1. Introduction
Marine macroalgae are known to contain structurally diverse natural compounds with a range of different biological activities [
1]. One class of compounds derived from macroalgae are known as bromophenols (BPs). The BPs are biosynthesized from polyphenols by bromoperoxidases in the presence of hydrogen peroxide and bromide [
2]. They are common marine secondary metabolites [
3], although the algal content seems to vary with season [
4] and tide [
2]. Different biological activities have been reported for the BPs; among these are antioxidant, antimicrobial, anticancer, anti-diabetic and anti-thrombotic effects, which make them interesting compounds in the development of new pharmaceutical agents [
3].
Structurally, the BPs contain one or several benzene rings and a varying number of bromine and hydroxyl-substituents [
3]. The relationship between the molecular structure of some BPs and their antioxidant effect have previously been investigated [
5], and the trend is that the activity is improved by both an increased number of hydroxyl groups and conjugations [
6,
7,
8]. According to the review of Liu and co-workers, the 1,4-dihydroxy arrangement seems to be favorable for antioxidant activity, while bromination appears to be of little importance [
3]. When natural BPs were compared to their corresponding debrominated compounds, it was discovered that bromination even decreased the antioxidant effect [
9].
The first marine BP was isolated from the red alga
Polysiphonia morrowii in 1955 [
10], and subsequently several BPs have been isolated and identified from red, brown and green algae [
3]. Some species of red algae have concentrations of BPs up to 2590 ng/g [
2], and more than 30 monoaryl and diaryl BPs have been discovered in the
Rhodomelaceae family [
11]. Marine alga is not the only marine source of these metabolites, as they are also found in ascidians and sponges [
3]. The red alga
Vertebrata lanosa is a member of the family
Rhodomelaceae. Several synonyms are known for this alga, the name
Polysiphonia lanosa has also been widely used. The alga is commonly distributed in Europe, and it is also found in North America, Canada, tropical and subtropical Western Atlantic [
12]. Crude extracts of
V. lanosa have been reported to show high antioxidant activity in biochemical assays in addition to having a high phenolic content [
13].
Antioxidants serve as a defense against free radicals, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS and RNS form naturally during many metabolic processes, when well regulated, they contribute toward maintaining homeostasis in normal healthy cells and work as signaling molecules [
14]. However, the level of free radicals can increase if this balance is lost, which can happen in response to xenobiotics or environmental stress. When the balance is shifted towards pro-oxidants, a state of oxidative stress occurs, this condition can be a contributing factor to the development of several medical conditions, such as cardiovascular diseases, including atherosclerosis, various types of cancer, diabetes and neurodegenerative diseases, like Parkinson’s and Alzheimer’s disease. Cells have several protective mechanisms against the harmful effects of ROS and RNS, both enzymatic (e.g., superoxide dismutases, catalase and glutathione peroxidase) and nonenzymatic (e.g., GSH, NADPH, α-tocopherol and ascorbic acid) [
14,
15,
16]. These antioxidant mechanisms work to prevent, intercept and repair the damage caused by the free radicals [
14].
Recently, a study was published where the antioxidant activities of 19 naturally occurring BPs were reported, six of which were new, from the alga
Rhodomela confervoides. Most of the isolated compounds showed a significant antioxidant activity against 2,2-Diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) radicals [
5]. At present, approximately 30 BPs with antioxidant activity have been reported from marine algae. The published studies have only investigated the activity
in vitro, mainly determined by the DPPH radical scavenging method [
3].
Thus, the purpose of the present study was to further examine the antioxidant potential of one new and three known BPs isolated form V. lanosa. In addition to a biochemical assay, Oxygen Radical Absorbance Capacity (ORAC), two cellular assays were included; Cellular Antioxidant Activity (CAA) assay and Cellular Lipid Peroxidation Antioxidant Activity (CLPAA) assay. The two latter methods allowed us to explore the bioavailability of the compounds, particularly if they were able to pass cellular membranes.
3. Experimental Section
3.1. Material
Compounds 1–4 were all isolated from the red alga Vertebrata lanosa, collected by the marine biobank Marbank in Oldervik, Ullsfjorden (Norway, spring 2010). The alga was freeze-dried and ground before extraction with water, followed by extraction of the remaining organic material using dichloromethane:MeOH 1:1. The extracts were stored at −24 °C until usage. Only the organic extract was used, due to a larger content of the target compounds.
3.2. Sample Preparation
The extract was dissolved in hexane and partitioned twice against MeOH at room temperature. The MeOH phase was reduced to dryness in vacuo at 40 °C, resuspended in purified water and partitioned twice against EtOAc at room temperature, followed by the removal of volatiles in vacuo at 40 °C and SpeedVac. The EtOAc phase contained a larger concentration of the target compounds and was, thus, used further in the purification process.
