2.1. APS8 Induces Cytotoxicity in Lung Cancer (LC) Cells
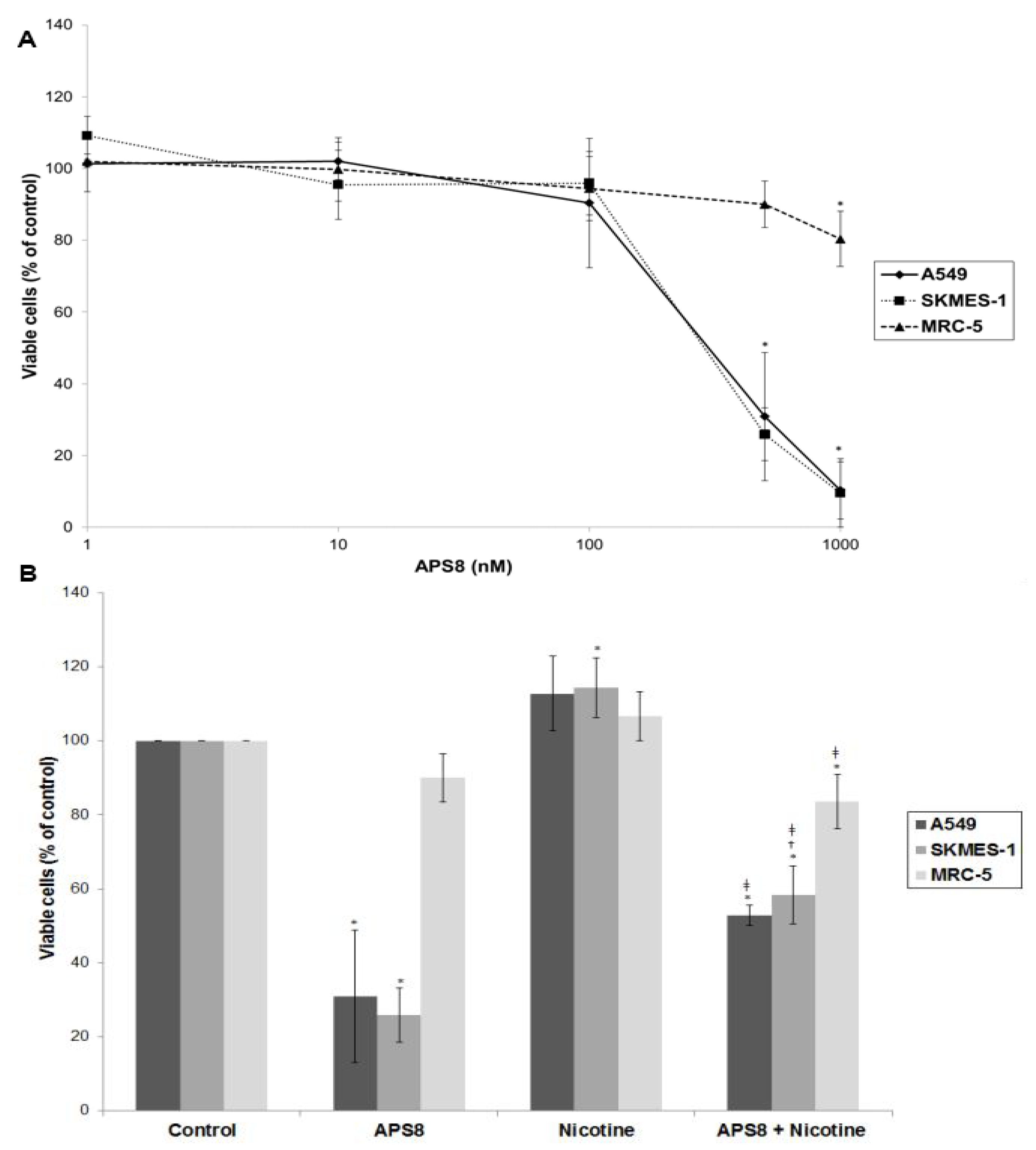
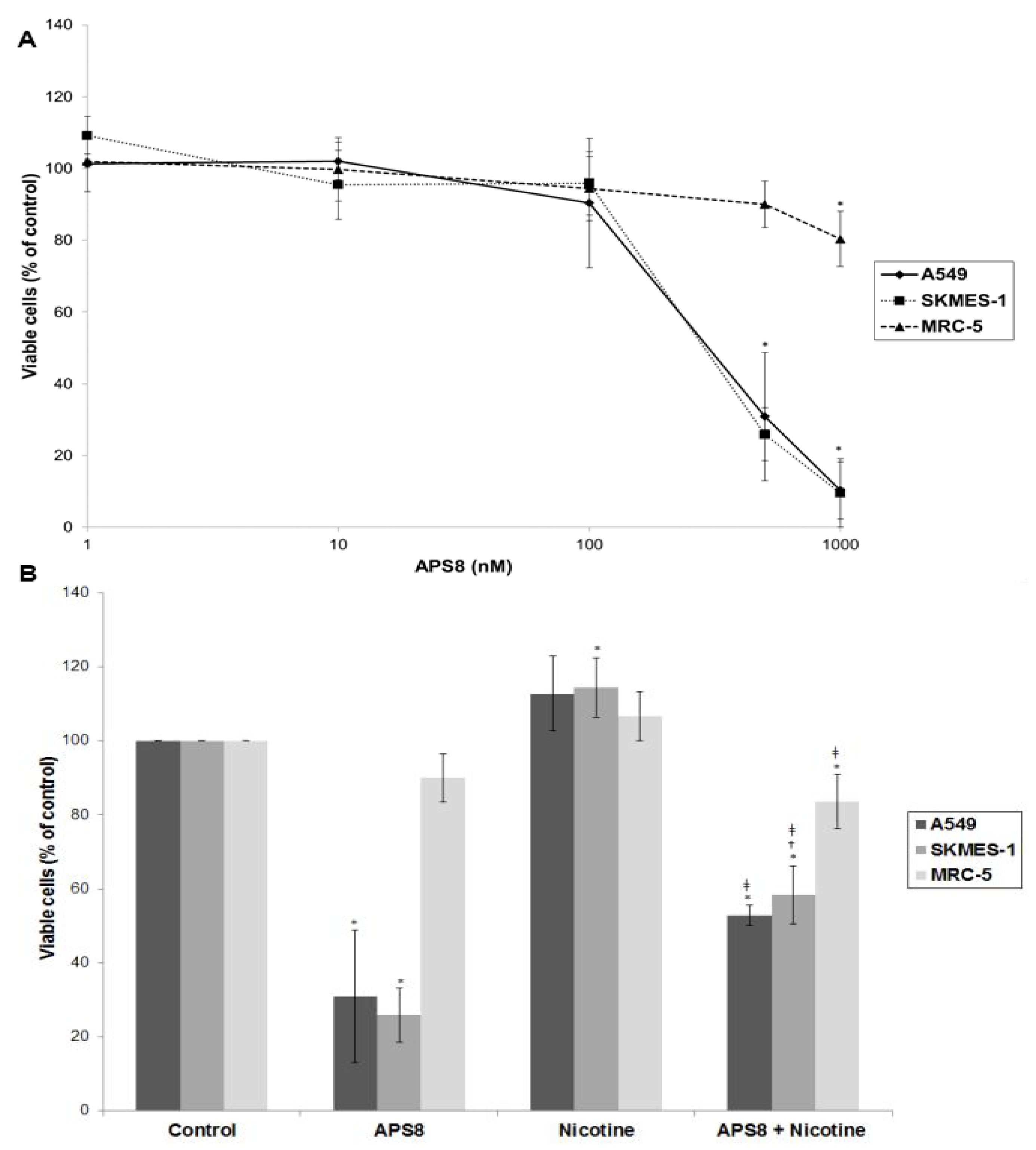
In order to examine if APS8 is cytotoxic to LC cells the SKMES-1 and A549 cell lines were treated with various concentrations of APS8 for 48 h and analyzed for cell viability by MTT-assay (
Figure 2A). The effect on normal lung fibroblasts was also examined. APS8 in a concentration dependent manner strongly decreased viability of LC cell lines (IC
50 375 ± 4.89 nM for A549 cells and 362 ± 9.29 nM for SKMES-1 cells). Lung fibroblast cell line MRC-5 was largely unaffected thus incubation of these cells for 48 h with APS8 only resulted in a 20% decrease in cell viability at the highest concentration (1 μM). Next, the effect of APS8 on nicotine response was examined. Nicotine alone slightly enhanced cell survival of both A549 and SKMES-1 (13% for A549 and 14% for SKMES-1) (
p < 0.05) while only a minor effect was observed with MRC-5 normal fibroblasts (6%) (
Figure 2B). Importantly, APS8 significantly counteracted nicotine-induced effects in both LC cells (about 50%) while MRC-5 normal cells were much less affected. As compared to the APS8 only treatment, a combination of APS8 with nicotine caused a statistically significant (
p < 0.05) increase of viable SKMES-1 cells (for 28%) and statistically insignificant increase of viable A549 cells (for 22%), while normal cells were not affected.
