Protease Inhibitors from Marine Venomous Animals and Their Counterparts in Terrestrial Venomous Animals

Abstract
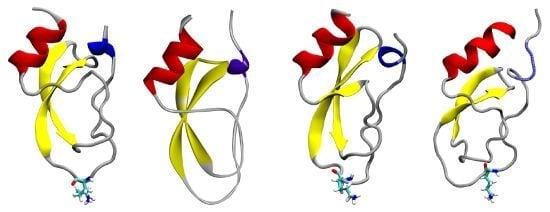
:
1. Introduction
2. Protease Inhibitors from Sea Anemones

{kind=link}
{kind=link}
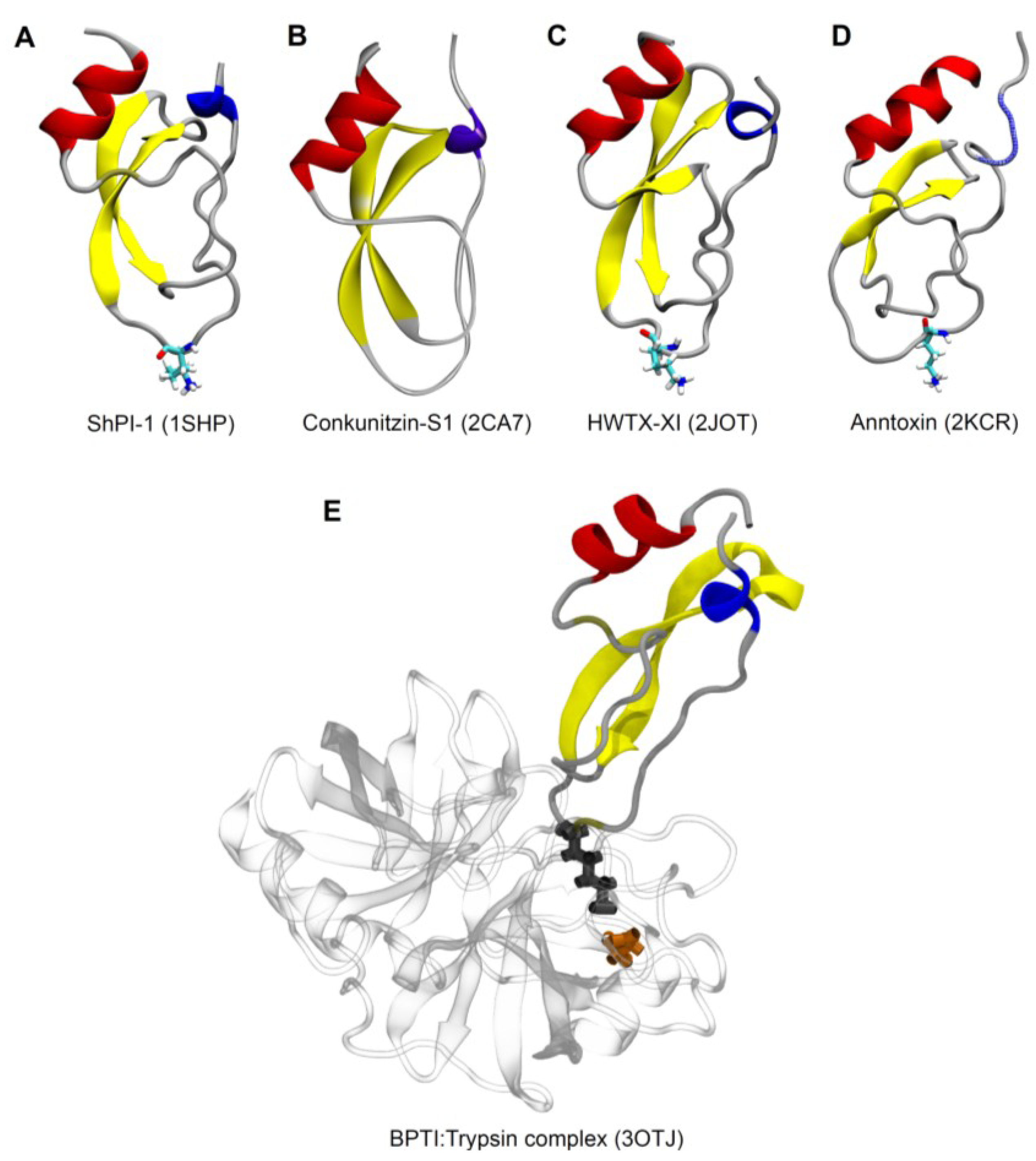
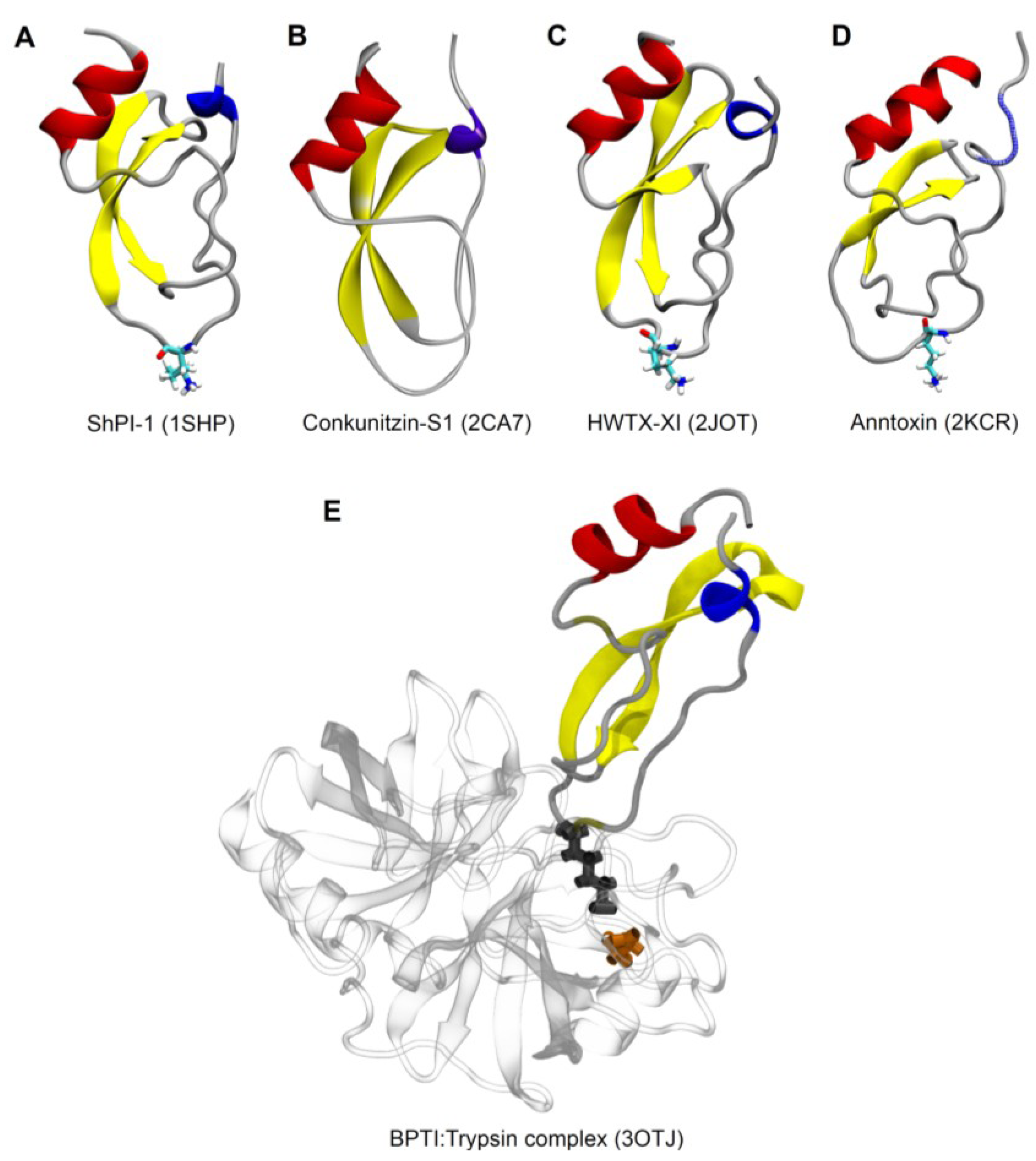{kind=link}
| Specie | Toxin | UniProt KB | AA a | Protease inhibited | Inhibitory activity b–g | References |
|---|---|---|---|---|---|---|
| # Actinia equina | ♦ AEPI-I | - | 59 III | - | - | [26] |
| ♦ AEPI-II | - | 59 III | - | - | [26] | |
| ● Equistatin | P81439 | 199 III | Cathepsin L *, | 0.051 (CL), | [25,43] | |
| papain, | 0.57 (P), | |||||
| cathepsin B | 1.4 (CB) b | |||||
| ♦ AEAPI | - | 57 III | Trypsin *, plasmin | 700 IU/mg (T) c | [24] | |
| # Anemonia sulcata | ■ Elastase inhibitor | P16895 | 48 III | Porcine elastase | 1.0 d | [28,44] |
| ♦ Kalicludin-1 or AsKC1 | Q9TWG0 | 58 III | Trypsin | <30 d | [27] | |
| ♦ Kalicludin-2 or AsKC2 | Q9TWF9 | 58 III | Trypsin | <30 d | [27] | |
| ♦ Kalicludin-3 or AsKC3 | Q9TWF8 | 59 III | Trypsin | <30 d | [27] | |
| ♦ SA5 II | P10280 | 62 III | Kallikrein, Trypsin | 0.3 (T) d | [29,32] | |
| # Anthopleura aff. xanthogrammica | ♦ AXPI-I or AXAPI | P81547 | 58 III | Trypsin *, chymotrypsin, elastase, thermolysin | 1900 IU/mg (T) c | [24,41] |
| ♦ AXPI-II | P81548 | 58 III | Trypsin *, chymotrypsin | 490 IU/mg (T) c | [41] | |
| # Anthopleura elegantissima | ♦ APEKTx1 | P86862 | 65 III | Trypsin | 124 d | [42] |
| # Anthopleura fuscoviridis | ♦ AFAPI-I | - | 56 III | Trypsin *, plasmin | 950 IU/mg (T) d | [24] |
| ♦ AFAPI-III | - | 56 III | Trypsin *, plasmin | 900 IU/mg (T) d | [24] | |
| # Heteractis crispa | ♦ Jn-IV | P16344 | 56 III | Trypsin | 9.6 d | [32,33] |
| ♦ InhVJ | - | 56 III | Trypsin *, | 2.49 (T), | [45,46,47] | |
| chymotrypsin | 21.7 (C) d | |||||
| ♦ APHC1 | B2G331 | 56 III | Trypsin, | 1000 (T), | [34] | |
| chymotrypsin | ~5000 (C) d | |||||
| # Rhodactis rhodostoma | ♦ Inhibitor 4 | - | 48 II | Trypsin, kallikrein *, chymotrypsin | 0.95 (T), 0.49 (K), 33 (C) d | [31] |
| # Stichodactyla haddoni | ♦ SHTX-3 | B1B5I8 | 62 III | Trypsin | 203 IU/mg c | [40] |
| # Stoichactis helianthus | ♦ ShPI-1 | P31713 | 55III | Serine *, | 0.11 (T), | [36,37] |
| cysteine, | 2.3 (C), | |||||
| aspartic proteases | 2.7 (Pl) d | |||||
| ♦ ShPI-2 | P81129 | 55 III | Serine, cysteine, aspartic proteases | n.f. | [38] | |
| + Dendroaspis polylepis polylepis | ♦ Dendrotoxin E | P00984 | 59 III | Trypsin *, | 1.0 (T), | [48] |
| chymotrypsin | 100 (C) d | |||||
| + Daboia siamensis | ♦ BBPTI-1 | - | 60 III | Chymotrypsin | 4.77 b | [49] |
| ♦ CBPTI-1 | A8Y7N4 | 66 III | Trypsin | 407 b | [50] | |
| ♦ CBPTI-2 | A8Y7N5 | 60 III | Trypsin | 666 b | [50] | |
| ♦ CBPTI-3 | A8Y7N6 | 60 III | Chymotrypsin | 2.55 b | [50] | |
| + Hemachatus haemachatus | ♦ HHV inhibitor II | P00985 | 57 III | Trypsin, chymotrypsin, kallikrein, plasmin | - | [51] |
| + Naja nivea | ♦ NNV inhibitor II | P00986 | 57 III | Trypsin | - | [51] |
| + Vipera ammodytes | ♦ Trypsin inhibitor I | P00991 | 61 III | Trypsin *, chymotrypsin, kallikrein, plasmin | 0.34 (T), 270 (C) b | [52,53] |
| ♦ Trypsin inhibitor II | - | 62 III | Trypsin *, chymotrypsin, kallikrein, plasmin | 0.56 (T), 300 (C) b | [52] | |
| ♦ Chymotrypsin inhibitor | P00992 | 65 III | Chymotrypsin *, trypsin, plasma kallikrein | 4.3 (C), 5100 (T) b | [52] | |
| + Bungarus fasciatus | ♦ BF9 | P25660 | 65 III | Chymotrypsin | 58 d | [54] |
| ♦ Bungaruskunin | B2KTG1 | 59 III | Chymotrypsin *, trypsin, elastase | 6100 (C) b | [55] | |
| + Naja naja | ♦ Trypsin inhibitor | P20229 | 57 III | Trypsin | 0.0035 b | [56] |
| + Naja atra | ♦ NACI | Q5ZPJ7 | 57 III | Chymotrypsin | 25 b | [57,58] |
| + Ophiophagus hannah | ♦ Oh11-1 | P82966 | 58 III | Chymotrypsin | 3520 b | [59] |
| ♦ OH-TCI | B6RLX2 | 58 III | Chymotrypsin *, | 84.6 (C), | [60] | |
| trypsin | 391 (T) b | |||||
| + Bungarus multicinctus | ♦ PILP-1 | B4ESA2 | 58 III | Trypsin | 55.62 b | [61] |
| + Oxyuranus scutellatus scutellatus | ♦ TSPI | B7S4N9 | 62 III | Kallikrein *, trypsin, plasmin, elastase, factor Xa, α-factor XIIa | 0.057 (PK), 0.23 (TK), 0.31 (T), 6.1 (Pl), 201 (E), 871 (X), 2380 (XII) b | [62,63] |
| + Pseudonaja textilis textilis | ♦ Textilinin-1 | Q90WA1 | 59 III | Plasmin *, | 0.49 ± 0.02 (Pl), | [7,63] |
| trypsin | 0.76 ± 0.02 (T) b | |||||
| ♦ Textilinin-2 | Q90WA0 | 59 III | Plasmin | 2.2 b | [7] | |
| + Macrovipera lebetina transmediterranea | ♦ PIVL | I2G9B4 | 67 III | Trypsin | - | [64] |
| + Pseudechis australis | ♦ Pr-mulgin 1 | E7FL11 | 59 III | Metalloprotease 2 | 60 b | [65] |
| ♦ Pr-mulgin 2 | E7FL12 | 59 III | Trypsin *, | 5 (T), | [65] | |
| chymotrypsin, | 40 (C), | |||||
| plasmin | 40 (Pl) b | |||||
| ♦ Pr-mulgin 3 | E7FL13 | 59 III | Trypsin, | 5 (T), | [65] | |
| plasmin | 100 (Pl) b | |||||
| § Mesobuthus tamulus | ♦ Fraction IX-1-a | - | - | Trypsin *, | 19.2 IU/mg (T), | [66] |
| kallikrein | 87 IU/mg (K) c | |||||
| § Hadrurus gertschi | ♦ rHg1 | P0C8W3 | 67 III | Trypsin | 107 d | [67,68] |
| § Lychas mucronatus | ♦ rLmKTT-1a | P0DJ46 | 59 III | Trypsin | 140 d | [16,68] |
| ♦ rLmKTT-1b or SdPI | P0DJ45 | 59 III | Trypsin | 160 d | [16,68] | |
| ♦ rLmKTT-1c | P0DJ48 | 59 III | Trypsin | 124 d | [68] | |
| § Mesobuthus martensii | ♦ rBmKTT-1 | P0DJ49 | 59 III | Trypsin | 136 d | [68] |
| ♦ rBmKTT-2 | P0DJ50 | 58 IV | Trypsin | 420 d | [68] | |
| ♦ rBmKTT-3 | P0DJ47 | 70 III | Trypsin | 760 d | [68] | |
| § Scorpiops jendeki | ◊ rSjAPI | - | 64 V | Chymotrypsin *, | 97.1 (C), | [69] |
| elastase | 3700 (E) b | |||||
| ¥ Ornithoctonus huwena | ♦ HWTX-XI | P68425 | 55 III | Trypsin | 0.23 d | [14] |
| ¥ Araneus ventricosus | ♦ AvKTI | K7YYJ2 | 57 III | Trypsin, | 7.34 (T), | [70] |
| chymotrypsin, | 37.75 (C), | |||||
| plasmin *, | 4.89 (Pl), | |||||
| neutrophil elastase | 169.07 (E) b | |||||
| ♦ AvCI | L7X735 | 70 IV | Chymotrypsin, | 49.85 (C), | [71] | |
| subtilisin A, | 20.51 (SA), | |||||
| proteinase K, | 65.42 (ptK), | |||||
| neutrophil elastase, | 8.74 (nE), | |||||
| pancreatic elastase | 11.32 (pE) b | |||||
| ¤ Bombina bombina | ◊ BSTI | Q90248 | 60 V | Trypsin, | 80–100 (T), | [72] |
| thrombin | 1300 (Th) b | |||||