3.3. Purification
HPLC purification was performed using a Waters (Milford, MA, USA) purification system controlled by MassLynx version 4.1. The purification system consisted of a Waters 600 pump, a Waters 3100 mass spectrometer (in negative mode, with an ESI-electrospray source), a Waters 2996 photo diode array detector and a Waters 2767 sample manager. Samples were injected in 1:1 H2O/can, and fraction collection was triggered when the intensity of the target mass exceeded the threshold specified in the method. The compounds were isolated on a Waters XTerra MS-C18 (10 × 250 mm, 5 µm) column and a Waters XSELECT CSH (10 × 250 mm, 5 µm) column. Gradients of H2O with 0.1% FA (A) and acetonitrile with 0.1% FA (B) were used at a flow rate of 6 mL/min and optimized for each compound (compound 1: 50%–55% B over 10 min; compounds 2 and 4: 50%–60% B over 10 min; compound 3: 65%–85% B over 25 min).
3.4. Accurate Mass Determination and Elemental Composition of Compounds 1–4 Using High Resolution ESI-MS
Accurate mass determination was performed using a Waters (Milford, MA, USA) UPLC-ToF-MS system controlled by MassLynx version 4.1. The UPLC-ToF-MS system consisted of a Waters LCT Premier and a Waters Acquity UPLC. The compounds were separated on an Acquity UPLC® BEH C18 (2.1 × 50 mm, 1.7 μm) column. Gradients of H2O with 0.1% FA (A) and acetonitrile with 0.1% FA (B) were used at a flow rate of 0.350 mL/min (20%–100% B over 3.5 min).
Possible elemental compositions of compounds 1–4 were calculated using accurate MS, while ChemDraw Pro V 12.0.2 was used to determine the calculated accurate masses.
The calculated elemental compositions were compared with the literature and the information used in combination with NMR data to elucidate the structures of compounds 1–4.
Compound 1: m/z 292.8448 [M − H]+, calculated for C7H4Br2O3 292.8448. Compound 2: m/z 494.8067 [M − H]+, calculated for C14H11Br3O5 494.8078. Compound 3: m/z 572.7182 [M − H]+, calculated for C14H10Br4O5 571.7103. Compound 4: m/z 972.6160 [M − H]+, calculated for C28H20Br6O9 972.6129.
3.5. Identification of Compounds 1–4 by NMR
NMR data were used for structure confirmation of compounds
1–
3 and elucidation of compound
4. The NMR experiments were acquired on an Agilent (Varian) Inova spectrometer operating at 599.934 and 150.863 MHz for
1H and
13C, respectively, using a cryogenically cooled inverse detection HCN probe with enhanced proton channel (2nd generation). Typically, 16k data points were acquired for the 1D