Figure 2.
Viability of NSCLC (A549, SKMES-1) and normal lung fibroblast MRC-5 cells. (A) Viability of A549, SKMES-1 and MRC-5 cells treated with 0, 1, 10, 100, 500, and 1000 nM APS8 for 48 h was assessed by MTT assay. Each point represents the mean value of three independent experiments ± SE. Statistical analysis was performed by Student’s t-test. * P < 0.05; (B) Viability of A549, SKMES-1 and MRC-5 cells treated with APS8 (500 nM), nicotine (1 μM) or a combination of both compounds for 48 h. The MTT assay was used. Each point represents the mean value of three independent experiments ± SE. Statistical analysis was performed by ANOVA/Tukey-Kramer multiple comparison. * P < 0.05, compared with control; † P < 0.05, compared with APS8 treatment; ‡ P < 0.05, compared with nicotine treatment.
Figure 2.
Viability of NSCLC (A549, SKMES-1) and normal lung fibroblast MRC-5 cells. (A) Viability of A549, SKMES-1 and MRC-5 cells treated with 0, 1, 10, 100, 500, and 1000 nM APS8 for 48 h was assessed by MTT assay. Each point represents the mean value of three independent experiments ± SE. Statistical analysis was performed by Student’s t-test. * P < 0.05; (B) Viability of A549, SKMES-1 and MRC-5 cells treated with APS8 (500 nM), nicotine (1 μM) or a combination of both compounds for 48 h. The MTT assay was used. Each point represents the mean value of three independent experiments ± SE. Statistical analysis was performed by ANOVA/Tukey-Kramer multiple comparison. * P < 0.05, compared with control; † P < 0.05, compared with APS8 treatment; ‡ P < 0.05, compared with nicotine treatment.
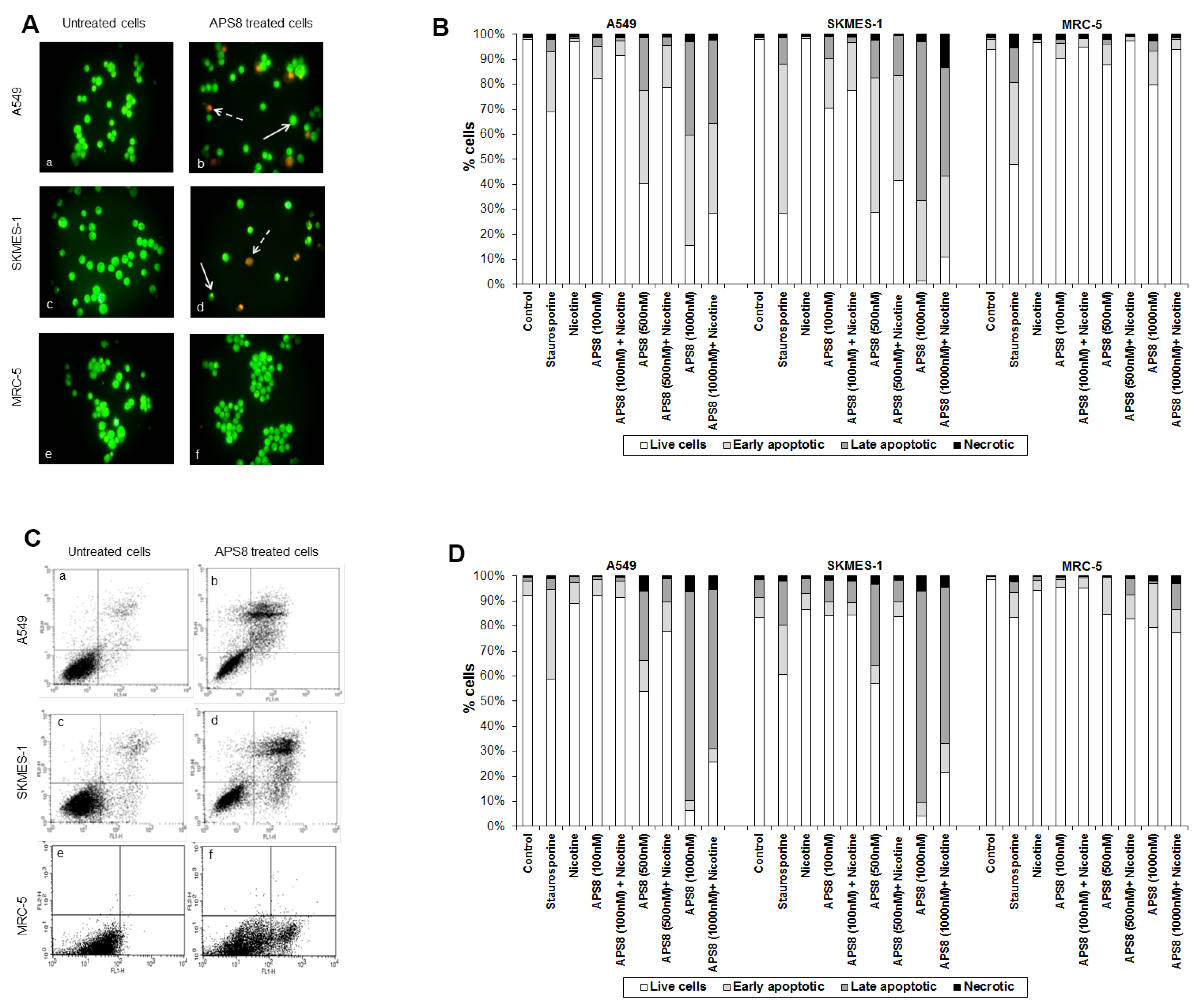
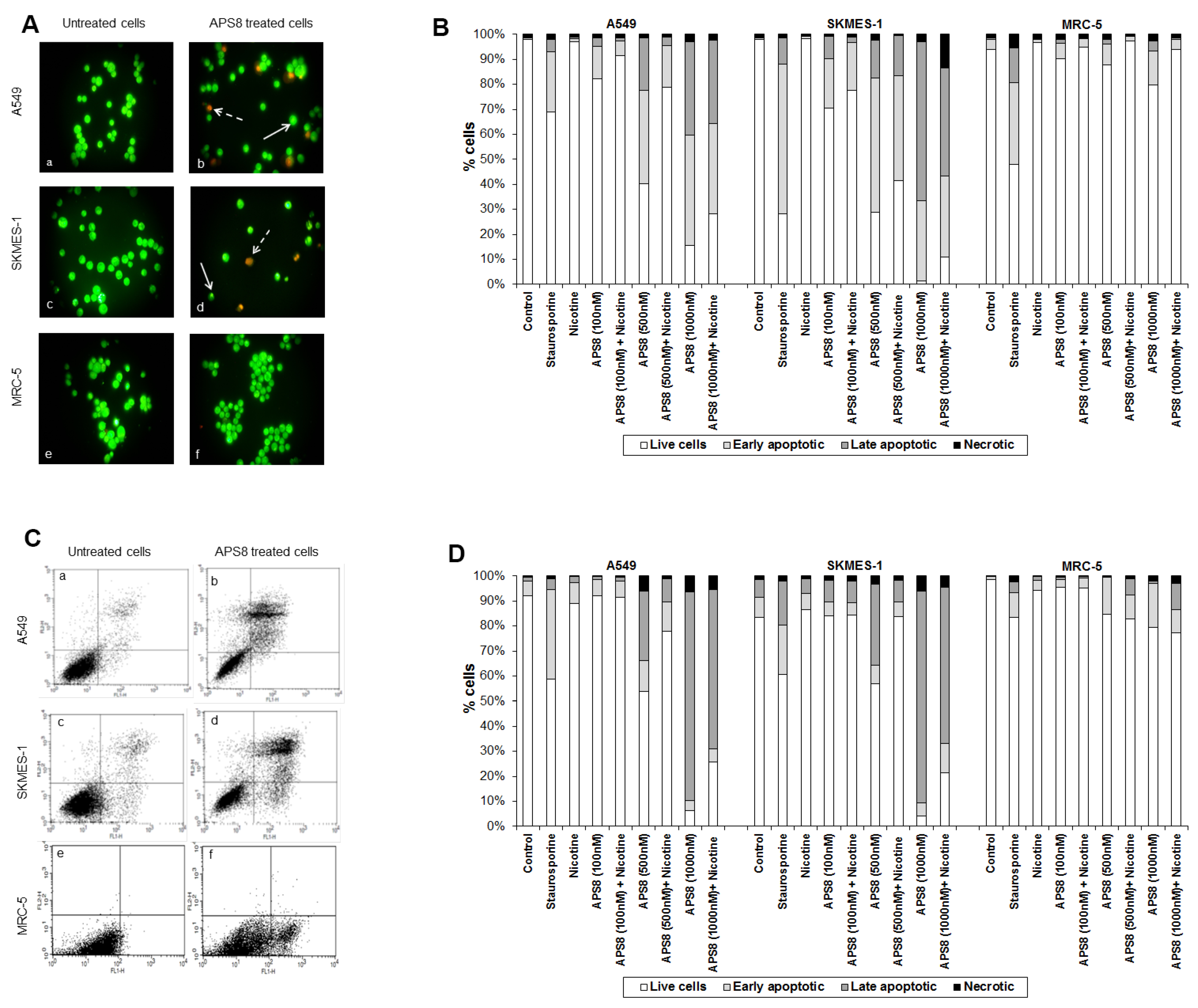
APS8 caused a prominent induction of apoptotic cell morphology in both A549 and SKMES-1 LC cells (
Figure 3A, panel b and d). Quantification of APS8-induced apoptosis revealed a statistically significant (
p < 0.05) and comparable response in A549 and SKMES-1 cells where about 40% of cells were found to be apoptotic after exposure to 500 nM of APS8 for 48 h (
Figure 3B). Importantly, no induction of apoptosis was seen in normal fibroblasts MRC-5, which displayed the same nuclear morphology in the presence or absence of APS8 (
Figure 3A, panel f and
Figure 3B), thus corroborating a cancer cell specific apoptotic effect of APS8. The positive control staurosporine induced apoptosis in all cell types examined with the A549 cell line being least affected with only a 30% induction of apoptosis.