| ¤ Bombina maxima | ◊ BMTI | Q8QFP3 | 60 V | Trypsin | 60 b | [73] |
| ¤ Rana areolata | ◊ Trypsin inhibitor | - | 61 V | Trypsin | ~20,000 e | [74] |
| ¤ Bombina orientalis | ◊ BOTI | Q800F0 | 60 V | - | - | [75] |
| ¤ Bombina variegata | ◊ BVTI | Q800E9 | 60 V | - | - | [75] |
| ¤ Bombina microdeladigitora | ◊ BMSI 1 | B1P2F8 | 60 V | Trypsin, thrombin | 20 (T), 150 (Th) b | [76] |
| ◊ BMSI 2 | B1P2F9 | 60 V | - | - | [76] | |
| ¤ Hyla simplex | Hylaserpin S1 | H6SWK9 | 392 | Trypsin, | 55 (T), | [77] |
| chymotrypsin | 310 (C) b | |||||
| ◊ Hylaserpin S2 | H6SWL0 | 56 V | Trypsin | 72 b | [77] | |
| ¤ Bufo andrewsi | BATI | - | - | Trypsin | 14 b | [78] |
| Trypsin | 4.6 × 106 (T), | |||||
| Baserpin | - | - | chymotrypsin, | 8.9 × 106 (C), | [79] | |
| elastase | 6.8 × 106 (E) f | |||||
| ¤ Kaloula pulchra hainana | KPHTI | - | - | Trypsin | 27 c | [80] |
| ¤ Phyllomedusa sauvagii | ■ PSKP-1 | P83578 | 58 III | Prolyl endopeptidase | 124 ± 56 g | [81] |
| ■ PSKP-2 | P83579 | 58 III | - | - | [81] | |
| ¤ Phyllomedusa nordestina | PI01 | K9N0E2 | 78 III | - | - | [82] |
| PI02 | K9N1K5 | 77 III | - | - | [82] | |
| PI03 | K9N2T9 | 53 III | - | - | [82] | |
| ¤ Agalychnis callidryas | ■ ACKTI | I7J523 | 52 III | Trypsin | 1.9 b | [83] |
| ¤ Dyscophus guineti | ♦ Kunitz-type PI | J9UVV9 | 57 III | Trypsin | - | [17] |
| ¤ Kassina senegalensis | ♦ KSCI | F8K9Q3 | 62 III | Chymotrypsin | - | [84] |
| ¤ Hyla annectans | ♦ Anntoxin | C7AR58 | 60 II | Trypsin | 25 b | [85] |
| ¤ Odorrana grahami | OGTI | - | 17 II | Trypsin | 400 b | [86] |
| ¤ Huia versabilis | HV-BBI | B1VC43 | 18 I | Trypsin | 18.8 + 1.8 b | [87] |
| ø Vespa bicolor Fabricius | ♦ Bicolin | C0LNR2 | 54 III | Trypsin *, thrombin | 550 (T), 26,000 (Th) b | [88] |
| ø Bombus ignitus | ♦ Bi-KTI | G3LH89 | 58 III | Plasmin | 43.53 g, 3.6 b | [6] |
| ø Bombus terrestris | ♦ Bt-KTI | D8KY58 | 58 III | Plasmin | 2.01 b | [89] |
| Source | Peptide | IC50 (nM) | Target | Ref. |
|---|---|---|---|---|
| Sea anemone | APEKTx1 * | 0.9 ± 0.1 | Kv1.1 | [42] |
| AsKC1 * | 2800 | Kv1.2 | [27] | |
| AsKC2 * | 1100 | Kv1.2 | [27] | |
| AsKC3 * | 1300 | Kv1.2 | [27] | |
| SHTX-3 * | 650 | Synap a | [40] | |
| Cone snail | Conk-S1 | 1.33 ± 0.5 | Shaker | [94] |
| Snake | α-DTX | 0.4–150 | Kv1.1, Kv1.2, Kv1.6 | [95] |
| DTX-I | 0.13–50 | Kv1.1, Kv1.2, Kv1.6 | [95] | |
| DTX-K | 0.03 | Kv1.1 | [96] | |
| δ-DTX | 0.029–1.8 | Kv1.1 | [96,97] | |
| DaE | 300 | Kv1.1 | [98] | |
| Scorpion | Hg1 * | 6.2 ± 1.2 | Kv1.3 | [68] |
| LmKTT-1a * | 1580 | Kv1.3 | [68,99] | |
| LmKTT-1b * | >1000 | Kv1.3 | [68] | |
| LmKTT-1c * | >1000 | Kv1.3 | [68] | |
| BmKTT-1 * | 129.7 ± 31.3 | Kv1.3 | [68] | |
| BmKTT-2 * | 371.3 ± 82.1 | Kv1.3 | [68] | |
| BmKTT-3 * | >1000 | Kv1.3 | [68] | |
| Spider | HWTX-XI * | 2570 | Kv1.1 | [14] |
3. PIs from Conus Venoms
4. PIs from Terrestrial Venomous Animals
4.1. PIs from Snake Venoms
4.2. PIs from Scorpion Venoms
4.3. PIs from Spider Venoms
4.4. PIs from the Skin Secretion of Anurans
4.5. PIs from Hymenopterans’ Venoms
5. PIs and Potassium Channel Activity

6. Molecular Diversity
7. Therapeutic Potential
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Di Cera, E. Serine proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef]
- Lingaraju, M.H.; Gowda, L.R. A Kunitz trypsin inhibitor of Entada scandens seeds: Another member with single disulfide bridge. Biochim. Biophys. Acta 2008, 1784, 850–855. [Google Scholar] [CrossRef]
- Puente, X.S.; Sanchez, L.M.; Gutierrez-Fernandez, A.; Velasco, G.; Lopez-Otin, C. A genomic view of the complexity of mammalian proteolytic systems. Biochem. Soc. Trans. 2005, 33, 331–334. [Google Scholar] [CrossRef]
- López-Otín, C.; Overall, C.M. Protease degradomics: A new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002, 3, 509–519. [Google Scholar] [CrossRef]
- Oliva, M.L.; Souza-Pinto, J.C.; Batista, I.F.; Araujo, M.S.; Silveira, V.F.; Auerswald, E.A.; Mentele, R.; Eckerskorn, C.; Sampaio, M.U.; Sampaio, C.A. Leucaena leucocephala serine proteinase inhibitor: Primary structure and action on blood coagulation, kinin release and rat paw edema. Biochim. Biophys. Acta 2000, 1477, 64–74. [Google Scholar] [CrossRef]
- Choo, Y.M.; Lee, K.S.; Yoon, H.J.; Qiu, Y.; Wan, H.; Sohn, M.R.; Sohn, H.D.; Jin, B.R. Antifibrinolytic role of a bee venom serine protease inhibitor that acts as a plasmin inhibitor. PLoS One 2012, 7, e32269. [Google Scholar]
- Masci, P.P.; Whitaker, A.N.; Sparrow, L.G.; de Jersey, J.; Winzor, D.J.; Watters, D.J.; Lavin, M.F.; Gaffney, P.J. Textilinins from Pseudonaja textilis textilis. Characterization of two plasmin inhibitors that reduce bleeding in an animal model. Blood Coagul. Fibrinolysis 2000, 11, 385–393. [Google Scholar] [CrossRef]
- Salvador, L.A.; Taori, K.; Biggs, J.S.; Jakoncic, J.; Ostrov, D.A.; Paul, V.J.; Luesch, H. Potent elastase inhibitors from cyanobacteria: Structural basis and mechanisms mediating cytoprotective and anti-inflammatory effects in bronchial epithelial cells. J. Med. Chem. 2013, 56, 1276–1290. [Google Scholar] [CrossRef]
- Kennedy, A.R. Chemopreventive agents: Protease inhibitors. Pharmcol. Ther. 1998, 78, 167–209. [Google Scholar] [CrossRef]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Abbenante, G.; Fairlie, D.P. Protease inhibitors in the clinic. Med. Chem. 2005, 1, 71–104. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Krowarsch, D.; Cierpicki, T.; Jelen, F.; Otlewski, J. Canonical protein inhibitors of serine proteases. Cell Mol. Life Sci. 2003, 60, 2427–2444. [Google Scholar] [CrossRef]
- Yuan, C.H.; He, Q.Y.; Peng, K.; Diao, J.B.; Jiang, L.P.; Tang, X.; Liang, S.P. Discovery of a distinct superfamily of Kunitz-type toxin (KTT) from tarantulas. PLoS One 2008, 3, e3414. [Google Scholar] [CrossRef]
- Isaeva, M.P.; Chausova, V.E.; Zelepuga, E.A.; Guzev, K.V.; Tabakmakher, V.M.; Monastyrnaya, M.M.; Kozlovskaya, E.P. A new multigene superfamily of Kunitz-type protease inhibitors from sea anemone Heteractis crispa. Peptides 2012, 34, 88–97. [Google Scholar] [CrossRef]
- Zhao, R.; Dai, H.; Qiu, S.; Li, T.; He, Y.; Ma, Y.; Chen, Z.; Wu, Y.; Li, W.; Cao, Z. SdPI, the first functionally characterized Kunitz-type trypsin inhibitor from scorpion venom. PLoS One 2011, 6, e27548. [Google Scholar]