1H experiments using 90° reading pulses, 5 seconds of relaxation delay and 64 transients. Data matrices of 1440 × 200 and 1440 × 256 complex points were acquired for the gradient selected edited HSQC and absolute value HMBC, respectively, using 32 transients. For carbon 1D spectra, 32k complex data points were acquired, using 45° reading pulses, 1-second relaxation delay and 20,000 transients, applying
1H decoupling during both relaxation and acquisition time. All spectra were acquired at 298 K in methanol-
d4 or CDCl
3 (Sigma-Aldrich) using sweep widths of 9600 and 30,200–37,800 Hz for
1H and
13C, respectively. All experiments were referenced to the residual solvent peak; 3.34 ppm for methanol-
d4 and 7.26 ppm for CDCl
3 [
27].
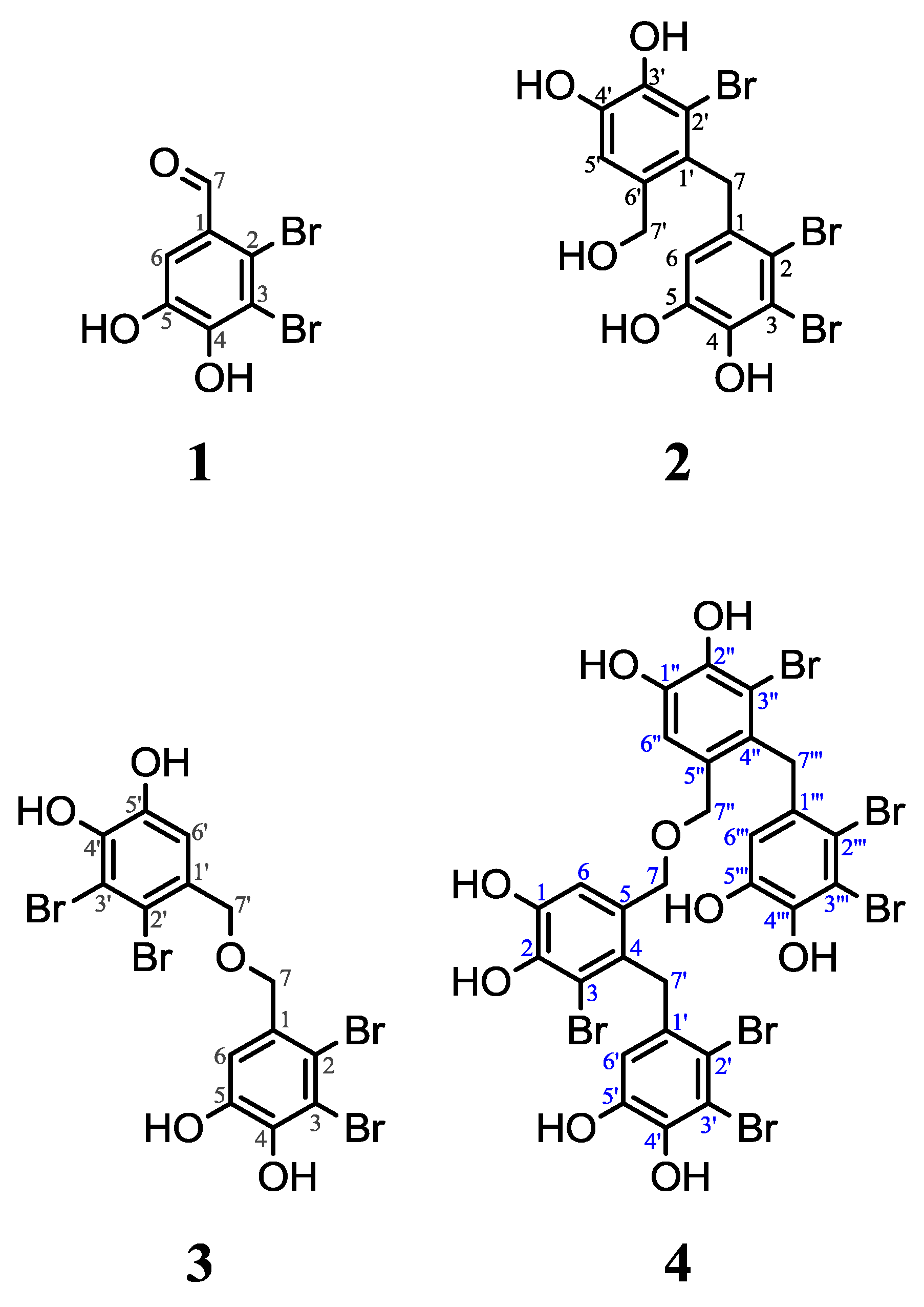
Compound
1 was identified as 2,3-dibromo-4,5-dihydroxybenzylaldehyde, published for the first time by Katsui and co-workers [
28].
1H NMR (600 MHz, methanol-
d4) δ
H 7.34 (s, 1H), 10.12 (s, 1H).
13C NMR (600 MHz, methanol-
d4) δ
C n/a (s, C-3), 152.70 (s, C-4), 146.37 (s, C-5), 114.02 (d, C-6), 126.81 (s, C-1), 192.38 (d, C-7), 122.04 (s, C-2). Impurities of other related compounds (15 mol%) and lipids (25–50 mol%) were present.
Compound
2 was identified as 2,2′,3-tribromo-3′,4,4′,5-tetrahydroxy-6′-hydroxymethyldiphenylmethane, published for the first time by Fan and co-workers [
26]. For
1H and
13C NMR, see
Table 1. It existed in a mixture with compound
4 in the ratio of ~4:1
(1H and
13C NMR values for compound
4 are presented in
Table 1). Impurities of other related compounds (less than 5 mol%) and lipid (6–14 mol%) were present.
Compound
3 was identified as bis(2,3-dibromo-4,5-dihydroxylbenzyl) ether, first published by Kurihara and coworkers [
29].
1H NMR (600 MHz, CDCl
3) δ
H 5.46 (s, OH), 5.60 (s, OH), 7.16 (s, 1H), 4.61 (s, 1H).
13C NMR (600 MHz, CDCl
3) δ
C 113.20 (s, C-3′), 140.60 (s, C-4′), 143.41 (s, C-5′), 115.11 (d, C-6′), 131.67 (s, C-1′), 72.64 (d, C-7′), 113.80 (s, C-2′). Impurities of other related compounds (10 mol%) and lipid (less than 5%) were present.
Compound
4 was identified as 5,5″-oxybis(methylene)bis(3-bromo-4-(2′,3′-dibromo-4′,5′-dihydroxylbenzyl)benzene-1,2-diol) and not reported previously.
13C,
1H and HMBC data are presented in
Table 1. High resolution MS spectra (Figure S1) and the following NMR spectra are attached in the supplementary material:
1H (Figure S2),
13C (Figure S3), superimposed HSQC and HMBC (Figure S4). Compound
4 existed in a mixture with compound
2 in the ratio of ~1:1.5. Impurities of lipid (20–40 mol%) were present.