Figure 3.
APS8 induces apoptosis in NSCLC but not in normal fibroblasts. (A) Apoptosis after APS8 treatment (500 nM, 48 h) in A549, SKMES-1, and MRC-5 were assessed by staining with acridine orange and ethidium bromide and analysis by fluorescence microscope. Photos were taken at 400× magnification. Dashed arrows indicate cells in early apoptosis and full arrows point to late apoptotic cells. Green cells are alive; (B) Induction of apoptosis in A549, SKMES-1, and MRC-5 lines as measured by dual staining. Cells were treated with staurosporine (2 μM), APS8 (100 nM, 500 nM, and 1000 nM), nicotine (1 μM) or combination of APS8 and nicotine. The graph indicates the percentage of cells in the single cell populations. Each point is the mean of three independent experiments. The protective effect of nicotine was significant only for A549 cancer cells treated with 500 nM of APS8 (* P < 0.05); (C) APS8 induction of apoptosis in A549, SKMES-1, and MRC-5 cell lines was measured by flow cytometric analysis of annexin V and propidium iodide stained cells at 48 h. Controls (a, c, and e) and APS8 (500 nM) treated cells (b, d, and f); (D) Induction of apoptosis in A549, SKMES-1 and MRC-5 lines by flow cytometry. Cells were treated with staurosporine (2 μM), APS8 (100 nM, 500 nM, and 1000 nM), nicotine (1 μM) or combination of APS8 and nicotine. The graph indicates the percentage of gated cells in each cell population. Each point is the mean of three independent experiments.
Figure 3.
APS8 induces apoptosis in NSCLC but not in normal fibroblasts. (A) Apoptosis after APS8 treatment (500 nM, 48 h) in A549, SKMES-1, and MRC-5 were assessed by staining with acridine orange and ethidium bromide and analysis by fluorescence microscope. Photos were taken at 400× magnification. Dashed arrows indicate cells in early apoptosis and full arrows point to late apoptotic cells. Green cells are alive; (B) Induction of apoptosis in A549, SKMES-1, and MRC-5 lines as measured by dual staining. Cells were treated with staurosporine (2 μM), APS8 (100 nM, 500 nM, and 1000 nM), nicotine (1 μM) or combination of APS8 and nicotine. The graph indicates the percentage of cells in the single cell populations. Each point is the mean of three independent experiments. The protective effect of nicotine was significant only for A549 cancer cells treated with 500 nM of APS8 (* P < 0.05); (C) APS8 induction of apoptosis in A549, SKMES-1, and MRC-5 cell lines was measured by flow cytometric analysis of annexin V and propidium iodide stained cells at 48 h. Controls (a, c, and e) and APS8 (500 nM) treated cells (b, d, and f); (D) Induction of apoptosis in A549, SKMES-1 and MRC-5 lines by flow cytometry. Cells were treated with staurosporine (2 μM), APS8 (100 nM, 500 nM, and 1000 nM), nicotine (1 μM) or combination of APS8 and nicotine. The graph indicates the percentage of gated cells in each cell population. Each point is the mean of three independent experiments.
![Marinedrugs 11 02574 g003]()
Next, we investigated whether APS8 is able to induce apoptosis in nicotine treated LC and fibroblasts (