- Conlon, J.M.; Kim, J.B. A protease inhibitor of the Kunitz family from skin secretions of the tomato frog, Dyscophus guineti (Microhylidae). Biochem. Biophys. Res. Commun. 2000, 279, 961–964. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Berndt, K.D.; Guntert, P.; Orbons, L.P.; Wuthrich, K. Determination of a high-quality nuclear magnetic resonance solution structure of the bovine pancreatic trypsin inhibitor and comparison with three crystal structures. J. Mol. Biol. 1992, 227, 757–775. [Google Scholar] [CrossRef]
- Wagner, G.; Braun, W.; Havel, T.F.; Schaumann, T.; Go, N.; Wuthrich, K. Protein structures in solution by nuclear magnetic resonance and distance geometry. The polypeptide fold of the basic pancreatic trypsin inhibitor determined using two different algorithms, DISGEO and DISMAN. J. Mol. Biol. 1987, 196, 611–639. [Google Scholar] [CrossRef]
- Deisenhofer, J.; Steigemann, W. Crystallographic refinement of the structure of bovine pancreatic trypsin inhibitor at l.5 A resolution. Acta Cryst. 1975, 31, 238–250. [Google Scholar] [CrossRef]
- Fritz, H.; Brey, B.; Beress, L. Polyvalent isoinhibitors for trypsin, chymotrypsin, plasmin and kallikreins of sea anemones (Anemonia sulcata), isolation, inhibitory behavior and amino acid composition. Hoppe Seyler Z. Physiol. Chem. 1972, 353, 19–30. [Google Scholar] [CrossRef]
- Wunderer, G.; Beress, L.; Machleidt, W.; Fritz, H. Broad-specificity inhibitors from sea anemones. Methods Enzymol.. 1976, 45, 881–888. [Google Scholar] [CrossRef]
- Minagawa, S.; Sugiyama, M.; Ishida, M.; Nagashima, Y.; Shiomi, K. Kunitz-type protease inhibitors from acrorhagi of three species of sea anemones. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 240–245. [Google Scholar] [CrossRef]
- Lenarcic, B.; Ritonja, A.; Strukelj, B.; Turk, B.; Turk, V. Equistatin, a new inhibitor of cysteine proteinases from Actinia equina, is structurally related to thyroglobulin type-1 domain. J. Biol. Chem. 1997, 272, 13899–13903. [Google Scholar]
- Ishida, M.; Minagawa, S.; Miyauchi, K.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Amino acid sequences of Kunitz-type protease inhibitors from the sea anemone Actinia equina. Fish. Sci. 1997, 63, 794–798. [Google Scholar] [CrossRef]
- Schweitz, H.; Bruhn, T.; Guillemare, E.; Moinier, D.; Lancelin, J.M.; Beress, L.; Lazdunski, M. Kalicludines and kaliseptine. Two different classes of sea anemone toxins for voltage sensitive K+ channels. J. Biol. Chem. 1995, 270, 25121–25126. [Google Scholar]
- Tschesche, H.; Kolkenbrock, H.; Bode, W. The covalent structure of the elastase inhibitor from Anemonia sulcata—A “non-classical” Kazal-type protein. Biol. Chem. Hoppe Seyler 1987, 368, 1297–1304. [Google Scholar] [CrossRef]
- Wunderer, G.; Machleidt, W.; Fritz, H. The broad-specificity proteinase inhibitor 5 II from the sea anemone Anemonia sulcata. Methods Enzymol. 1981, 88, 816–820. [Google Scholar] [CrossRef]
- Hemmi, H.; Kumazaki, T.; Yoshizawa-Kumagaye, K.; Nishiuchi, Y.; Yoshida, T.; Ohkubo, T.; Kobayashi, Y. Structural and functional study of an Anemonia elastase inhibitor, a “nonclassical” Kazal-type inhibitor from Anemonia sulcata. Biochemistry 2005, 44, 9626–9636. [Google Scholar] [CrossRef]
- Mebs, D.; Liebrich, M.; Reul, A.; Samejima, Y. Hemolysins and proteinase inhibitors from sea anemones of the Gulf of Aqaba. Toxicon 1983, 21, 257–264. [Google Scholar]
- Sokotun, I.N.; Il’ina, A.P.; Monastyrnaya, M.M.; Leychenko, E.V.; Es’kov, A.A.; Anastuk, S.D.; Kozlovskaya, E.P. Proteinase inhibitors from the tropical sea anemone Radianthus macrodactylus: Isolation and characteristic. Biochemistry. Biokhimiia 2007, 72, 301–306. [Google Scholar] [CrossRef]
- Zykova, T.A.; Vinokurov, L.M.; Markova, L.F.; Kozlovskaya, E.P.; Elyakov, G.B. Amino acid sequence of trypsin inhibitor IV from Radianthus macrodactylus. Bioorg. Khim. 1985, 11, 293–301. [Google Scholar]
- Andreev, Y.A.; Kozlov, S.A.; Koshelev, S.G.; Ivanova, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E.V. Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 2008, 283, 23914–23921. [Google Scholar] [CrossRef]
- Mebs, D.; Gebauer, E. Isolation of proteinase inhibitory, toxic and hemolytic polypeptides from a sea anemone, Stoichactis sp. Toxicon 1980, 18, 97–106. [Google Scholar] [CrossRef]
- Antuch, W.; Berndt, K.D.; Chavez, M.A.; Delfin, J.; Wuthrich, K. The NMR solution structure of a Kunitz-type proteinase inhibitor from the sea anemone Stichodactyla helianthus. Eur. J. Biochem. 1993, 212, 675–684. [Google Scholar] [CrossRef]
- Delfín, J.; Martínez, I.; Antuch, W.; Morera, V.; González, Y.; Rodríguez, R.; Márquez, M.; Saroyán, A.; Larionova, N.; Díaz, J.; Padrón, G.; Chávez, M. Purification, characterization and immobilization of proteinase inhibitors from Stichodactyla helianthus. Toxicon 1996, 34, 1367–1376. [Google Scholar] [CrossRef]
- Díaz, J.; Morera, V.; Delfin, J.; Huerta, V.; Lima, G.; Rodríguez de la Vega, M.; García, B.; Padrón, G.; Assfalg-Machleidt, I.; Machleidt, W.; Chávez, M. Purification and partial characterization of a novel proteinase inhibitor from the sea anemone Stichodactyla helianthus. Toxicon 1998, 36, 1275–1276. [Google Scholar]
- García-Fernández, R.; Pons, T.; Perbandt, M.; Valiente, P.A.; Talavera, A.; González-González, Y.; Rehders, D.; Chávez, M.A.; Betzel, C.; Redecke, L. Structural insights into serine protease inhibition by a marine invertebrate BPTI Kunitz-type inhibitor. J. Struct. Biol. 2012, 180, 271–279. [Google Scholar] [CrossRef]
- Honma, T.; Kawahata, S.; Ishida, M.; Nagai, H.; Nagashima, Y.; Shiomi, K. Novel peptide toxins from the sea anemone Stichodactyla haddoni. Peptides 2008, 29, 536–544. [Google Scholar] [CrossRef]
- Minagawa, S.; Ishida, M.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Isolation and amino acid sequences of two Kunitz-type protease inhibitors from the sea anemone Anthopleura aff. xanthogrammica. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 118, 381–386. [Google Scholar] [CrossRef]
- Peigneur, S.; Billen, B.; Derua, R.; Waelkens, E.; Debaveye, S.; Beress, L.; Tytgat, J. A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties. Biochem. Pharmacol. 2011, 82, 81–90. [Google Scholar] [CrossRef]