3.6. Preparation of Solutions
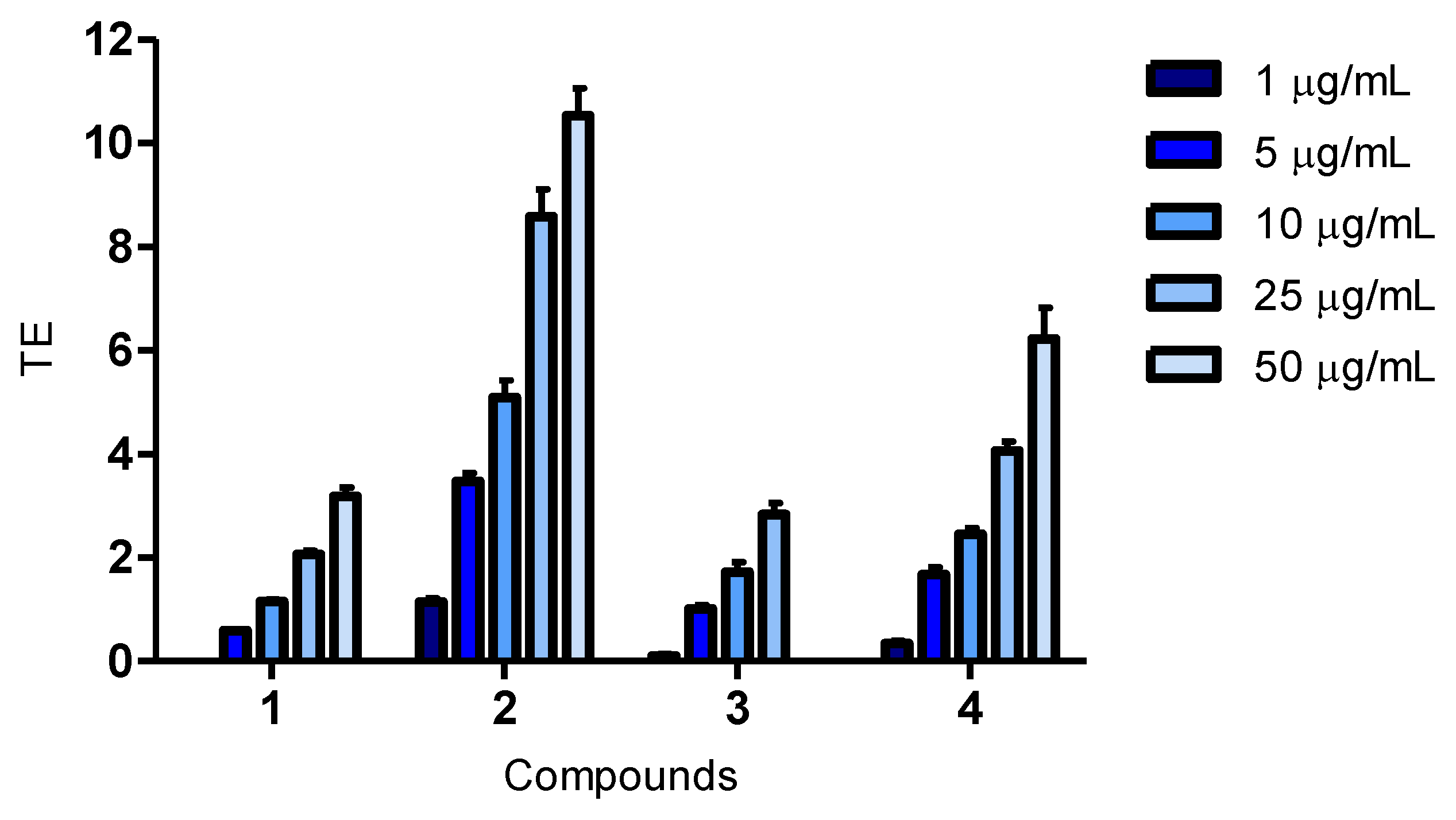
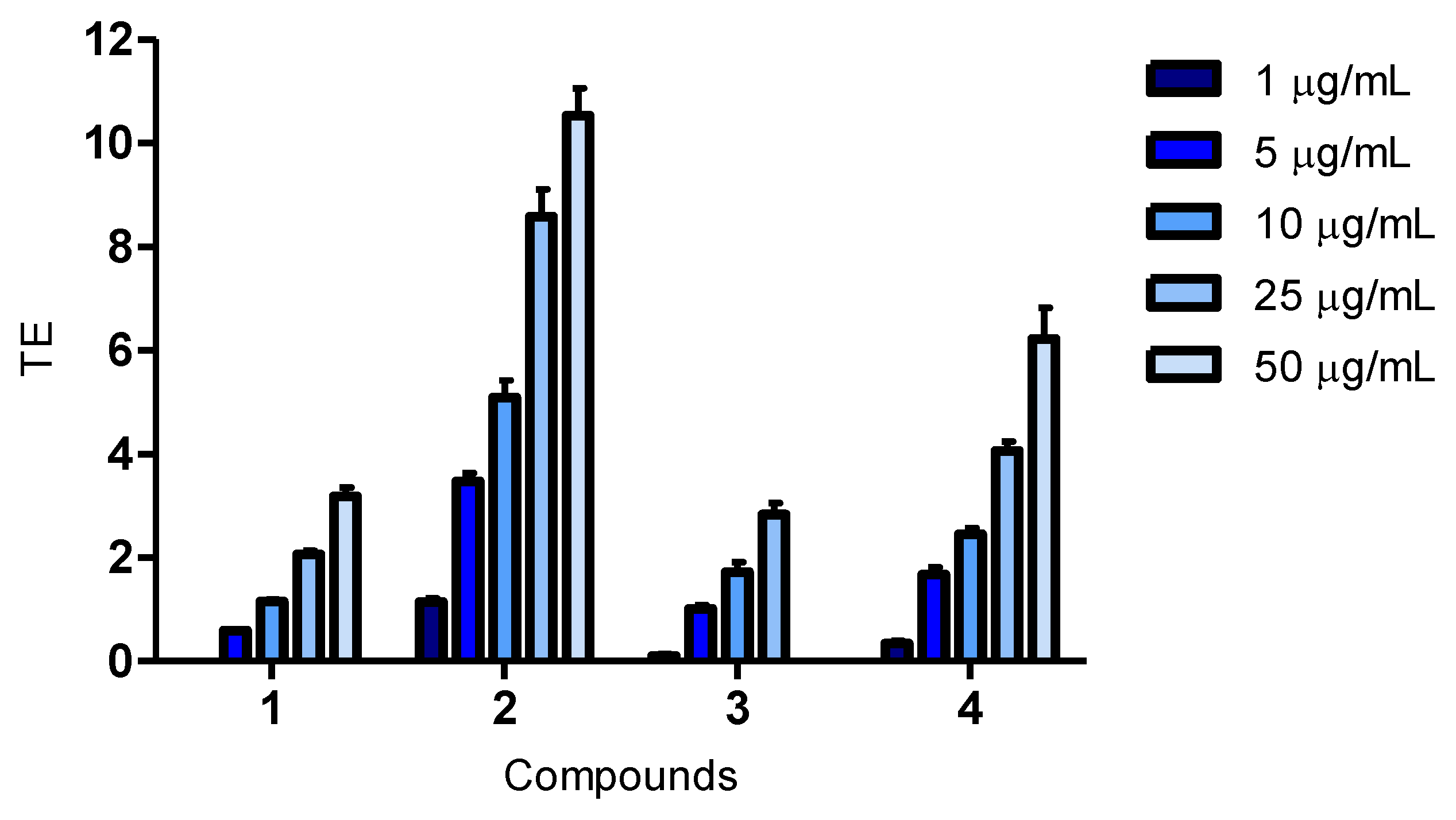
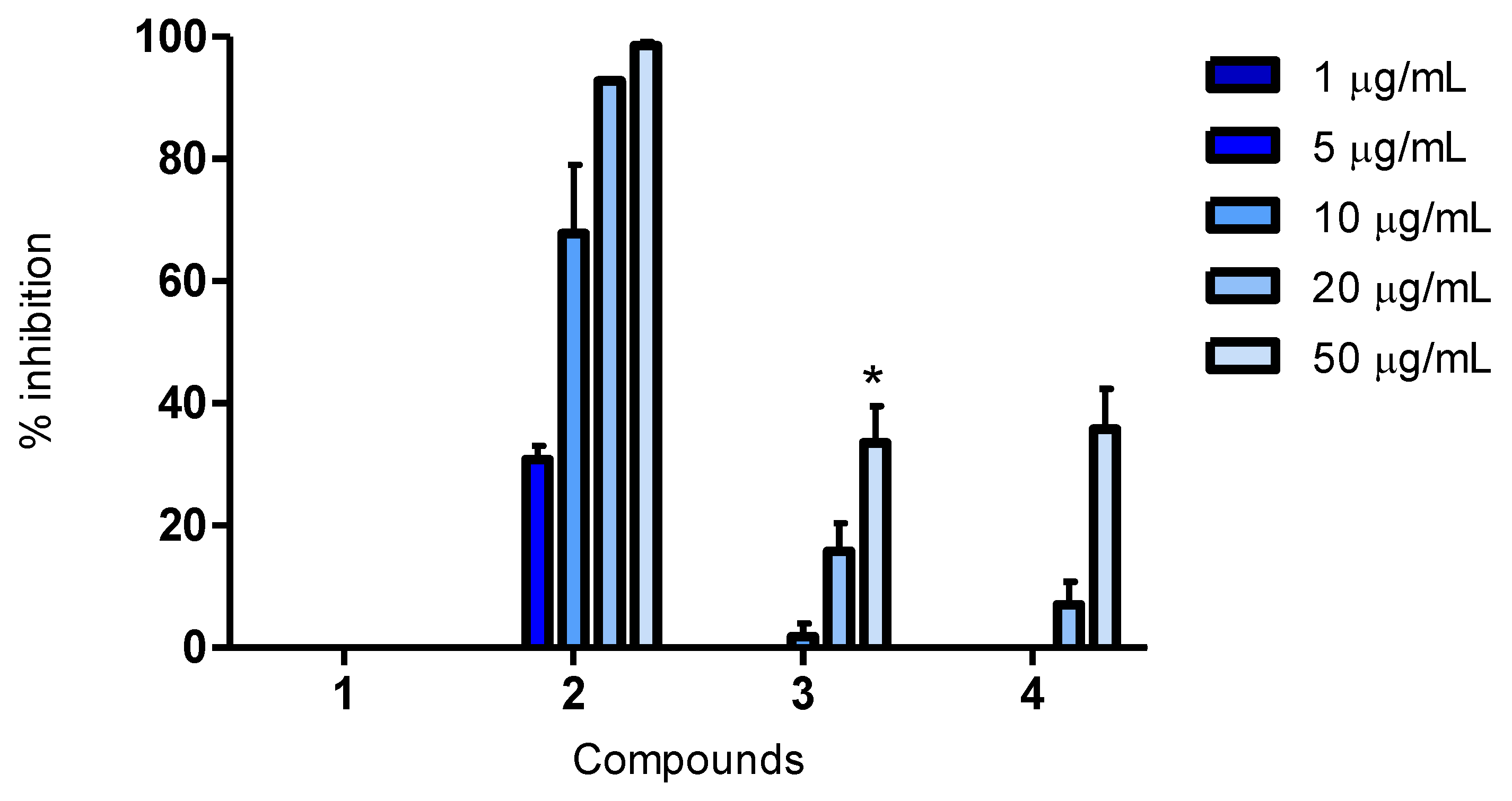
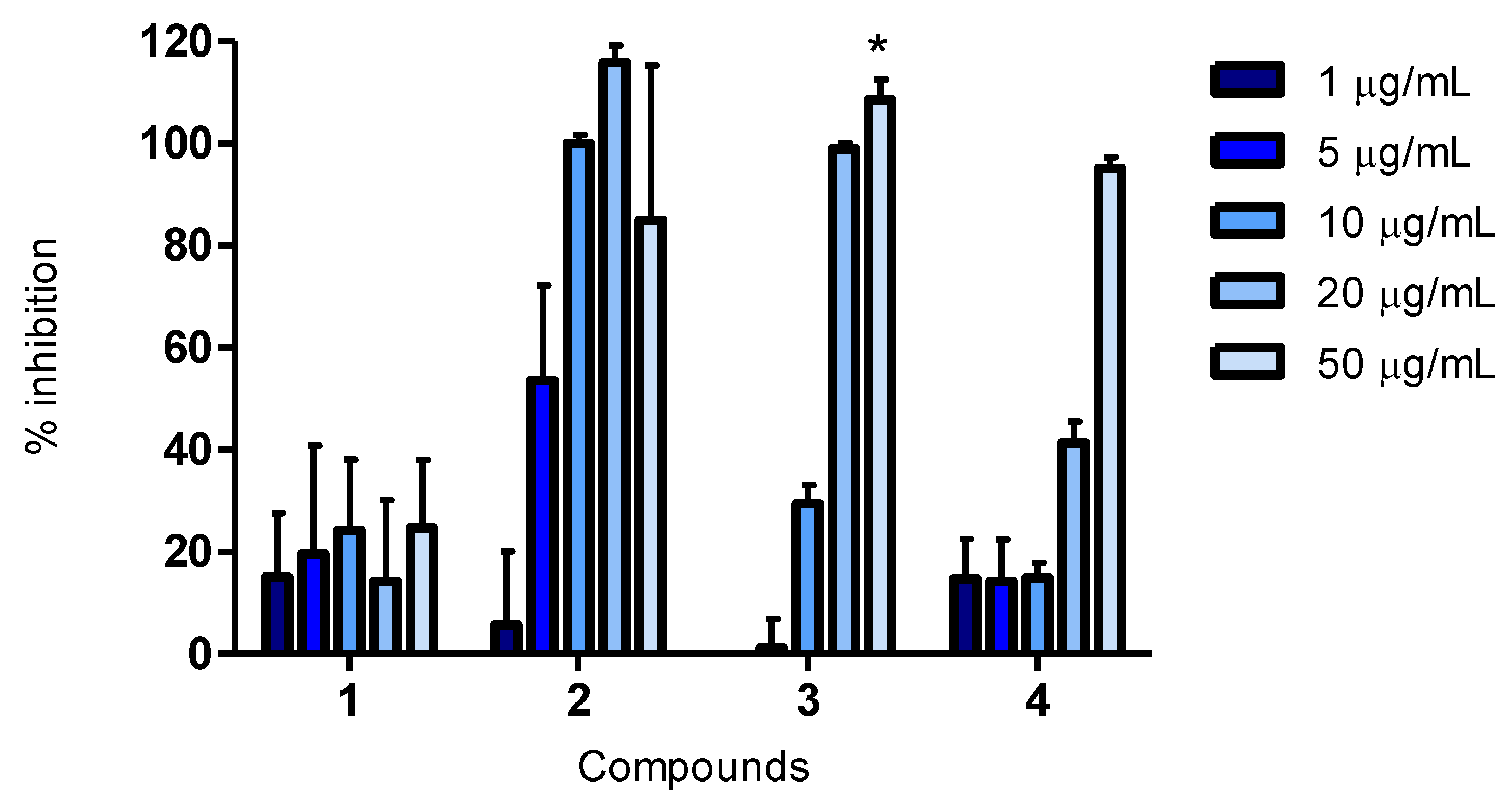
A 20 mg/mL stock solution in DMSO was prepared for each compound. Working solutions were diluted in RPMI-1640 for the cell viability assay, while in MilliQ water for the remaining assays. Final test concentrations for the cell viability assay were 5, 25 and 50 μg/mL. In the ORAC assay, final test concentrations were 1, 5, 10, 25 and 50 μg/mL, while they were 1, 5, 10, 20 and 50 μg/mL for CAA and CLPAA. An exception was compound 3, where 30 μg/mL was the highest concentration in the antioxidant assays.