Figure 3B). As expected, nicotine alone did not trigger an apoptotic response in any of the cell types examined. LC cells treated with a combination of nicotine and APS8 displayed a greater resistance to apoptosis as compared to those treated only with APS8. Moreover, a greater sensitization was observed in SKMES-1 cells relative to A549 cells. In MRC-5 cells only the highest dose of APS8 induced limited apoptosis and this was reduced by the simultaneous exposure to nicotine.
The apoptotic properties of APS8 were also examined using annexin-V/PI staining. Exposure of A549 or SKMES-1 cells to APS8 resulted in typical apoptotic cells, evident as a shift to the right quadrants of the flow diagram (
Figure 3C, panels b and d). Quantification of cell populations demonstrated a concentration dependent induction of apoptosis in both A549 and SKMES-1 cells (
Figure 3D). Importantly, no induction of annexin-V was observed in normal MRC-5 fibroblasts (
Figure 3C, panel f). Even at the highest concentration of APS8 used (1 μM), 80% of MRC-5 cells remained non-apoptotic (
Figure 3D).
We also analyzed whether nicotine attenuates APS8 induced apoptosis using this assay (
Figure 3D). Although nicotine slightly reduced APS8-induced apoptosis in A549 cells, apoptosis was still evident. A minor protective effect of nicotine was also evident in SKMES-1 cells. APS8 in any of the concentrations used did not induce apoptosis in MRC-5 fibroblasts or influenced their response to nicotine. Hence, our results support that APS8 has capacity to trigger an apoptotic response in LC cells whereas normal cells remain unaffected.
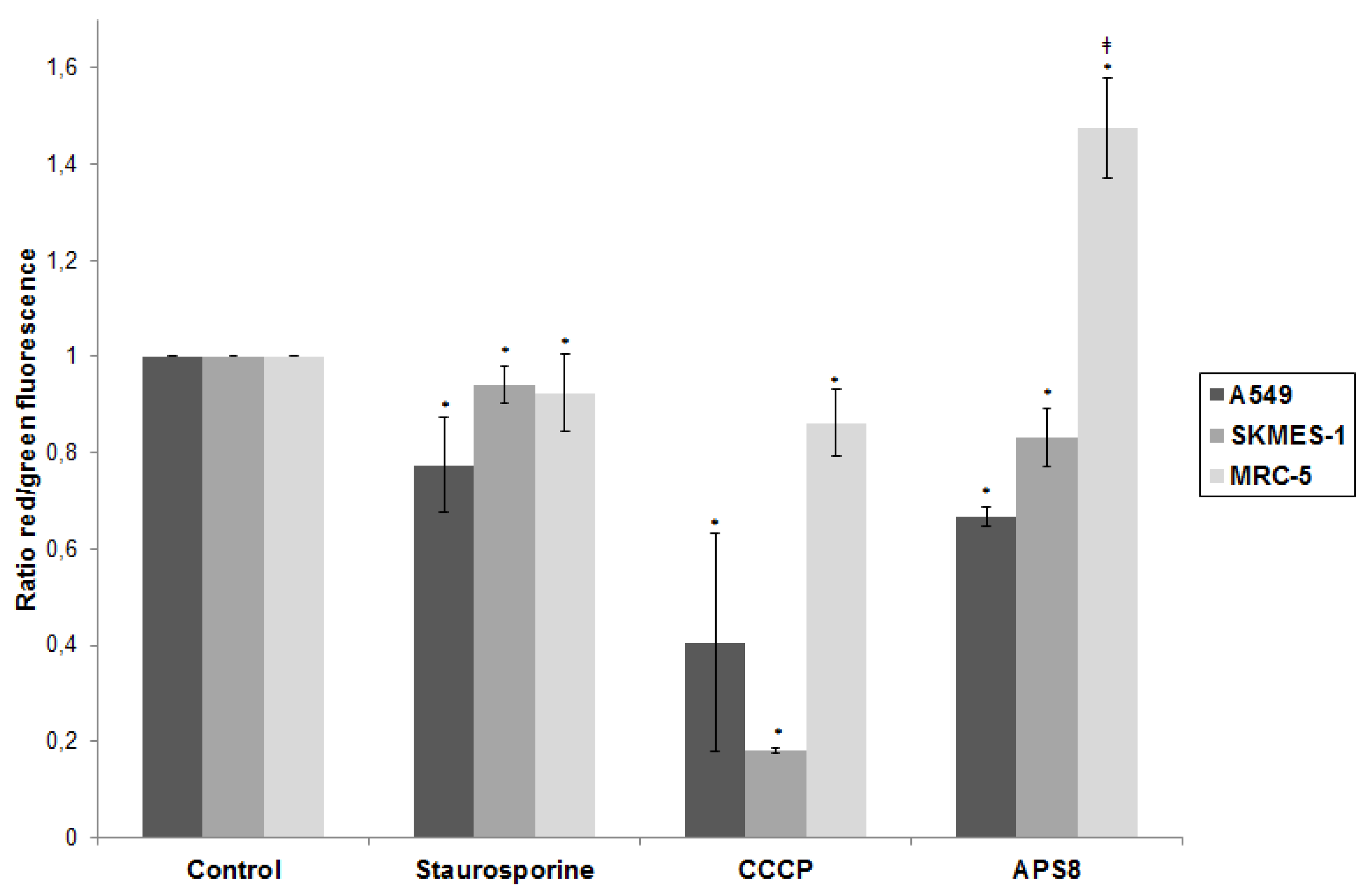
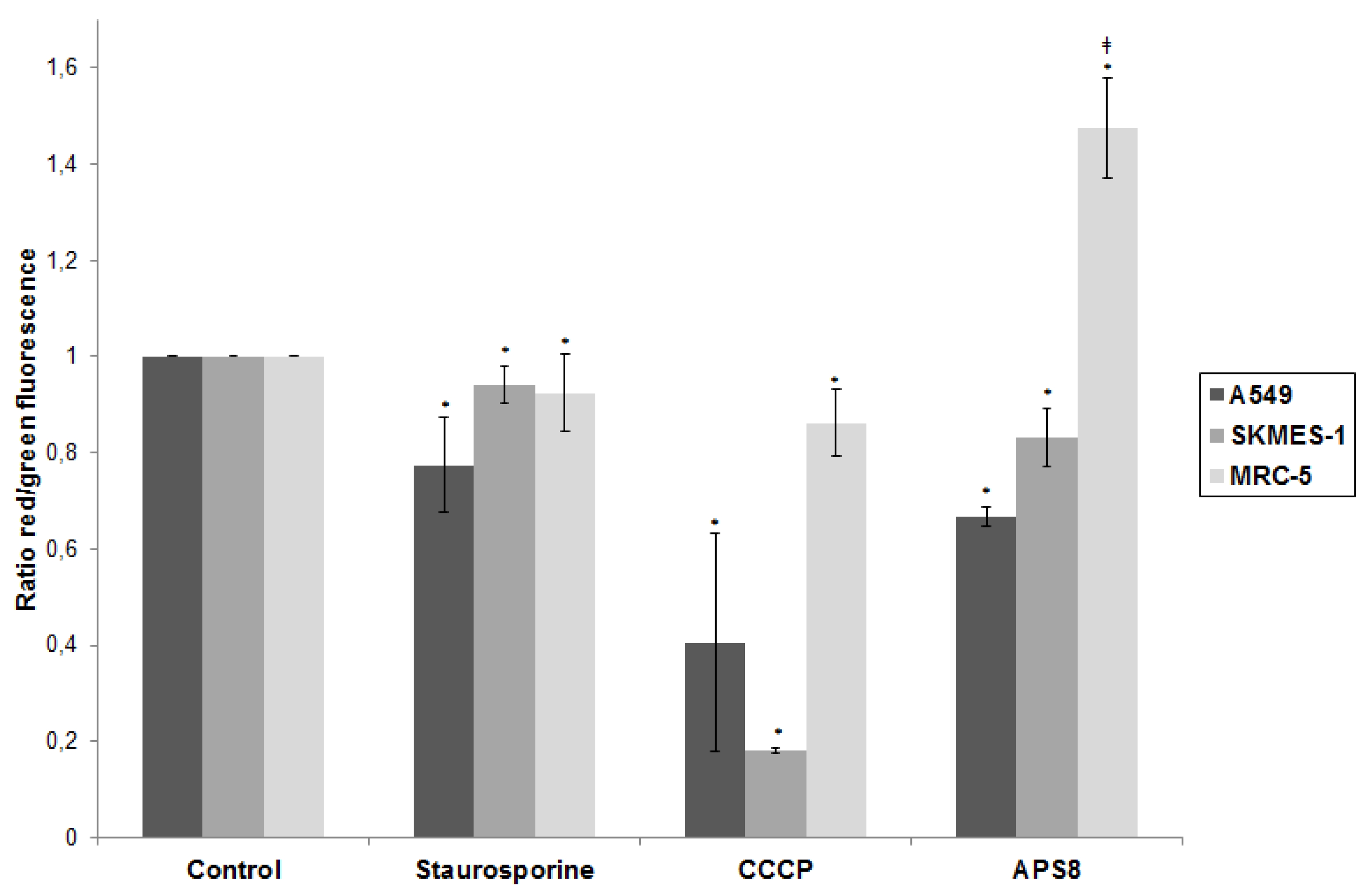
Figure 4.
APS8 causes depolarization of mitochondria in NSCLC cells but not in normal fibroblasts. Ratio of red and green fluorescence following exposure to staurosporine (1 μM), CCCP (50 mM), APS8 (500 nM) for 48 h. Each point is the mean of three independent experiments ± SE. Statistical analysis was performed by ANOVA/Tukey-Kramer multiple comparison. * P < 0.05, compared with control; ‡ P < 0.05, compared with staurosporine treatment.
Figure 4.
APS8 causes depolarization of mitochondria in NSCLC cells but not in normal fibroblasts. Ratio of red and green fluorescence following exposure to staurosporine (1 μM), CCCP (50 mM), APS8 (500 nM) for 48 h. Each point is the mean of three independent experiments ± SE. Statistical analysis was performed by ANOVA/Tukey-Kramer multiple comparison. * P < 0.05, compared with control; ‡ P < 0.05, compared with staurosporine treatment.
2.3. APS8 Treatment Results in Increased Expression of Pro-Apoptotic Proteins and Down-Regulation of Anti-Apoptotic Proteins in LC Cells
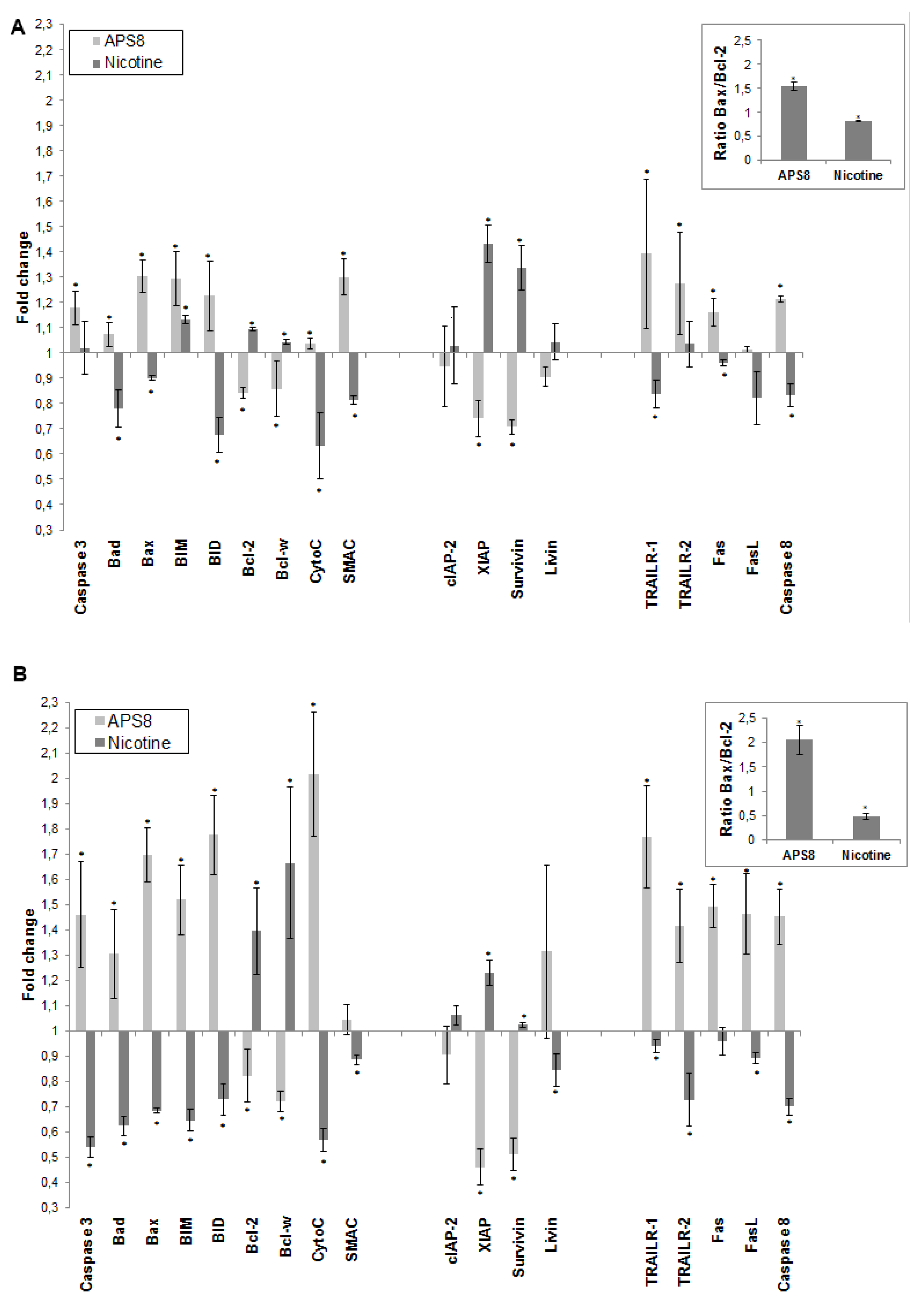
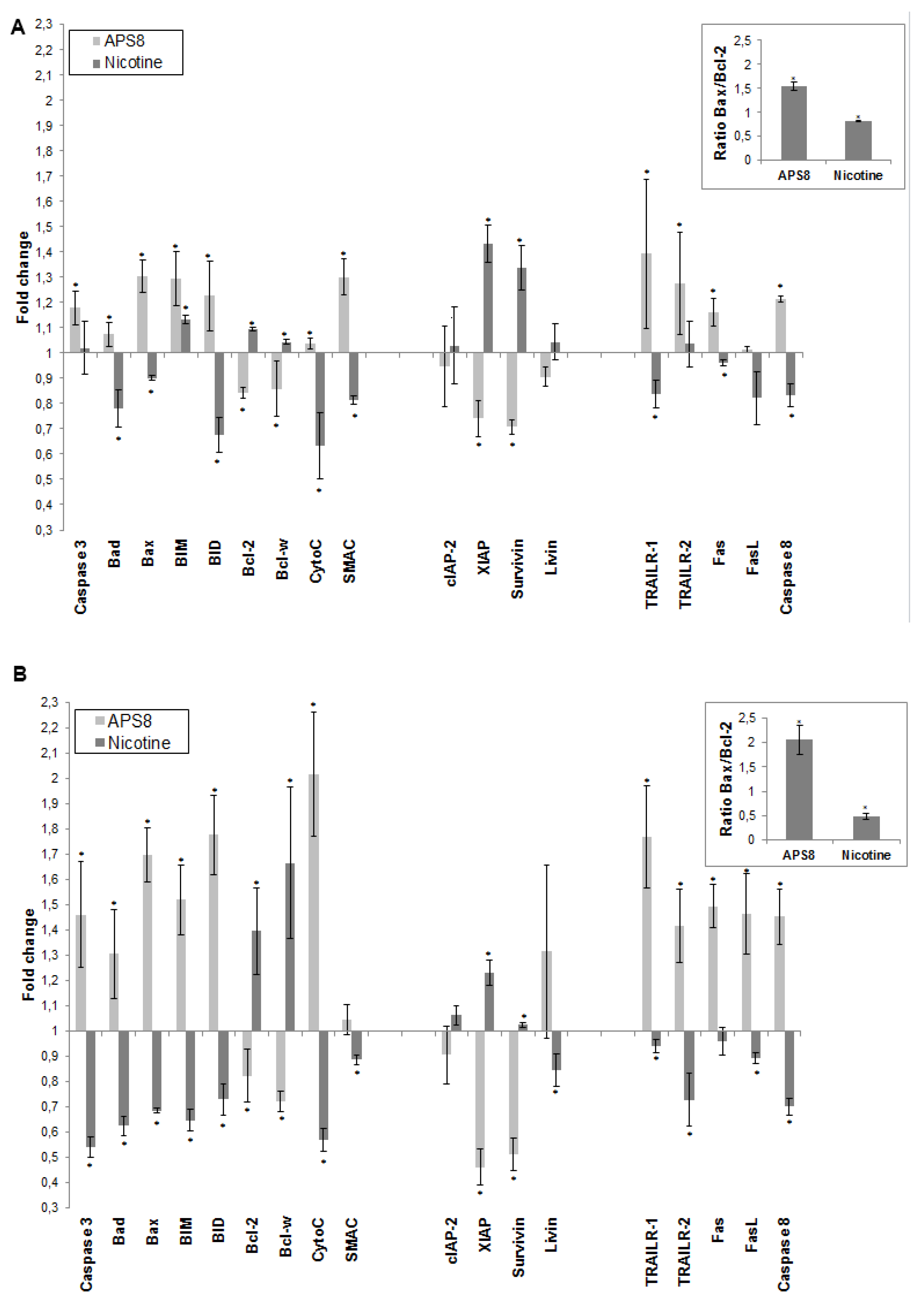
To further understand how APS8 influenced the apoptotic propensity of LC cells we examined the effect of APS8 on the expression of a number of pro-and anti-apoptotic proteins using the Human Apoptosis Antibody Array. APS8 (500 nM) treatment of A549 cells for 48 h resulted in up-regulation of several pro-apoptotic proteins,
i.e., bad, bax, bim and bid, cytochrome C, SMAC, TRAIL-R1 and TRAIL-R2, Fas, FasL, caspase-8, and caspase-3 albeit to the different extent (
