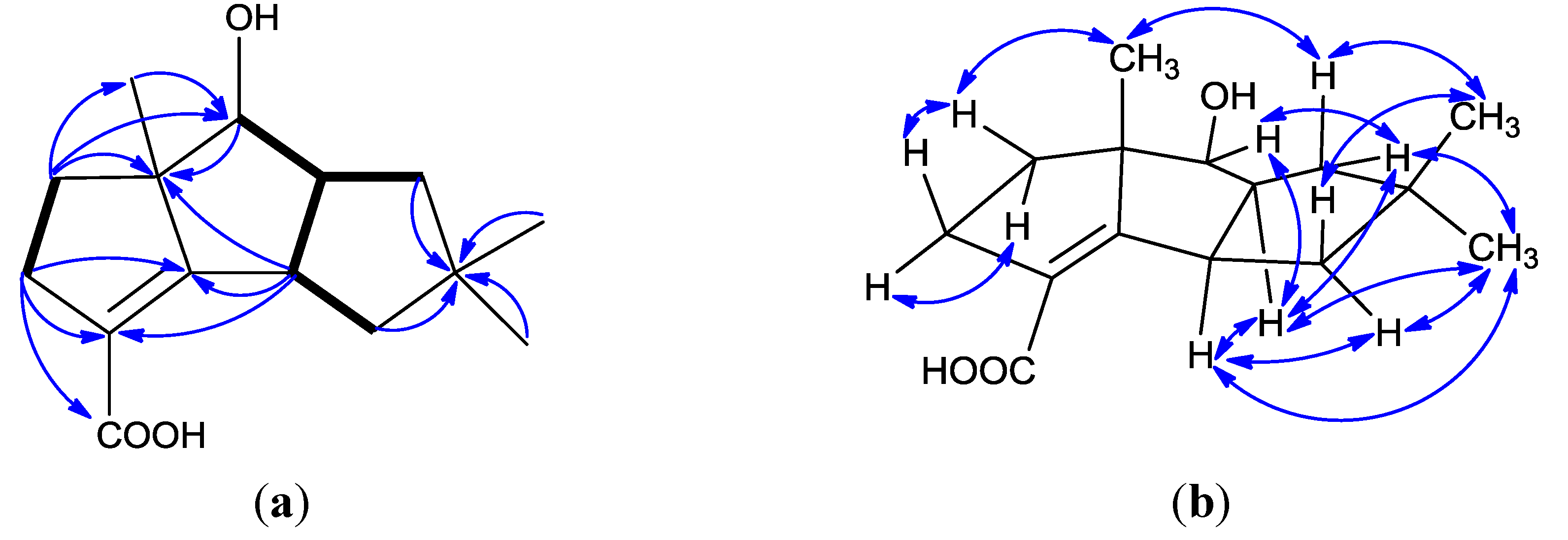
| Position | δC, Type a | δH, Mult., (J) a | δCb | δH, Mult., (J) b |
|---|---|---|---|---|
| 1 | 42.1, CH | 3.55, ddd (9.6, 9.6, 9.6) | 42.6 | 3.53, ddd (9.6, 9.6, 9.2) |
| 2 | 175.6, C | 173.7 | ||
| 3 | 123.4, C | 124.5 | ||
| 4 | 33.8, CH2 | α: 2.86, ddd (15.6, 9.6, 6.4); β: 2.71, dd (15.6, 6.0) | 34.9 | α: 2.80, ddd (15.2, 10.8, 6.8); β: 2.62, dd (15.2, 8.0) |
| 5 | 41.2, CH2 | α: 1.84, m; β: 1.75, m | 42.3 | α: 1.75, m; β: 1.70, m |
| 6 | 61.4, C | 62.1 | ||
| 7 | 79.0, CH | 4.05, d (9.6) | 79.1 | 3.99, d (9.2) |
| 8 | 51.9, CH | 3.21, dddd (9.6, 9.6, 9.6, 6.8) | 53.0 | 3.17, dddd (9.2, 9.2, 9.2,9.2) |
| 9 | 42.1, CH2 | α: 1.81, dd (12.0, 6.8); β: 1.48, ddd (12.0, 9.6, 2.0) | 43.3 | α: 1.97, dd (12.0, 9.2); β: 1.42, ddd (12.0, 9.2, 2.0) |
| 10 | 41.9, C | 42.2 | ||
| 11 | 46.6, CH2 | α: 2.00, ddd (12.8, 9.6, 2.0); β: 1.38, dd (12.8, 9.6) | 47.6 | α: 1.92, ddd (12.8, 9.6, 2.0); β: 1.38, dd (12.8, 9.6) |
| 12 | 170.6, C | 166.7 | ||
| 13 | 18.5, CH3 | 1.25, s | 19.2 | 1.24, s |
| 14 | 28.7, CH3 | 1.11, s | 29.3 | 1.09, s |
| 15 | 26.6, CH3 | 0.97, s | 27.2 | 0.95, s |
| 7-OH | 4.92, brs | 3.84, brs | ||
| 12-OH | 10.53, brs | 10.47, brs |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Strain and Culture Method
3.3. Extraction and Isolation
 −17.73 (c 0.015, MeOH); UV (MeOH) λmax (ε) 244 nm (9310), 219 nm (7344); IR (KBr) υmax 2956, 2920, 2851, 1736, 1673, 1648, 1464, 1434, 1371, 1334, 1281, 1266, 1209, 1119, 1077, 927, 905, 763, 719, 711 cm−1; 1H and 13C NMR data, see Table 1; LREIMS m/z 248, 233, 220, 204, 187, 174, 159, 147, 132, 119, 105, 77, 65, 55; HREIMS m/z 248.1406 [M]+ (calcd for C15H20O3, 248.1407). −9.11 (c 0.006, MeOH); UV (MeOH) λmax (ε) 237 nm (14829); IR (KBr) υmax 3372, 2952, 2924, 2866,1677, 1463, 1445, 1422, 1385, 1366, 1329, 1279, 1263, 1245, 1228, 1197, 1158, 1135, 1100, 1060, 1030, 1015, 971, 912, 811, 764, 691, 665 cm−1; 1H and 13C NMR data, see Table 2. LREIMS m/z 250, 232, 214, 204, 199, 187, 171, 145, 131, 119, 105, 91, 77, 69, 55; HREIMS m/z 250.1563 [M]+ (calcd for C15H22O3, 250.1563).
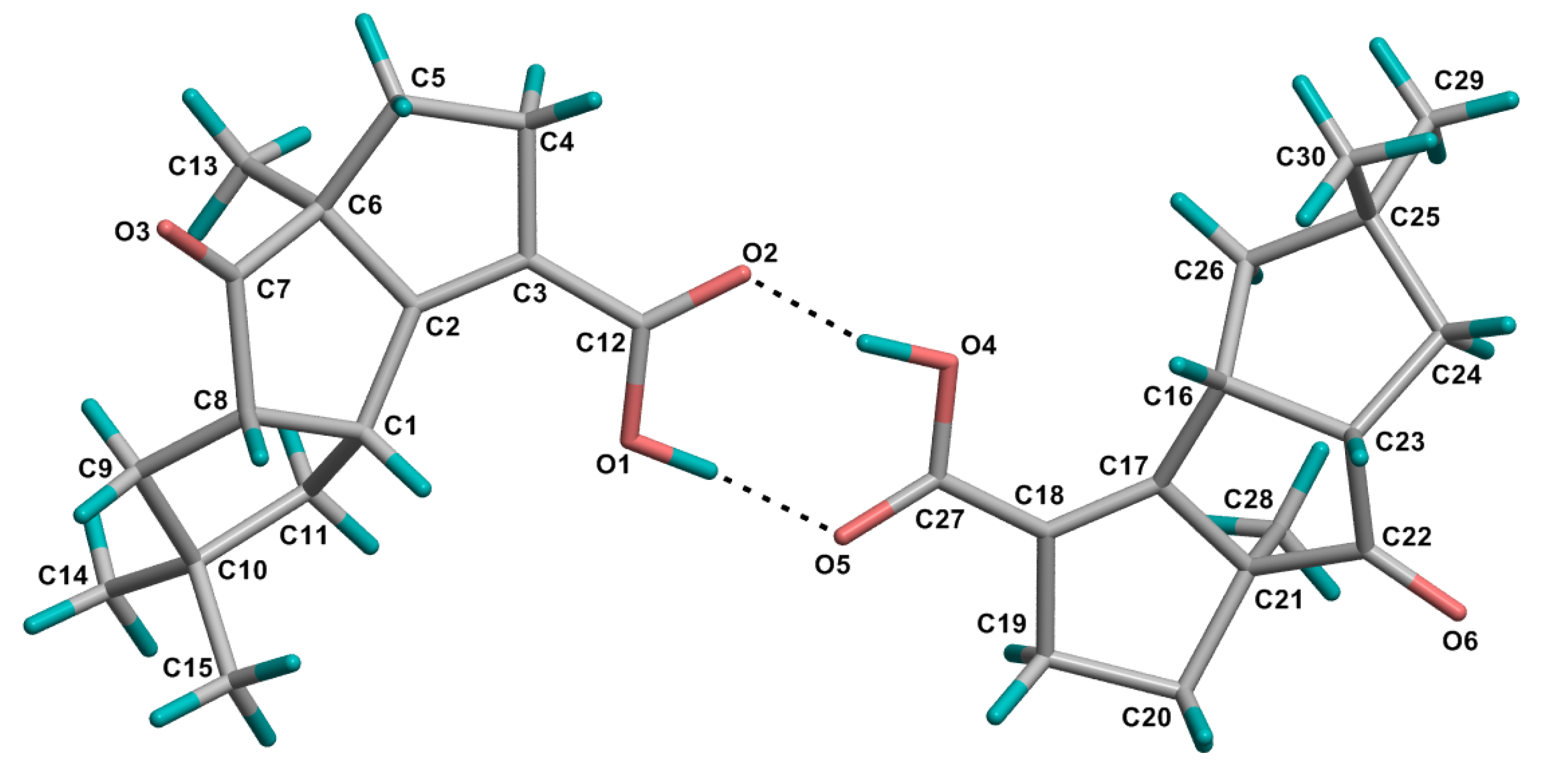
−17.73 (c 0.015, MeOH); UV (MeOH) λmax (ε) 244 nm (9310), 219 nm (7344); IR (KBr) υmax 2956, 2920, 2851, 1736, 1673, 1648, 1464, 1434, 1371, 1334, 1281, 1266, 1209, 1119, 1077, 927, 905, 763, 719, 711 cm−1; 1H and 13C NMR data, see Table 1; LREIMS m/z 248, 233, 220, 204, 187, 174, 159, 147, 132, 119, 105, 77, 65, 55; HREIMS m/z 248.1406 [M]+ (calcd for C15H20O3, 248.1407). −9.11 (c 0.006, MeOH); UV (MeOH) λmax (ε) 237 nm (14829); IR (KBr) υmax 3372, 2952, 2924, 2866,1677, 1463, 1445, 1422, 1385, 1366, 1329, 1279, 1263, 1245, 1228, 1197, 1158, 1135, 1100, 1060, 1030, 1015, 971, 912, 811, 764, 691, 665 cm−1; 1H and 13C NMR data, see Table 2. LREIMS m/z 250, 232, 214, 204, 199, 187, 171, 145, 131, 119, 105, 91, 77, 69, 55; HREIMS m/z 250.1563 [M]+ (calcd for C15H22O3, 250.1563).3.4. Crystal Structure Determination of 1
3.5. Cytotoxicity Assay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Li, H.J.; Chen, T.; Xie, Y.L.; Chen, W.D.; Zhu, X.F.; Lan, W.J. Isolation and structural elucidation of chondrosterins F-H from the marine fungus Chondrostereum sp. Mar. Drugs 2013, 11, 551–558. [Google Scholar] [CrossRef]