- Galesa, K.; Strukelj, B.; Bavec, S.; Turk, V.; Lenarcic, B. Cloning and expression of functional equistatin. Biol. Chem. 2000, 381, 85–88. [Google Scholar]
- Kolkenbrock, H.; Tschesche, H. A new inhibitor of elastase from the sea anemone (Anemonia sulcata). Biol. Chem. Hoppe Seyler 1987, 368, 93–99. [Google Scholar] [CrossRef]
- Sokotun, I.N.; Leichenko, E.V.; Vakorina, T.I.; Es’kov, A.A.; Il’ina, A.P.; Monastyrnaia, M.M.; Kozlovskaia, E.P. A serine protease inhibitor from the anemone Radianthus macrodactylus: Isolation and physicochemical characteristics (in Russian). Bioorg. Khim. 2007, 33, 448–455. [Google Scholar]
- Sokotun, I.N.; Gnedenko, O.V.; Leichenko, E.V.; Monastyrnaia, M.M.; Kozlovskaia, E.P.; Mol'nar, A.A.; Ivanov, A.C. Interaction investigation of trypsin inhibitor from sea anemone Radianthus macrodactylus with proteases (in Russian). Biomed. Khim. 2006, 52, 595–600. [Google Scholar]
- Gladkikh, I.; Monastyrnaya, M.; Leychenko, E.; Zelepuga, E.; Chausova, V.; Isaeva, M.; Anastyuk, S.; Andreev, Y.; Peigneur, S.; Tytgat, J.; Kozlovkaya, E. Atypical reactive center Kunitz-type inhibitor from the sea anemone Heteractis crispa. Mar. Drugs 2012, 10, 1545–1565. [Google Scholar] [CrossRef]
- Strydom, D.J. Snake venom toxins. Purification and properties of low-molecular-weight polypeptides of Dendroaspis polylepis polylepis (black mamba) venom. Eur. J. Biochem. 1976, 69, 169–176. [Google Scholar] [CrossRef]
- Guo, C.T.; McClean, S.; Shaw, C.; Rao, P.F.; Ye, M.Y.; Bjourson, A.J. Purification, characterization and molecular cloning of chymotrypsin inhibitor peptides from the venom of Burmese Daboia russelii siamensis. Peptides 2013, 43C, 126–132. [Google Scholar]
- Guo, C.T.; McClean, S.; Shaw, C.; Rao, P.F.; Ye, M.Y.; Bjourson, A.J. Trypsin and chymotrypsin inhibitor peptides from the venom of Chinese Daboia russellii siamensis. Toxicon 2013, 63, 154–164. [Google Scholar] [CrossRef]
- Hokama, Y.; Iwanaga, S.; Tatsuki, T.; Suzuki, T. Snake venom proteinase inhibitors. III. Isolation of five polypeptide inhibitors from the venoms of Hemachatus haemachatus (Ringhal’s corbra) and Naja nivea (Cape cobra) and the complete amino acid sequences of two of them. J. Biochem. 1976, 79, 559–578. [Google Scholar]
- Ritonja, A.; Turk, V.; Gubensek, F. Serine proteinase inhibitors from Vipera ammodytes venom. Isolation and kinetic studies. Eur. J. Biochem. 1983, 133, 427–432. [Google Scholar] [CrossRef]
- Ritonja, A.; Meloun, B.; Gubensek, F. The primary structure of Vipera ammodytes venom trypsin inhibitor I. Biochim. Biophys. Acta 1983, 748, 429–435. [Google Scholar] [CrossRef]
- Liu, C.S.; Wu, T.C.; Lo, T.B. Complete amino acid sequences of two protease inhibitors in the venom of Bungarus fasciatus. Int. J. Pept. Protein Res. 1983, 21, 209–215. [Google Scholar]
- Lu, J.; Yang, H.; Yu, H.; Gao, W.; Lai, R.; Liu, J.; Liang, X. A novel serine protease inhibitor from Bungarus fasciatus venom. Peptides 2008, 29, 369–374. [Google Scholar] [CrossRef]
- Shafqat, J.; Beg, O.U.; Yin, S.J.; Zaidi, Z.H.; Jornvall, H. Primary structure and functional properties of cobra (Naja naja naja) venom Kunitz-type trypsin inhibitor. Eur. J. Biochem. 1990, 194, 337–341. [Google Scholar] [CrossRef]
- Zhou, X.D.; Jin, Y.; Lu, Q.M.; Li, D.S.; Zhu, S.W.; Wang, W.Y.; Xiong, Y.L. Purification, characterization and primary structure of a chymotrypsin inhibitor from Naja atra venom. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 137, 219–224. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Yan, F.J.; Chang, L.S. Taiwan cobra chymotrypsin inhibitor: Cloning, functional expression and gene organization. Biochim. Biophys. Acta 2005, 1747, 213–220. [Google Scholar] [CrossRef]
- Chang, L.; Chung, C.; Huang, H.B.; Lin, S. Purification and characterization of a chymotrypsin inhibitor from the venom of Ophiophagus hannah (King Cobra). Biochem. Biophys. Res. Commun. 2001, 283, 862–867. [Google Scholar] [CrossRef]
- He, Y.Y.; Liu, S.B.; Lee, W.H.; Qian, J.Q.; Zhang, Y. Isolation, expression and characterization of a novel dual serine protease inhibitor, OH-TCI, from king cobra venom. Peptides 2008, 29, 1692–1699. [Google Scholar] [CrossRef]
- Chang, L.S.; Wang, J.J.; Cheng, Y.C.; Chou, W.M. Genetic organization of Bungarus multicinctus protease inhibitor-like proteins. Toxicon 2008, 51, 1490–1495. [Google Scholar] [CrossRef]
- Possani, L.D.; Martin, B.M.; Yatani, A.; Mochca-Morales, J.; Zamudio, F.Z.; Gurrola, G.B.; Brown, A.M. Isolation and physiological characterization of taicatoxin, a complex toxin with specific effects on calcium channels. Toxicon 1992, 30, 1343–1364. [Google Scholar] [CrossRef]
- Earl, S.T.; Richards, R.; Johnson, L.A.; Flight, S.; Anderson, S.; Liao, A.; de Jersey, J.; Masci, P.P.; Lavin, M.F. Identification and characterisation of Kunitz-type plasma kallikrein inhibitors unique to Oxyuranus sp. snake venoms. Biochimie 2012, 94, 365–373. [Google Scholar] [CrossRef]
- Morjen, M.; Kallech-Ziri, O.; Bazaa, A.; Othman, H.; Mabrouk, K.; Zouari-Kessentini, R.; Sanz, L.; Calvete, J.J.; Srairi-Abid, N.; El Ayeb, M.; et al. PIVL, a new serine protease inhibitor from Macrovipera lebetina transmediterranea venom, impairs motility of human glioblastoma cells. Matrix Biol. 2013, 32, 52–62. [Google Scholar] [CrossRef]
- Inagaki, H.; Kimoto, H.; Yamauchi, Y.; Toriba, M.; Kubo, T. Functional characterization of Kunitz-type protease inhibitor Pr-mulgins identified from New Guinean Pseudechis australis. Toxicon 2012, 59, 74–80. [Google Scholar] [CrossRef]
- Chhatwal, G.S.; Habermann, E. Neurotoxins, protease inhibitors and histamine releasers in the venom of the Indian red scorpion (Buthus tamulus): Isolation and partial characterization. Toxicon 1981, 19, 807–823. [Google Scholar] [CrossRef]
- Schwartz, E.F.; Diego-Garcia, E.; Rodríguez de la Vega, R.C.; Possani, L.D. Transcriptome analysis of the venom gland of the Mexican scorpion Hadrurus gertschi (Arachnida: Scorpiones). BMC Genomics 2007, 8, 119. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Hu, Y.T.; Yang, W.S.; He, Y.W.; Feng, J.; Wang, B.; Zhao, R.M.; Ding, J.P.; Cao, Z.J.; Li, W.X.; Wu, Y.L. Hg1, novel peptide inhibitor specific for Kv1.3 channels from first scorpion Kunitz-type potassium channel toxin family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, B.; Hu, J.; Yang, W.; Cao, Z.; Zhuo, R.; Li, W.; Wu, Y. SjAPI, the first functionally characterized Ascaris-type protease inhibitor from animal venoms. PLoS One 2013, 8, e57529. [Google Scholar] [CrossRef]