Figure 5A). Moreover, the majority of anti-apoptotic proteins, including bcl-2 and bcl-W, c-IAP-2, XIAP, survivin, and livin were down-regulated in A549 cells after APS8 exposure (
Figure 5A).
Similarly, treatment of SKMES-1 cells with APS8 (500 nM) also resulted in an increased expression of the pro-apoptotic proteins,
i.e., bad, bax, bim and bid, cytochrome C, SMAC, TRAIL-R1 and TRAIL-R2, Fas, caspase-8 and caspase-3, with a slighter higher expression than was observed in A549 cells (
Figure 5B). In accordance with results in A549 cells also SKMES-1 cells responded to APS8 with a down-regulated expression of the anti-apoptotic proteins bcl-2, bcl-W, cIAP-2, and XIAP (
Figure 5B). Importantly, treatment of normal MRC-5 fibroblasts did not show any significant change in the expression of any of the proteins known to be involved in apoptosis (data not shown).
In addition, we examined how nicotine treatment of A549 and SKMES-1 cells influenced the expression of these pro- and anti-apoptotic proteins (
Figure 5A,B). A majority of pro-apoptotic proteins, including bad, bax, bid, cytochrome C, SMAC, TRAIL-R1, Fas, FasL and caspase-8, were significantly down-regulated on nicotine exposure. Moreover, an increased expression of many proteins involved in signaling pathways known to prevent apoptosis, including bcl-2, bcl-W, cIAP-2, XIAP, surviving, and livin was evident. This was further illustrated when the bax/bcl-2 ratio was compared in ASP8 and nicotine treated cells. bax/bcl-2 ratio increased after APS8 treatment in both cell lines while it decreased after nicotine treatment (
Figure 5A,B, inserts). These results further emphasize the selective activation of pro-apoptotic signaling in LC cells by APS8.
Figure 5.
APS8 increases the expression of pro-apoptotic proteins and represses the expression of anti-apoptotic proteins. The change in expression of anti- and pro-apoptotic proteins was examined in A549 (A) and SKMES-1 (B) cells after treatment with APS8 (500 nM) or nicotine (1 μM) for 48 h using Human Apoptosis Antibody Array. Fold values relative to untreated cells are given. For presentation proteins are divided into three groups: proteins belonging to the intrinsic pathway, inhibitors of apoptosis (IAPs), and proteins belonging to the extrinsic pathway. Each point is the mean of three independent experiments ± SE. * P < 0.05 compared with control. The insert shows the ratio of main pro-apoptotic and main pro-survival proteins in APS8 and nicotine treated cells.
Figure 5.
APS8 increases the expression of pro-apoptotic proteins and represses the expression of anti-apoptotic proteins. The change in expression of anti- and pro-apoptotic proteins was examined in A549 (A) and SKMES-1 (B) cells after treatment with APS8 (500 nM) or nicotine (1 μM) for 48 h using Human Apoptosis Antibody Array. Fold values relative to untreated cells are given. For presentation proteins are divided into three groups: proteins belonging to the intrinsic pathway, inhibitors of apoptosis (IAPs), and proteins belonging to the extrinsic pathway. Each point is the mean of three independent experiments ± SE. * P < 0.05 compared with control. The insert shows the ratio of main pro-apoptotic and main pro-survival proteins in APS8 and nicotine treated cells.