- Li, H.J.; Xie, Y.L.; Xie, Z.L.; Chen, Y.; Lam, C.K.; Lan, W.J. Chondrosterins A–E, triquinane-type sesquiterpenoids from soft coral-associated fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627–638. [Google Scholar] [CrossRef]
- Li, H.J.; Lan, W.J.; Lam, C.K.; Yang, F.; Zhu, X.F. Hirsutane sesquiterpenoids from the marine-derived fungus Chondrostereum sp. Chem. Biodivers. 2011, 8, 317–324. [Google Scholar] [CrossRef]
- Yang, F.; Gao, Y.H.; Wu, K.W.; Deng, R.; Li, D.D.; Wei, Z.X.; Jiang, S.; Wu, X.Q.; Feng, G.K.; Li, H.J.; et al. A novel sesquiterpene hirsutanol A induces autophagical cell death in human hepatocellular carcinoma cells by increasing reactive oxygen species. Chin. J. Cancer 2010, 29, 655–660. [Google Scholar] [CrossRef]
- Yang, F.; Chen, W.D.; Deng, R.; Li, D.D.; Wu, K.W.; Feng, G.K.; Li, H.J.; Zhu, X.F. Hirsutanol A induces apoptosis and autophagy via reactive oxygen species accumulation in breast cancer MCF-7 cells. J. Pharmacol. Sci. 2012, 119, 214–220. [Google Scholar] [CrossRef]
- Yang, F.; Chen, W.D.; Deng, R.; Zhang, H.; Tang, J.; Wu, K.W.; Li, D.D.; Feng, G.K.; Lan, W.J.; Li, H.J.; et al. Hirsutanol A, a novel sesquiterpene compound from fungus Chondrostereum sp., induces apoptosis and inhibits tumor growth through mitochondrial-independent ROS production: Hirsutanol A inhibits tumor growth through ROS production. J. Transl. Med. 2013, 11, 32:1–32:10. [Google Scholar]
- Sheldrick, G.M. SADABS: PROGRAM for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL 5.10 for Windows NT: Structure Determination Software Programs; Bruker Analytical X-ray Systems: Madison, WI, USA, 1997. [Google Scholar]
- CCDC CIF Depository Request Form. Available online: http://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi (accessed on 30 December 2013).