3.7. Cell Culture and Seeding
MRC-5 (human fetal lung; ATCC, CCL-171™) cells were grown in RPMI-1640 and HepG2 (human hepatocellular liver carcinoma; ATCC HB-8065™) in MEM-Earle’s medium (F0325); both media were supplemented with fetal bovine serum (10%, S0115), non-essential amino acids (1%, K0293), sodium pyruvate (1 mM, L0473), l-alanyl-l-glutamine (2 mM, K0302) and gensumycin (10 μg/mL, A2712) and incubated at 37 °C with 5% CO2. All incubations were performed at 37 °C with 5% CO2. Media and supplements were all from Biochrom (Berlin, Germany).
3.8. Cytotoxicity
Cell viability was determined by a colorimetric [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) assay.
MRC-5 cells were seeded in 96-well microtiter plates (Nunc 167008) at a concentration of 2000 cells/well and incubated for 24 h. After the media was removed, RPMI-1640 with 10% FBS, in addition to the compounds in triplicate, were added. The plate was then incubated for 72 h at 37 °C in a humidified atmosphere of 5% CO2. At the end of the exposure time, CellTiter 96® AQueous One Solution Reagent (Promega, Madison, WI, USA) was added to each well and the plate incubated for 1 h. Absorbance was measured at 485 nm in a DTX 880 Multimode Detector (Beckman Coulter, CA, USA).
Cells in RPMI-1640 medium were used as negative control and cells treated with Triton® X-100 (Sigma-Aldrich) reagent as positive control. Relative cell survival was determined by using the measured optical density (OD), calculated as: cell survival (%) = (OD treated well − OD positive control)/(OD negative control − OD positive control) × 100.
3.9. Antioxidant Assay for Oxygen Radical Absorbance Capacity (ORAC)
The assay was performed based on the procedure of Huang
et al [
19].
The method was carried out in black 96-well plates (Nunc 7350004). Fluorescence was recorded using a 1420 Victor3 Plate Reader (Perkin Elmer, MA, USA, with the software, Workout 2.0, from Dazaq Solutions Ltd.) at 37 °C. The compounds, and Trolox (Sigma-Aldrich, 238813) at final concentrations of 0–17 μM included to construct a standard curve, were added in duplicate, followed by the addition of fluorescein (Fluka, 46960) (final concentration of 55 nM). Quercetin dehydrate (Sigma-Aldrich, 200595) and luteolin (Cayman #10004161) (final concentrations of 1, 5, 10, 25 and 50 μg/mL) were used as antioxidant controls. After a 15-minute incubation, AAPH (2,2′-azobis(2-methylpropioanamidine) dihydrochloride; Sigma-Aldrich, 440914) (final concentration of 16 mM) was added to each sample. Fluorescence was recorded 25 times with a 70-second delay between the repeats. Excitation (486 nm) and emission (520 nm) were used. Phosphate buffer (PB) was used as a blank for the 0 μM Trolox sample. The area under the curve (AUC) was calculated with the AUCblanc values subtracted. A standard curve was made using the Trolox values, and Trolox equivalents of the samples were calculated. Results were expressed as μM Trolox equivalents (TE). Total reaction volume was 210 µL (where 25 µL was the volume of the test compound). All reagents were dissolved in 75 nM PB (pH 7.4), and the incubations were at 37 °C.
3.10. Antioxidant Assay for Cellular Antioxidant Activity (CAA)
The assay was performed based on the procedure of Wolfe and Liu (2007) [
22].
The method was carried out in black 96-well plates (#3603, Corning, NY, USA) with an optical bottom. Fluorescence was recorded using the same plate reader, software and temperature as for the ORAC assay. HepG2 cells were seeded at a density of approximately 80,000 cells/well and incubated overnight. Cells were then incubated with DCFH-DA (2′7′-dichlorofluorescin diacetate; Fluka, 35847) (final concentration of 25 μM) and the test compounds in duplicate for 1 h. Quercetin and luteolin (final concentrations of 1, 5, 10, 20 and 50 (or 30) μg/mL) were used as antioxidant controls. After incubation, Hank’s saline solution without phenol red (Biochrom, BCHRL2035) supplemented with AAPH (final concentration of 600 μM) was added to all the wells, except for the negative control. To these wells was added Hank’s saline solution without AAPH. The plate was immediately placed in the plate reader and fluorescence recorded; excitation of 485 nm and emission of 520 nm were used. The plate was incubated for 1 h before a second reading. Cells were washed with PBS between the addition of new reagents. The total reaction volume was 100 μL (where 20 μL was the volume of the test compound). The incubations were at 37 °C in a humidified atmosphere of 5% CO2.
Cells in MEM Earle’s medium were used as negative control and cells treated with AAPH as positive control. The oxidative degeneration of DCFH-DA to DCF was calculated as: activity (%) = (fluorescence treated well − fluorescence positive control)/(fluorescence negative control − fluorescence positive control) × 100. Fluorescence value: fluorescence reading 2 − fluorescence reading 1. Inhibition (%) was calculated as: 100% − activation%. The results are presented as relative values compared to the positive and negative controls, with and without AAPH, respectively.