2.5. APS8 is a Negative Regulator of Human α7 nAChRs
In order to confirm that APS8 indeed is negative regulator of α7 nAChRs we examined its capacity to inhibit α7 nAChR activity
in vitro systems,
i.e.,
Xenopus oocytes and SHEP-1 cells expressing human α7 nAChR (
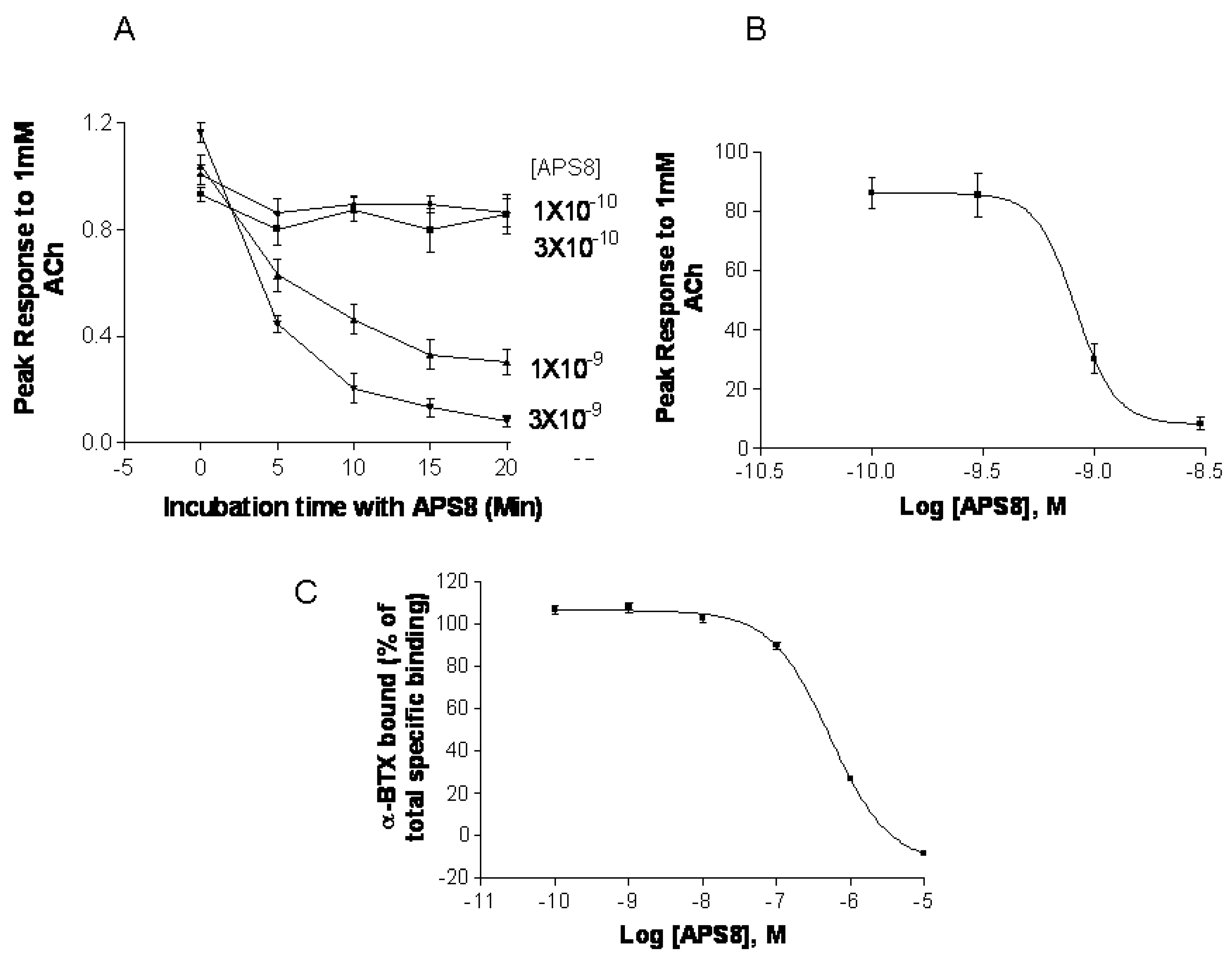
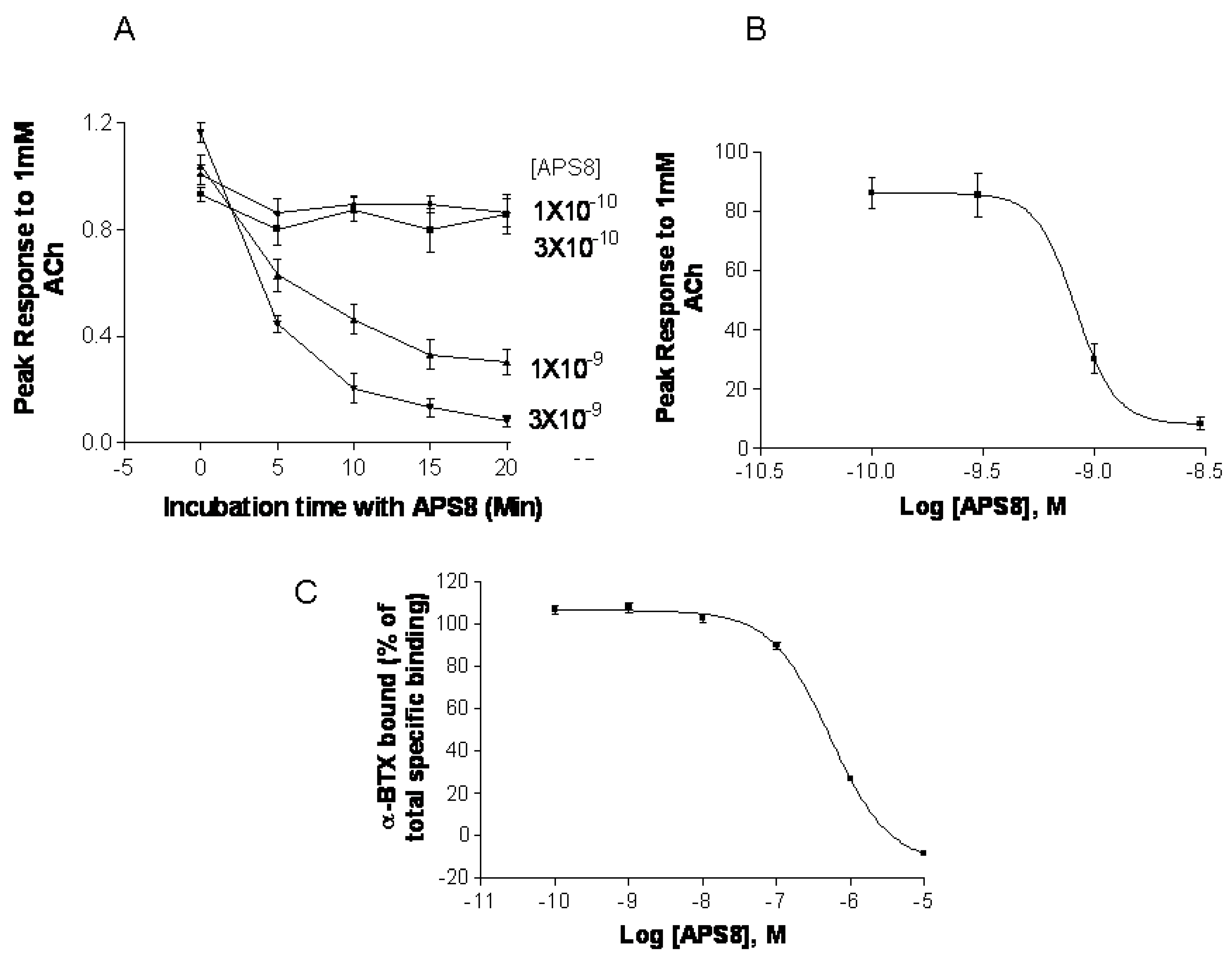
Figure 7). APS8 potently inhibited the responses of
Xenopus oocytes expressing human α7 nAChR in a time dependent manner (
Figure 7A,B). The IC
50 was approximately 1 nM. The action of APS8 could at least partially be slowly reversed by prolonged washing of the oocyte with frog Ringer solution (results not shown). APS8 was also able to inhibit the specific binding of the α7 nAChR antagonist
125I-α-BTX in SHEP-1 cells system, but only at concentrations which were at least two orders of magnitude higher than was required to functionally inhibit α7 nAChR when expressed in
Xenopus oocytes (
Figure 7C). In summary, these results demonstrate that APS8 indeed is an antagonist of α7 nAChRs.
Figure 7.
APS8 inhibits the response of human α7 nAChRs expressed in the Xenopus oocyte to a near maximally activating concentration of ACh (points represent mean values for five oocytes). In part (A) the time course of inhibition is shown; in part (B) the concentration dependence of APS8 inhibition at 20 min incubation is shown; Part (C) shows APS8 inhibition of the specific binding of [125I]-α-bungarotoxin to human α7 nAChRs expressed in cultured SHEP-1 cells. Each point is a mean value for four replicates.
Figure 7.
APS8 inhibits the response of human α7 nAChRs expressed in the Xenopus oocyte to a near maximally activating concentration of ACh (points represent mean values for five oocytes). In part (A) the time course of inhibition is shown; in part (B) the concentration dependence of APS8 inhibition at 20 min incubation is shown; Part (C) shows APS8 inhibition of the specific binding of [125I]-α-bungarotoxin to human α7 nAChRs expressed in cultured SHEP-1 cells. Each point is a mean value for four replicates.
In the current study we analyzed the potential of the Reniera sarai derived synthetic compound APS8 as a LC antitumor agent. We show that it holds potential to selectively trigger a cytotoxic and pro-apoptotic response in NSCLC cells whereas normal fibroblasts remained unaffected. Moreover, we show that APS8 is an antagonist of the α7 nAChRs.