- Wan, H.; Lee, K.S.; Kim, B.Y.; Zou, F.M.; Yoon, H.J.; Je, Y.H.; Li, J.; Jin, B.R. A spider-derived Kunitz-type serine protease inhibitor that acts as a plasmin inhibitor and an elastase inhibitor. PLoS One 2013, 8, e53343. [Google Scholar]
- Wan, H.; Lee, K.S.; Kim, B.Y.; Yuan, M.; Zhan, S.; You, H.; Li, J.; Jin, B.R. A spider (Araneus ventricosus) chymotrypsin inhibitor that acts as an elastase inhibitor and a microbial serine protease inhibitor. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2013, 165, 36–41. [Google Scholar] [CrossRef]
- Mignogna, G.; Pascarella, S.; Wechselberger, C.; Hinterleitner, C.; Mollay, C.; Amiconi, G.; Barra, D.; Kreil, G. BSTI, a trypsin inhibitor from skin secretions of Bombina bombina related to protease inhibitors of nematodes. Protein Sci. 1996, 5, 357–362. [Google Scholar]
- Lai, R.; Liu, H.; Lee, W.H.; Zhang, Y. Identification and cloning of a trypsin inhibitor from skin secretions of Chinese red-belly toad Bombina maxima. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2002, 131, 47–53. [Google Scholar] [CrossRef]
- Ali, M.F.; Lips, K.R.; Knoop, F.C.; Fritzsch, B.; Miller, C.; Conlon, J.M. Antimicrobial peptides and protease inhibitors in the skin secretions of the crawfish frog, Rana areolata. Biochim. Biophys. Acta 2002, 1601, 55–63. [Google Scholar] [CrossRef]
- Chen, T.; Shaw, C. Identification and molecular cloning of novel trypsin inhibitor analogs from the dermal venom of the Oriental fire-bellied toad (Bombina orientalis) and the European yellow-bellied toad (Bombina variegata). Peptides 2003, 24, 873–880. [Google Scholar] [CrossRef]
- Lu, X.; Ma, Y.; Wu, J.; Lai, R. Two serine protease inhibitors from the skin secretions of the toad, Bombina microdeladigitora. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 149, 608–612. [Google Scholar] [CrossRef]
- Wu, J.; Liu, H.; Yang, H.; Yu, H.; You, D.; Ma, Y.; Ye, H.; Lai, R. Proteomic analysis of skin defensive factors of tree frog Hyla simplex. J. Proteome Res. 2011, 10, 4230–4240. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, Y.; Lee, W.H.; Zhang, Y. Isolation and preliminary characterization of a 22-kDa protein with trypsin inhibitory activity from toad Bufo andrewsi skin. Toxicon 2005, 46, 277–281. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, Y.; Wei, S.S.; Lee, W.H.; Zhang, Y. Purification and characterization of an irreversible serine protease inhibitor from skin secretions of Bufo andrewsi. Toxicon 2005, 46, 635–640. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, M.; Wei, S. Isolation and characterization of a trypsin inhibitor from the skin secretions of Kaloula pulchra hainana. Toxicon 2010, 56, 502–507. [Google Scholar] [CrossRef]
- Gebhard, L.G.; Carrizo, F.U.; Stern, A.L.; Burgardt, N.I.; Faivovich, J.; Lavilla, E.; Ermacora, M.R. A Kazal prolyl endopeptidase inhibitor isolated from the skin of Phyllomedusa sauvagii. Eur. J. Biochem. 2004, 271, 2117–2126. [Google Scholar] [CrossRef]
- Neiva, M.; Vargas, D.C.; Conceição, K.; Rádis-Baptista, G.; Assakura, M.T.; Jared, C.; Hayashi, M.A. Gene expression analysis by ESTs sequencing of the Brazilian frog Phyllomedusa nordestina skin glands. Toxicon 2013, 61, 139–150. [Google Scholar] [CrossRef]
- Li, R.; Wang, H.; Jiang, Y.; Yu, Y.; Wang, L.; Zhou, M.; Zhang, Y.; Chen, T.; Shaw, C. A novel Kazal-type trypsin inhibitor from the skin secretion of the Central American red-eyed leaf frog, Agalychnis callidryas. Biochimie 2012, 94, 1376–1381. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhou, M.; Yang, M.; Ma, C.; Chen, T.; Zhang, Y.; Zeller, M.; Hornshaw, M.; Shaw, C. Functional peptidomics of amphibian skin secretion: A novel Kunitz-type chymotrypsin inhibitor from the African hyperoliid frog, Kassina senegalensis. Biochimie 2012, 94, 891–899. [Google Scholar] [CrossRef]
- You, D.; Hong, J.; Rong, M.; Yu, H.; Liang, S.; Ma, Y.; Yang, H.; Wu, J.; Lin, D.; Lai, R. The first gene-encoded amphibian neurotoxin. J. Biol. Chem. 2009, 284, 22079–22086. [Google Scholar]
- Li, J.; Wu, J.; Wang, Y.; Xu, X.; Liu, T.; Lai, R.; Zhu, H. A small trypsin inhibitor from the frog of Odorrana grahami. Biochimie 2008, 90, 1356–1361. [Google Scholar] [CrossRef]
- Song, G.; Zhou, M.; Chen, W.; Chen, T.; Walker, B.; Shaw, C. HV-BBI—A novel amphibian skin Bowman-Birk-like trypsin inhibitor. Biochem. Biophys. Res. Commun. 2008, 372, 191–196. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Lu, Z.; Zhai, L.; Jiang, J.; Liu, J.; Yu, H. A novel serine protease inhibitor from the venom of Vespa bicolor Fabricius. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009, 153, 116–120. [Google Scholar] [CrossRef]
- Qiu, Y.; Lee, K.S.; Choo, Y.M.; Kong, D.; Yoon, H.J.; Jin, B.R. Molecular cloning and antifibrinolytic activity of a serine protease inhibitor from bumblebee (Bombus terrestris) venom. Toxicon 2013, 63, 1–6. [Google Scholar] [CrossRef]
- Lenarcic, B.; Turk, V. Thyroglobulin type-1 domains in equistatin inhibit both papain-like cysteine proteinases and cathepsin D. J. Biol. Chem. 1999, 274, 563–566. [Google Scholar] [CrossRef]
- Strukelj, B.; Lenarcic, B.; Gruden, K.; Pungercar, J.; Rogelj, B.; Turk, V.; Bosch, D.; Jongsma, M.A. Equistatin, a protease inhibitor from the sea anemone Actinia equina is composed of three structural and functional domains. Biochem. Biophys. Res. Commun. 2000, 269, 732–736. [Google Scholar] [CrossRef]
- Honma, T.; Shiomi, K. Peptide toxins in sea anemones: Structural and functional aspects. Mar. Biotechnol. (NY) 2006, 8, 1–10. [Google Scholar] [CrossRef]
- Frazão, B.; Vasconcelos, V.; Antunes, A. Sea anemone (Cnidaria, Anthozoa, Actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar]
- Bayrhuber, M.; Graf, R.; Ferber, M.; Zweckstetter, M.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Becker, S. Production of recombinant Conkunitzin-S1 in Escherichia coli. Protein Expr. Purif. 2006, 47, 640–644. [Google Scholar] [CrossRef]