3.11. Cellular Lipid Peroxidation Antioxidant Activity (CLPAA) Assay
The assay was performed based on the procedure of Pap
et al. [
23].
The method was carried out in the same plates as in the CAA assay. Fluorescence was recorded using the same plate reader, software and temperature as for the ORAC and CAA assays. HepG2 cells were seeded and incubated overnight, as described in the CAA assay. The cells were labelled with 5 µM C11-BODIPY (#D3861, Invitrogen, Eugene, OR, USA) for 30 min, following the addition of compounds at various concentrations and incubating for 1 h. Quercetin and luteolin (final concentrations of 1, 5, 10, 20 and 50 (or 30) μg/mL) were used as antioxidant controls. Cumene hydroperoxide (cumOOH, 247502, Sigma-Aldrich, St. Louis, MO, USA) (at a final concentration of 50 µM) in Hank’s saline solution without phenol red was added to initiate lipid peroxidation. The addition was done to all the wells, except the ones for negative control, to which were added Hank’s saline solution without phenol red and without cumOOH. The plate was immediately placed in the plate reader. Both red [590 (excitation)/632 (emission)] and green [485 (excitation)/520 (emission)] fluorescence was recorded during ~1 h at 37 °C. Cells were washed with PBS between additions of new reagents. The total reaction volume was 100 μL (where 20 μL was the volume of the test compound). All incubations were carried out at 37 °C in a humidified atmosphere of 5% CO2.
Cells in MEM Earle’s medium were used as negative control and cells treated with cumOOH as positive control. The oxidative degeneration of C11-BODIPY was calculated based on the increase of green fluorescence as: activity (%) = (fluorescence treated well − fluorescence positive control)/(fluorescence negative control − fluore scence positive control) × 100. Inhibition (%) was calculated as: 100% − activation%. The results are presented as relative values compared to the positive and negative controls, with and without cumOOH, respectively.
4. Conclusions
Cellular antioxidant effects for the bromophenol, 2,2′,3-tribromo-3′,4,4′,5-tetrahydroxy-6′-hydroxymethyl-diphenylmethane (compound 2), have been reported for the first time. The fact that the compound was active in the CAA assay demonstrates that it enters cells, and as it was also active in the CLPAA assay, it shows the ability to decrease lipid peroxidation in cell membranes. Compound 2 was a better antioxidant than luteolin in both cellular assays and of quercetin in the CLPAA assay, and it was active well below the cytotoxic concentration in all assays.
The structure of compound
4 and its activity in biological assays is reported for the first time, and compared to quercetin and luteolin, it showed a weak antioxidant activity at 50 μg/mL in all the assays (
Figure 1,
Figure 2,
Figure 3). The possibility of being an artefact of the extraction and purification process has been discussed.
Only a few studies have been reported on structure-activity relationships (SARs) for BPs, and the ones published are all from
in vitro biochemical assays [
3]. Hydroxyl substitution, a 1,4-dihydroxy arrangement, conjugation and bromination are all factors believed to influence the antioxidant activity of BPs in these assays. Results show that, more extensively, hydroxyl substitution increases antioxidant activity of BPs in DPPH radical scavenging assays [
3,
6,
7,
8,
30,
31]. This corresponds well with the results from our cellular assays, where compound
2, having one hydroxyl substituent more, had a greater antioxidant activity than compound
3. However, compound
4 contained three hydroxyl substituents more than compound
2, but was found to be less active than the latter. An explanation for this might be that compound
4 is a larger molecule, which can influence the cellular uptake. The 1,4-dihydroxy arrangement has been reported to be very suitable for antioxidant activity, and compound
2 resembles such an arrangement, having a hydroxyl and a hydroxymethyl in
para position to each other [
3,
32]. The degree of conjugation is proposed as an important factor in increasing the antioxidant activity of BPs [
3]. Nevertheless, compound
2 does not have a higher degree of conjugation, so this does not explain the results in the present study. Bromination of BPs has also been investigated in DPPH radical scavenging assays. The antioxidant activity was reported both as decreasing, when compared to corresponding debrominated compounds, and as slightly increasing, when compared to corresponding chlorinated compounds. Liu and co-workers, therefore, concluded that bromination seemed to be of little importance for the antioxidant activity of BPs [
3,
9,
31]. Halogenation of polar molecules is a known way to increase their lipophilicity and, consequently, increase their bioavailability [
33]. Thus, although bromination was found not to be of importance for antioxidant activity in biochemical assays, it might influence the bioavailability of compounds. To be active in cellular assays, the compounds have to penetrate the cell membrane, and the ability to do so is influenced by their size, solubility, polarity and charge. We believe that the poor activity of compound
4 in the cellular assays is due to its larger size compared to compound
2.
This present identification of cellular antioxidant activity of structurally related BPs can be used as a starting point for SAR studies, which are more relevant to in vivo conditions, taking into account factors like bioactivity and membrane permeability. The cellular assays can be used to, at an early stage, exclude compounds lacking the ability of penetrating membranes and, hence, do not have an effect in vivo.




{kind=link}
{kind=link}
{kind=link}
{kind=link}