A variety of mAChR and nAChR subtypes are found on normal cells [
3,
14] but have an altered expression pattern in normal bronchial epithelial cells and airway fibroblasts of smokers, ex-smokers, and non-smokers [
15]. Importantly, many cancer cell lines and primary cancer cells show a higher expression of mAChR and nAChR subtypes. Why LC cells have an increased expression of mAChR and nAChR is not completely clear but it might reflect adaption of the tissue to nicotine and other compounds inhaled by tobacco smoke [
15]. Hence, α7 nAChRs might be a therapeutic target and in light of this our findings on the selective cytotoxic effects of APS8 to LC cells but not to normal lung fibroblasts are interesting.
Cytotoxicity for lung adenocarcinoma or squamous carcinoma NSCLC cell lines and the resistance of MRC-5 cells to natural poly-APS could be explained by the lack of nAChRs on the MRC-5 cells. Albeit, we did not evaluate the expression of α7 nAChRs in A549 or SKMES-1 cells, immunohistochemistry analyses of this receptor in A549 cells by the human protein atlas project demonstrated a high expression [
32]. When preparing this manuscript, Lau
et al., in fact, demonstrated by ELISA-based assay that A549 cells, as well as multiple other bronchioalveolar carcinoma (BAC) cell lines, do express α7 nAChRs [
33]. Although our results reveal a therapeutic window for APS8, siRNA experiment towards α7 nAChRs would be required in order to confirm that these receptors are the specific APS8 target.
The ability to resist apoptosis is a hallmark of almost all types of tumor cells, including NSCLC, and this hampers the efficacy of chemotherapeutics [
34]. ACh and the nAChR selective agonist nicotine have been found to protect NSCLC, SCLC, and some other tumor cells expressing α7 nAChRs against apoptosis induced by chemotherapeutic agents [
16,
35,
36,
37]. How the anti-apoptotic effect in NSCLC is achieved by ACh, other nAChR agonists, and the two main carcinogenic metabolites of nicotine
N-nitrosonornicotine (NNN) and (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), was recently revealed by Schuller [
7]. In smokers, nicotine and its metabolites act predominantly through the activation of Ca
2+ permeable α7 nAChRs, possibly because the heteromeric nAChR subtypes are already desensitized by low concentrations of these compounds during smoking. The influx of Ca
2+ and also Na
+ ions provokes membrane depolarization, which in turn activates voltage-gated Ca
2+ channels, leading to an additional influx of Ca
2+ [
7]. The increased intracellular concentration of calcium triggers signal-transduction cascades involved in the regulation of numerous cellular processes: proliferation, angiogenesis, migration, differentiation, and apoptosis [
7,
8,
38,
39,
40]. Thus, the α7 nAChR appears to be a promising target for the therapy of NSCLC or at least BAC as recently was proposed. One possible therapeutical approach would be the use of α7 nAChR antagonists either alone or in combination with other chemotherapeutic drugs. Such a combination would enhance apoptosis but also impede proliferation and metastasis of tumor cells [
1,
20]. Indeed, snake venom polypeptide α-toxins,
i.e., α-cobratoxin and α-bungarotoxin, which are potent natural ACh antagonists of muscle and several other nAChR subtypes were recently proposed as possible candidates to achieve such a goal [
18]. However, due to their blocking actions on neuromuscular nAChRs, these peptide toxins are also highly toxic and therefore could be problematic for clinical treatment of cancer patients. Interestingly, APS8 (i.v. LD
50 is about 8 mg/kg in mice, [
31]) is significantly less toxic than snake α-toxins and APS8 possesses a more stable chemical scaffold suggesting that this compound might be suitable as an
in vivo probe to test the therapeutic potential of α7 nAChR antagonists.
We show here that APS8 is an extremely potent α7 nAChR antagonist. The response of human α7 nAChRs expressed in Xenopus oocytes to 1 mM ACh, that alone produces a near maximal response, was completely blocked by about 3 nM APS8 after 20 min of incubation. The recovery from this blockade was very slow, requiring several hours (not shown). APS8 was only able to inhibit the specific binding of the snake toxin α-bungarotoxin to human α7 nAChRs at concentrations over 100× the function-inhibiting concentrations. These results point to a noncompetitive mode of APS8 mediated inhibition and binding of APS8 outside the actual ACh binding sites on this nAChR subtype.
We anticipated that APS8
in vitro would inhibit proliferation and trigger apoptosis in tumor cell lines, but not in normal fibroblasts. Our hypothesis was based on our earlier observation that the parental compound from which APS8 was derived, the natural poly-APSs from the marine sponge
Reniera sarai, can induce apoptosis of NSCLC cells, yet being well tolerated by normal cells [
28]. Exposure to APS8 induced apoptosis in both lung cancer cell lines while normal fibroblasts MRC-5 did not undergo any significant apoptosis. These results were consistent when using different methods and confirmed our hypothesis.
The cytotoxic effect is probably exerted by inducing apoptosis through the binding to α7 nAChRs, which are expressed to a greater extent by cancer than by most normal cells. As expected from previous studies in a number of laboratories, nicotine promoted cell survival of LC cells. Therefore, we expected that nicotine in combination with APS8 would reduce the APS8 cytotoxic effect on cancer cell lines. Our results show that nicotine in combination with APS8 had only moderate effect on survival of A549 cells, while slightly stronger protection was evident on SKMES-1 cells. These results are in accordance with a previous study that suggested that nicotine has higher influence on proliferation of squamous carcinomas than of adenocarcinomas [