- Harvey, A.L. Twenty years of dendrotoxins. Toxicon 2001, 39, 15–26. [Google Scholar] [CrossRef]
- Owen, D.G.; Hall, A.; Stephens, G.; Stow, J.; Robertson, B. The relative potencies of dendrotoxins as blockers of the cloned voltage-gated K+ channel, mKv1.1 (MK-1), when stably expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1997, 120, 1029–1034. [Google Scholar] [CrossRef]
- Hopkins, W.F. Toxin and subunit specificity of blocking affinity of three peptide toxins for heteromultimeric, voltage-gated potassium channels expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 1998, 285, 1051–1060. [Google Scholar]
- Tytgat, J.; Vandenberghe, I.; Ulens, C.; van Beeumen, J. New polypeptide components purified from mamba venom. FEBS Lett. 2001, 491, 217–221. [Google Scholar] [CrossRef]
- Chen, Z.; Luo, F.; Feng, J.; Yang, W.; Zeng, D.; Zhao, R.; Cao, Z.; Liu, M.; Li, W.; Jiang, L.; Wu, Y. Genomic and structural characterization of Kunitz-type peptide LmKTT-1a highlights diversity and evolution of scorpion potassium channel toxins. PLoS One 2013, 8, e60201. [Google Scholar] [CrossRef]
- Monastyrnaya, M.M.; Zykova, T.A.; Apalikova, O.V.; Shwets, T.V.; Kozlovskaya, E.P. Biologically active polypeptides from the tropical sea anemone Radianthus macrodactylus. Toxicon 2002, 40, 1197–1217. [Google Scholar] [CrossRef]
- Takahashi, H.; Iwanaga, S.; Suzuki, T. Isolation of a novel inhibitor of kallikrein, plasmin and trypsin from the venom of Russell’s viper (Vipera russelli). FEBS Lett. 1972, 27, 207–210. [Google Scholar] [CrossRef]
- Anderluh, G.; Podlesek, Z.; Macek, P. A common motif in proparts of Cnidarian toxins and nematocyst collagens and its putative role. Biochim. Biophys. Acta 2000, 1476, 372–376. [Google Scholar] [CrossRef]
- Craig, A.G.; Bandyopadhyay, P.; Olivera, B.M. Post-translationally modified neuropeptides from Conus venoms. Eur. J. Biochem. 1999, 264, 271–275. [Google Scholar] [CrossRef]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Vijayan, V.; Ferber, M.; Graf, R.; Korukottu, J.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Zweckstetter, M.; Becker, S. Conkunitzin-S1 is the first member of a new Kunitz-type neurotoxin family. Structural and functional characterization. J. Biol. Chem. 2005, 280, 23766–23770. [Google Scholar] [CrossRef]
- Elliger, C.A.; Richmond, T.A.; Lebaric, Z.N.; Pierce, N.T.; Sweedler, J.V.; Gilly, W.F. Diversity of conotoxin types from Conus californicus reflects a diversity of prey types and a novel evolutionary history. Toxicon 2011, 57, 311–322. [Google Scholar] [CrossRef]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Characterization of the Conus bullatus genome and its venom-duct transcriptome. BMC Genomics 2011, 12, 60. [Google Scholar] [CrossRef]
- Strydom, D.J. Protease inhibitors as snake venom toxins. Nat. New Biol. 1973, 243, 88–89. [Google Scholar] [CrossRef]
- Harvey, A.L.; Karlsson, E. Dendrotoxin from the venom of the green mamba, Dendroaspis angusticeps. Naunyn Schmiedebergs Arch. Pharmacol. 1980, 312, 1–6. [Google Scholar] [CrossRef]
- Harvey, A.L.; Robertson, B. Dendrotoxins: Structure-activity relationships and effects on potassium ion channels. Curr. Med. Chem. 2004, 11, 3065–3072. [Google Scholar] [CrossRef]
- Schweitz, H.; Heurteaux, C.; Bois, P.; Moinier, D.; Romey, G.; Lazdunski, M. Calcicludine, a venom peptide of the Kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for L-type channels in cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 878–882. [Google Scholar] [CrossRef]
- Gilquin, B.; Lecoq, A.; Desne, F.; Guenneugues, M.; Zinn-Justin, S.; Menez, A. Conformational and functional variability supported by the BPTI fold: Solution structure of the Ca2+ channel blocker calcicludine. Proteins 1999, 34, 520–532. [Google Scholar] [CrossRef]
- Kondo, K.; Toda, H.; Narita, K.; Lee, C.Y. Amino acid sequence of β2-bungarotoxin from Bungarus multicinctus venom. The amino acid substitutions in the B chains. J. Biochem. 1982, 91, 1519–1530. [Google Scholar]
- Wu, P.F.; Wu, S.N.; Chang, C.C.; Chang, L.S. Cloning and functional expression of B chains of β-bungarotoxins from Bungarus multicinctus (Taiwan banded krait). Biochem. J. 1998, 334, 87–92. [Google Scholar]
- Takahashi, H.; Iwanaga, S.; Kitagawa, T.; Hokama, Y.; Suzuki, T. Snake venom proteinase inhibitors. II. Chemical structure of inhibitor II isolated from the venom of Russell’s viper (Vipera russelli). J. Biochem. 1974, 76, 721–733. [Google Scholar]
- Zupunski, V.; Kordis, D.; Gubensek, F. Adaptive evolution in the snake venom Kunitz/BPTI protein family. FEBS Lett. 2003, 547, 131–136. [Google Scholar] [CrossRef]
- Siddiqi, A.R.; Zaidi, Z.H.; Jornvall, H. Purification and characterization of a Kunitz-type trypsin inhibitor from Leaf-nosed viper venom. FEBS Lett. 1991, 294, 141–143. [Google Scholar] [CrossRef]
- Chen, C.; Hsu, C.H.; Su, N.Y.; Lin, Y.C.; Chiou, S.H.; Wu, S.H. Solution structure of a Kunitz-type chymotrypsin inhibitor isolated from the elapid snake Bungarus fasciatus. J. Biol. Chem. 2001, 276, 45079–45087. [Google Scholar]
- Shafqat, J.; Zaidi, Z.H.; Jornvall, H. Purification and characterization of a chymotrypsin Kunitz inhibitor type of polypeptide from the venom of cobra (Naja naja naja). FEBS Lett. 1990, 275, 6–8. [Google Scholar] [CrossRef]
- Doorty, K.B.; Bevan, S.; Wadsworth, J.D.; Strong, P.N. A novel small conductance Ca2+-activated K+ channel blocker from Oxyuranus scutellatus taipan venom. Re-evaluation of taicatoxin as a selective Ca2+ channel probe. J. Biol. Chem. 1997, 272, 19925–19930. [Google Scholar]
- Filippovich, I.; Sorokina, N.; Masci, P.P.; de Jersey, J.; Whitaker, A.N.; Winzor, D.J.; Gaffney, P.J.; Lavin, M.F. A family of textilinin genes, two of which encode proteins with antihaemorrhagic properties. Br. J. Haematol. 2002, 119, 376–384. [Google Scholar] [CrossRef]
- Flight, S.; Johnson, L.; Trabi, M.; Gaffney, P.; Lavin, M.; de Jersey, J.; Masci, P. Comparison of textilinin-1 with aprotinin as serine protease inhibitors and as antifibrinolytic agents. Pathophysiol. Haemost. Thromb. 2005, 34, 188–193. [Google Scholar] [CrossRef]