- Mellows, G.; Mantle, P.G.; Feline, T.C.; Williams, D.J. Sesquiterpenoid metabolites from Stereum complicatum. Phytochemistry 1973, 12, 2717–2720. [Google Scholar] [CrossRef]
- Kupka, J.; Anke, T.; Giannetti, B.M.; Steglich, W. Antibiotics from basidiomycetes. XIV. Isolation and biological characterization of hypnophilin, pleurotellol, and pleurotellic acid from Pleurotellus hypnophilus (Berk.) Sacc. Arch. Microbiol. 1981, 130, 223–227. [Google Scholar] [CrossRef]
- Stadler, M.; Anke, T.; Dasenbrock, J.; Steglich, W. Antibiotics from basidiomycetes. XLII. Phellodonic acid, a new biologically active hirsutane derivative from Phellodon melaleucus (Thelephoraceae, Basidiomycetes). Z. Naturforsch C 1993, 48, 545–549. [Google Scholar]
- Liermann, J.C.; Schüffler, A.; Wollinsky, B.; Birnbacher, J.; Kolshorn, H.; Anke, T.; Opatz, T. Hirsutane-type sesquiterpenes with uncommon modifications from three basidiomycetes. J. Org. Chem. 2010, 75, 2955–2961. [Google Scholar]
- Comer, F.W.; McCapra, F.; Qureshi, I.H.; Scott, A.I. Structure and chemistry of hirsutic acid. Tetrahedron 1967, 23, 4761–4768. [Google Scholar] [CrossRef]
Supplementary Files
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.-J.; Jiang, W.-H.; Liang, W.-L.; Huang, J.-X.; Mo, Y.-F.; Ding, Y.-Q.; Lam, C.-K.; Qian, X.-J.; Zhu, X.-F.; Lan, W.-J. Induced Marine Fungus Chondrostereum sp. as a Means of Producing New Sesquiterpenoids Chondrosterins I and J by Using Glycerol as the Carbon Source. Mar. Drugs 2014, 12, 167-175. https://doi.org/10.3390/md12010167
Li H-J, Jiang W-H, Liang W-L, Huang J-X, Mo Y-F, Ding Y-Q, Lam C-K, Qian X-J, Zhu X-F, Lan W-J. Induced Marine Fungus Chondrostereum sp. as a Means of Producing New Sesquiterpenoids Chondrosterins I and J by Using Glycerol as the Carbon Source. Marine Drugs. 2014; 12(1):167-175. https://doi.org/10.3390/md12010167
Chicago/Turabian StyleLi, Hou-Jin, Wen-Han Jiang, Wan-Ling Liang, Jia-Xin Huang, Yu-Fei Mo, Yan-Qing Ding, Chi-Keung Lam, Xiao-Jun Qian, Xiao-Feng Zhu, and Wen-Jian Lan. 2014. "Induced Marine Fungus Chondrostereum sp. as a Means of Producing New Sesquiterpenoids Chondrosterins I and J by Using Glycerol as the Carbon Source" Marine Drugs 12, no. 1: 167-175. https://doi.org/10.3390/md12010167
APA StyleLi, H.-J., Jiang, W.-H., Liang, W.-L., Huang, J.-X., Mo, Y.-F., Ding, Y.-Q., Lam, C.-K., Qian, X.-J., Zhu, X.-F., & Lan, W.-J. (2014). Induced Marine Fungus Chondrostereum sp. as a Means of Producing New Sesquiterpenoids Chondrosterins I and J by Using Glycerol as the Carbon Source. Marine Drugs, 12(1), 167-175. https://doi.org/10.3390/md12010167