19]. Nicotine in combination with APS8 somewhat also protected LC cells from apoptosis suggesting antagonistic effect of APS8 to nicotine.
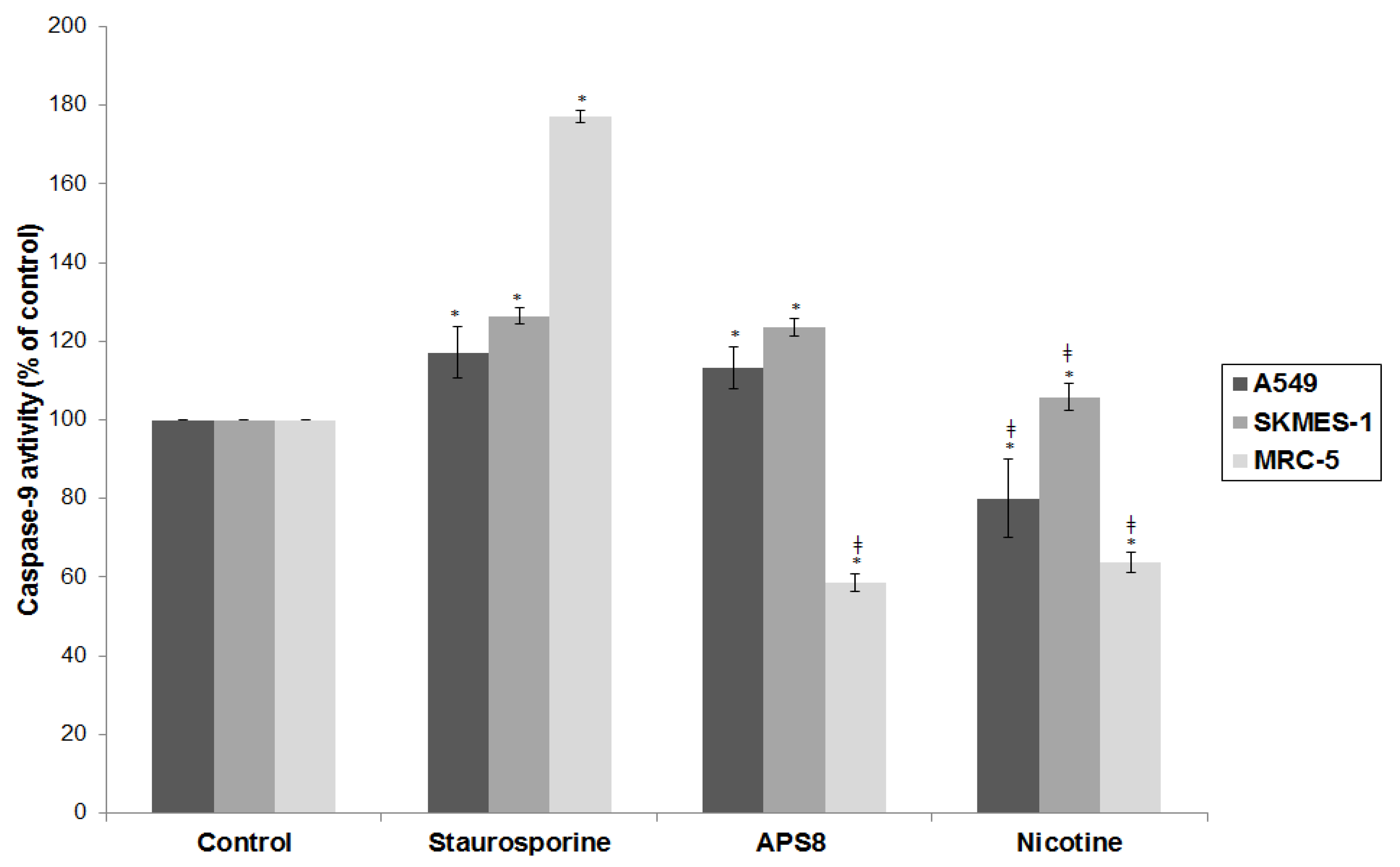
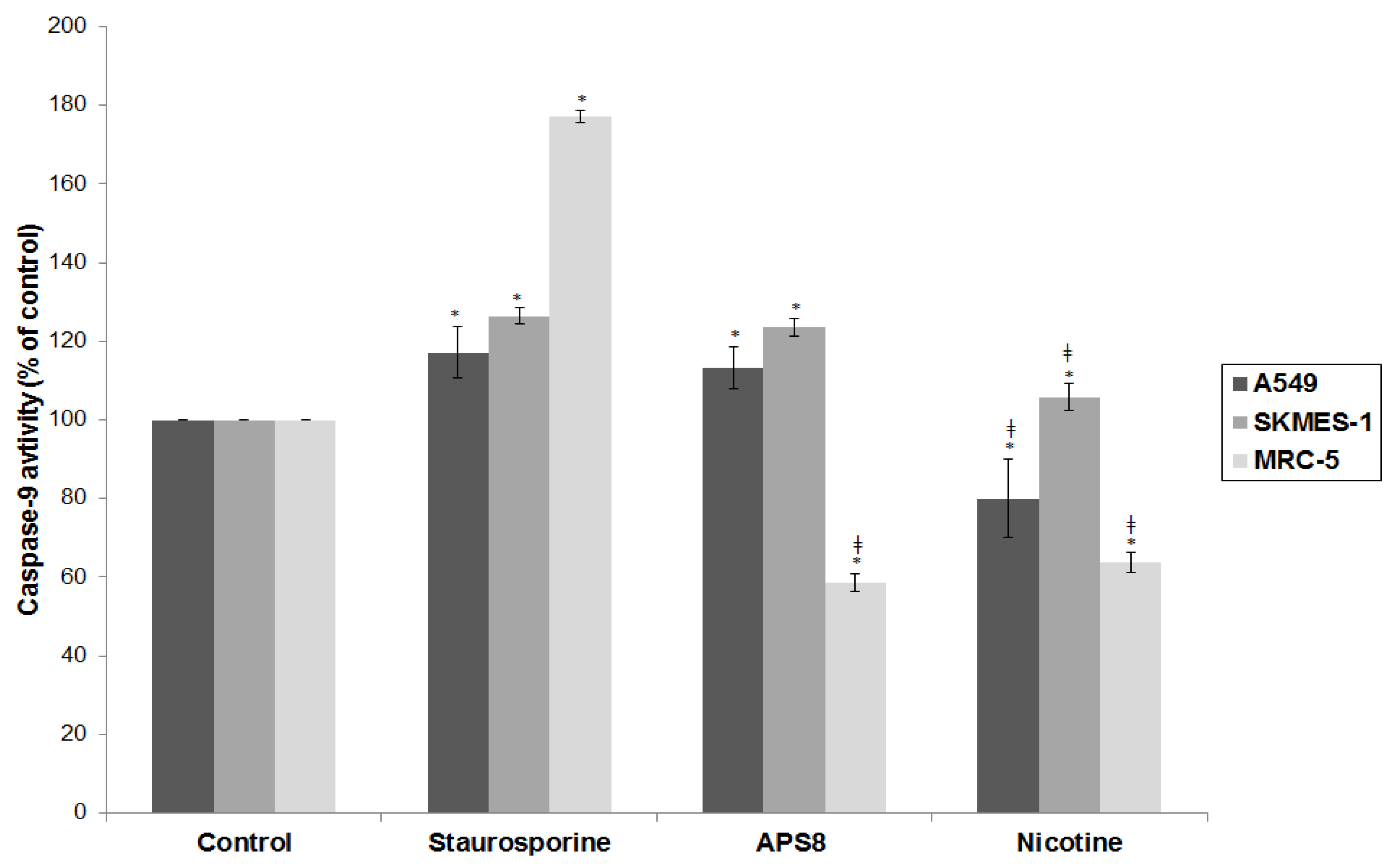
Mitochondrial permeabilization is one of the early stages of apoptosis, which leads to the release of several pro-apoptotic factors, including cytochrome c and SMAC, which in turn facilitate the activation of the apoptosome and downstream caspase-9 and caspase-3 activation. Our results show that APS8 treatment of A549 and SKMES-1 causes a mitochondrial depolarization, suggesting that APS8 in part can induce apoptosis via the apoptosome-mediated pathway. Indeed, after exposure to APS8 we observed an activation of caspase-9, a typical initiator caspase of the intrinsic apoptotic pathway.
To further test this hypothesis we used the RayBio
® Human Apoptosis Antibody Array kit. Treatment of A549 and SKMES-1 cells with APS8 (500 nM) increased the expression of caspase-3 which is an effector caspase activated by both intrinsic and extrinsic apoptotic pathways. The intrinsic pathway is activated by cellular stress, involves disruption of mitochondria and is controlled by bcl-2 family proteins. Induction of apoptosis relies on the ratio between pro-apoptotic (bax, bak, bcl-Xs, bad, bid) and anti-apoptotic (bcl-2, bcl-XL, bcl-W, mcl-1, A1) members of the bcl-2 family. Upon intrinsic apoptotic signal BH3 only proteins (bid, bad, PUMA, and NOXA) interact with the bcl-2 family, leading to inhibition of anti-apoptotic proteins and activation of pro-apoptotic proteins. Bax and bak proteins insert into the outer mitochondrial membrane causing the release of cytochrome c, bim, and SMAC from mitochondria. Released proteins then induce initiator caspase-9 activation and activation of effector caspase-3 [
41,
42]. The ratio between the levels of bax and bcl-2 proteins, at least in part, thus determines whether apoptosis will be induced with an increase in the bax/bcl-2 ratio causing a switch towards mitochondrial membrane permeabilization and activation of apoptosis. We show that treatment of both A549 and SKMES-1 cells expressing α7 and some other nAChR subtypes with APS8 triggers an increase in the expression of pro-apoptotic proteins bax, bad, bim and bid with a concomitant decrease in the levels of anti-apoptotic proteins bcl-W and bcl-2. Hence the bax/bcl-2 is increased, which may contribute to the observed increased apoptotic response of LC cells upon APS8 exposure. The data also revealed that both cytochrome c and SMAC, which are released from mitochondria during apoptosis, have an increased expression after APS8 treatment further increasing a pro-apoptotic capacity of the NSCLC cells. In summary, these results imply that APS8 induces apoptosis through the intrinsic pathway.
Our results also show that nicotine up-regulates bcl-2 and down-regulates bax, lowering the bax/bcl-2 ratio, which would favor a reduced apoptotic propensity of the NSCLC cells. Indeed, it was earlier suggested that inactivation of bax may be an essential step in anti-apoptotic mechanism induced by nicotine [
39]. Our data show strong up-regulation of bax and down regulation of bcl-2 by APS8, quite opposite effects as compared to nicotine, which suggests that APS8 may at least in part counteract the protective effect of nicotine by increasing the bax/bcl-2 ratio. This effect was more prominent in SKMES-1 than in the A549 cancer cell line.
Another large group of proteins involved in regulation of the apoptosome activity are inhibitors of apoptosis proteins (IAPs), a family of pro-survival proteins [
43]. In our experiments, all tested IAPs (cIAP-2, livin, XIAP and survivin) with the sole exception of livin in SKMES-1 cells, were down-regulated after exposure to APS8, whereas an increased expression was observed after nicotine treatment. Moreover, as mentioned earlier, SMAC displayed an increased expression in APS8 treated cells. This protein is an endogenous IAP antagonist and binds to IAP proteins, preventing their binding and inactivation of caspases, and thus promoting cell death. As was shown earlier, nicotine up-regulates IAPs, particularly XIAP, and in this way enhances resistance to anti-cancer treatments [
44]. Our results suggest that APS8 can circumvent nicotine-induced resistance to apoptosis by interfering with IAPs allowing proper apoptosome function and downstream caspase activation to take place.
The Fas, TNF-R1, TNF-R2, and TRAIL (TRAIL-R1, -R2, -R3, and -R4) cell membrane receptors are key molecules of the extrinsic pathway of apoptosis and are activated upon binding appropriate ligands (FasL, TNF, and TRAIL) resulting in downstream caspase-8 activation followed by effector caspases (3, 6, and 7) activation [
45]. Caspase-8 activation can trigger cleavage and activation of the BH3 only protein bid, which in turn can interact with bcl-2 proteins in mitochondria, resulting in intrinsic apoptotic pathway signaling and thereby activating effector caspases [
46]. In the present study we show that TRAIL-R1 and TRAIL-R2 expression are enhanced following APS8 treatment while a decreased expression was observed after nicotine treatment. Although APS8 markedly increased the expression levels of Fas in A549 cells, no significant changes of FasL were noted. In SKMES-1 cell line both Fas and FasL showed a marked increased expression following the APS8 treatment. On the contrary, nicotine in both cancer cell lines slightly decreased the levels of Fas/FasL and TRAIL receptors. These results suggest that APS8 can activate components of death receptor signaling which at least in part might be cell type specific but, nevertheless, are oppositely regulated by nicotine.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}