- Flight, S.M.; Johnson, L.A.; Du, Q.S.; Warner, R.L.; Trabi, M.; Gaffney, P.J.; Lavin, M.F.; de Jersey, J.; Masci, P.P. Textilinin-1, an alternative anti-bleeding agent to aprotinin: Importance of plasmin inhibition in controlling blood loss. Br. J. Haematol. 2009, 145, 207–211. [Google Scholar] [CrossRef]
- Mangano, D.T.; Tudor, I.C.; Dietzel, C. The risk associated with aprotinin in cardiac surgery. N. Engl. J. Med. 2006, 354, 353–365. [Google Scholar] [CrossRef]
- St Pierre, L.; Earl, S.T.; Filippovich, I.; Sorokina, N.; Masci, P.P.; De Jersey, J.; Lavin, M.F. Common evolution of waprin and kunitz-like toxin families in Australian venomous snakes. Cell Mol. Life Sci. 2008, 65, 4039–4054. [Google Scholar] [CrossRef]
- Chatrath, S.T.; Chapeaurouge, A.; Lin, Q.; Lim, T.K.; Dunstan, N.; Mirtschin, P.; Kumar, P.P.; Kini, R.M. Identification of novel proteins from the venom of a cryptic snake Drysdalia coronoides by a combined transcriptomics and proteomics approach. J. Proteome Res. 2011, 10, 739–750. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Lee, W.; Xu, X.; Zhang, Y.; Zhao, R.; Wang, W. Venom gland transcriptomes of two elapid snakes (Bungarus multicinctus and Naja atra) and evolution of toxin genes. BMC Genomics 2011, 12, 1. [Google Scholar] [CrossRef]
- Ramazanova, A.S.; Fil'kin, S.; Starkov, V.G.; Utkin Iu, N. Molecular cloning and analysis of cDNA sequences encoding serine proteinase and Kunitz type inhibitor in venom gland of Vipera nikolskii viper (in Russian). Bioorg. Khim. 2011, 37, 374–385. [Google Scholar]
- Corrêa-Netto, C.; Junqueira-de-Azevedo Ide, L.; Silva, D.A.; Ho, P.L.; Leitão-de-Araújo, M.; Alves, M.L.; Sanz, L.; Foguel, D.; Zingali, R.B.; Calvete, J.J. Snake venomics and venom gland transcriptomic analysis of Brazilian coral snakes, Micrurus altirostris and M. corallinus. J. Proteomics 2011, 74, 1795–1809. [Google Scholar] [CrossRef]
- Turk, V.; Bode, W. The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef]
- Richards, R.; St Pierre, L.; Trabi, M.; Johnson, L.A.; de Jersey, J.; Masci, P.P.; Lavin, M.F. Cloning and characterisation of novel cystatins from elapid snake venom glands. Biochimie 2011, 93, 659–668. [Google Scholar] [CrossRef]
- Evans, H.J.; Barrett, A.J. A cystatin-like cysteine proteinase inhibitor from venom of the African puff adder (Bitis arietans). Biochem. J. 1987, 246, 795–797. [Google Scholar]
- Brillard-Bourdet, M.; Nguyen, V.; Ferrer-di Martino, M.; Gauthier, F.; Moreau, T. Purification and characterization of a new cystatin inhibitor from Taiwan cobra (Naja naja atra) venom. Biochem. J. 1998, 331, 239–244. [Google Scholar]
- Mashiko, H.; Takahashi, H. Cysteine proteinase inhibitors in elapid and hydrophiid snake venoms. Toxicon 2002, 40, 1275–1281. [Google Scholar] [CrossRef]
- Ferreira, S.H. A bradykinin-potentiating factor (BFP) present in the venom of Bothrops jararca. Br. J. Pharmacol. Chemother. 1965, 24, 163–169. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Bartelt, D.C.; Greene, L.J. Isolation of bradykinin-potentiating peptides from Bothrops jararaca venom. Biochemistry 1970, 9, 2583–2593. [Google Scholar] [CrossRef]
- Stewart, J.M.; Ferreira, S.H.; Greene, L.J. Bradykinin potentiating peptide PCA-Lys-Trp-Ala-Pro: An inhibitor of the pulmonary inactivation of Bradykinin and conversion of angiotensin I to II. Biochem. Pharmacol. 1971, 20, 1557–1567. [Google Scholar] [CrossRef]
- Opie, L.H.; Kowolik, H. The discovery of captopril: From large animals to small molecules. Cardiovasc. Res. 1995, 30, 18–25. [Google Scholar]
- Higuchi, S.; Murayama, N.; Saguchi, K.; Ohi, H.; Fujita, Y.; da Silva, N.J., Jr.; de Siqueira, R.J.; Lahlou, S.; Aird, S.D. A novel peptide from the ACEI/BPP-CNP precursor in the venom of Crotalus durissus collilineatus. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2006, 144, 107–121. [Google Scholar] [CrossRef]
- Cidade, D.A.; Simão, T.A.; Davila, A.M.; Wagner, G.; Junqueira-de-Azevedo, I.L.; Ho, P.L.; Bon, C.; Zingali, R.B.; Albano, R.M. Bothrops jararaca venom gland transcriptome: Analysis of the gene expression pattern. Toxicon 2006, 48, 437–461. [Google Scholar] [CrossRef]
- Munawar, A.; Trusch, M.; Georgieva, D.; Spencer, P.; Frochaux, V.; Harder, S.; Arni, R.K.; Duhalov, D.; Genov, N.; Schluter, H.; Betzel, C. Venom peptide analysis of Vipera ammodytes meridionalis (Viperinae) and Bothrops jararacussu (Crotalinae) demonstrates subfamily-specificity of the peptidome in the family Viperidae. Mol. Biosyst. 2011, 7, 3298–3307. [Google Scholar] [CrossRef]
- Chi, C.W.; Wang, S.Z.; Xu, L.G.; Wang, M.Y.; Lo, S.S.; Huang, W.D. Structure-function studies on the bradykinin potentiating peptide from Chinese snake venom (Agkistrodon halys Pallas). Peptides 1985, 6, 339–342. [Google Scholar] [CrossRef]
- Higuchi, S.; Murayama, N.; Saguchi, K.; Ohi, H.; Fujita, Y.; Camargo, A.C.; Ogawa, T.; Deshimaru, M.; Ohno, M. Bradykinin-potentiating peptides and C-type natriuretic peptides from snake venom. Immunopharmacology 1999, 44, 129–135. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Clore, G.M.; Gronenborn, A.M. High-resolution structure of Ascaris trypsin inhibitor in solution: Direct evidence for a pH-induced conformational transition in the reactive site. Structure 1994, 2, 669–678. [Google Scholar] [CrossRef]
- Brazón, J.; D'Suze, G.; D'Errico, M.L.; Arocha-Piñango, C.L.; Guerrero, B. Discreplasminin, a plasmin inhibitor isolated from Tityus discrepans scorpion venom. Arch. Toxicol. 2009, 83, 669–678. [Google Scholar] [CrossRef]
- Silva, E.C.; Camargos, T.S.; Maranhao, A.Q.; Silva-Pereira, I.; Silva, L.P.; Possani, L.D.; Schwartz, E.F. Cloning and characterization of cDNA sequences encoding for new venom peptides of the Brazilian scorpion Opisthacanthus cayaporum. Toxicon 2009, 54, 252–261. [Google Scholar] [CrossRef]
- Gremski, L.H.; da Silveira, R.B.; Chaim, O.M.; Probst, C.M.; Ferrer, V.P.; Nowatzki, J.; Weinschutz, H.C.; Madeira, H.M.; Gremski, W.; Nader, H.B.; et al. A novel expression profile of the Loxosceles intermedia spider venomous gland revealed by transcriptome analysis. Mol. Biosyst. 2010, 6, 2403–2416. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Lai, R.; Lee, W.H.; Zhang, Y. Frog albumin is expressed in skin and characterized as a novel potent trypsin inhibitor. Protein Sci. 2005, 14, 2469–2477. [Google Scholar] [CrossRef]
- Hisada, M.; Satake, H.; Masuda, K.; Aoyama, M.; Murata, K.; Shinada, T.; Iwashita, T.; Ohfune, Y.; Nakajima, T. Molecular components and toxicity of the venom of the solitary wasp, Anoplius samariensis. Biochem. Biophys. Res. Commun. 2005, 330, 1048–1054. [Google Scholar] [CrossRef]
- Parkinson, N.M.; Conyers, C.; Keen, J.; MacNicoll, A.; Smith, I.; Audsley, N.; Weaver, R. Towards a comprehensive view of the primary structure of venom proteins from the parasitoid wasp Pimpla hypochondriaca. Insect Biochem. Mol. Biol. 2004, 34, 565–571. [Google Scholar] [CrossRef]
- Choo, Y.M.; Lee, K.S.; Yoon, H.J.; Kim, B.Y.; Sohn, M.R.; Roh, J.Y.; Je, Y.H.; Kim, N.J.; Kim, I.; Woo, S.D.; et al. Dual function of a bee venom serine protease: Prophenoloxidase-activating factor in arthropods and fibrin(ogen)olytic enzyme in mammals. PLoS One 2010, 5, e10393. [Google Scholar] [CrossRef]
- Kim, B.Y.; Lee, K.S.; Wan, H.; Zou, F.M.; Choi, Y.S.; Yoon, H.J.; Kwon, H.W.; Je, Y.H.; Jin, B.R. Anti-elastolytic activity of a honeybee (Apis cerana) chymotrypsin inhibitor. Biochem. Biophys. Res. Commun. 2013, 430, 144–149. [Google Scholar] [CrossRef]
- Skarzynski, T. Crystal structure of α-dendrotoxin from the green mamba venom and its comparison with the structure of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 1992, 224, 671–683. [Google Scholar] [CrossRef]
- Grissmer, S.; Nguyen, A.N.; Aiyar, J.; Hanson, D.C.; Mather, R.J.; Gutman, G.A.; Karmilowicz, M.J.; Auperin, D.D.; Chandy, K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994, 45, 1227–1234. [Google Scholar]
- Robertson, B.; Owen, D.; Stow, J.; Butler, C.; Newland, C. Novel effects of dendrotoxin homologues on subtypes of mammalian Kv1 potassium channels expressed in Xenopus oocytes. FEBS Lett. 1996, 383, 26–30. [Google Scholar] [CrossRef]
- Joubert, F.J.; Strydom, D.J. Snake venoms. The amino-acid sequence of trypsin inhibitor E of Dendroaspis polylepis polylepis (Black Mamba) venom. Eur. J. Biochem. 1978, 87, 191–198. [Google Scholar] [CrossRef]
- Harvey, A.L. Recent studies on dendrotoxins and potassium ion channels. Gen. Pharmacol. 1997, 28, 7–12. [Google Scholar]
- Harvey, A.L.; Rowan, E.G.; Vatanpour, H.; Engstrom, A.; Westerlund, B.; Karlsson, E. Changes to biological activity following acetylation of dendrotoxin I from Dendroaspis polylepis (black mamba). Toxicon 1997, 35, 1263–1273. [Google Scholar] [CrossRef]
- Gasparini, S.; Danse, J.M.; Lecoq, A.; Pinkasfeld, S.; Zinn-Justin, S.; Young, L.C.; de Medeiros, C.C.; Rowan, E.G.; Harvey, A.L.; Menez, A. Delineation of the functional site of α-dendrotoxin. The functional topographies of dendrotoxins are different but share a conserved core with those of other Kv1 potassium channel-blocking toxins. J. Biol. Chem. 1998, 273, 25393–25403. [Google Scholar] [CrossRef]
- Smith, L.A.; Reid, P.F.; Wang, F.C.; Parcej, D.N.; Schmidt, J.J.; Olson, M.A.; Dolly, J.O. Site-directed mutagenesis of dendrotoxin K reveals amino acids critical for its interaction with neuronal K+ channels. Biochemistry 1997, 36, 7690–7696. [Google Scholar] [CrossRef]
- Wang, F.C.; Bell, N.; Reid, P.; Smith, L.A.; McIntosh, P.; Robertson, B.; Dolly, J.O. Identification of residues in dendrotoxin K responsible for its discrimination between neuronal K+ channels containing Kv1.1 and 1.2 α subunits. Eur. J. Biochem. 1999, 263, 222–229. [Google Scholar] [CrossRef]
- Danse, J.M.; Rowan, E.G.; Gasparini, S.; Ducancel, F.; Vatanpour, H.; Young, L.C.; Poorheidari, G.; Lajeunesse, E.; Drevet, P.; Menez, R.; et al. On the site by which α-dendrotoxin binds to voltage-dependent potassium channels: Site-directed mutagenesis reveals that the lysine triplet 28–30 is not essential for binding. FEBS Lett. 1994, 356, 153–158. [Google Scholar] [CrossRef]
- Imredy, J.P.; MacKinnon, R. Energetic and structural interactions between δ-dendrotoxin and a voltage-gated potassium channel. J. Mol. Biol. 2000, 296, 1283–1294. [Google Scholar] [CrossRef]
- Lee, C.Y.; Chang, S.L.; Kau, S.T.; Luh, S.H. Chromatographic separation of the venom of Bungarus multicinctus and characterization of its components. J. Chromatogr. 1972, 72, 71–82. [Google Scholar]
- MacDermot, J.; Westgaard, R.H.; Thompson, E.J. β-Bungarotoxin. Separation of two discrete proteins with different synaptic actions. Biochem. J. 1978, 175, 271–279. [Google Scholar]
- Ueno, E.; Rosenberg, P. Mechanism of action of β-bungarotoxin, a presynaptically acting phospholipase A2 neurotoxin: Its effect on protein phosphorylation in rat brain synaptosomes. Toxicon 1996, 34, 1219–1227. [Google Scholar] [CrossRef]
- Shi, Y.; Gu, C.; Xu, Y. Four types of potassium currents in motor nerve terminals of snake. Sci. China C Life Sci. 1997, 40, 593–603. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Betz, H.; Rehm, H. Inhibition of β-bungarotoxin binding to brain membranes by mast cell degranulating peptide, toxin I, and ethylene glycol bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid. Biochemistry 1988, 27, 963–967. [Google Scholar] [CrossRef]
- Chu, S.T.; Chu, C.C.; Tseng, C.C.; Chen, Y.H. Met-8 of the β1-bungarotoxin phospholipase A2 subunit is essential for the phospholipase A2-independent neurotoxic effect. Biochem. J. 1993, 295, 713–718. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Goodstadt, L.; Ponting, C.P. CHROMA: Consensus-based colouring of multiple alignments for publication. Bioinformatics 2001, 17, 845–846. [Google Scholar] [CrossRef]
- Rodríguez de la Vega, R.C.; Merino, E.; Becerril, B.; Possani, L.D. Novel interactions between K+ channels and scorpion toxins. Trends Pharmacol. Sci. 2003, 24, 222–227. [Google Scholar] [CrossRef]
- Mouhat, S.; de Waard, M.; Sabatier, J.M. Contribution of the functional dyad of animal toxins acting on voltage-gated Kv1-type channels. J. Pept. Sci. 2005, 11, 65–68. [Google Scholar] [CrossRef]
- Jin, L.; Wu, Y. Molecular mechanism of δ-dendrotoxin-potassium channel recognition explored by docking and molecular dynamic simulations. J. Mol. Recognit. 2011, 24, 101–107. [Google Scholar] [CrossRef]
- Otlewski, J.; Jaskolski, M.; Buczek, O.; Cierpicki, T.; Czapinska, H.; Krowarsch, D.; Smalas, A.O.; Stachowiak, D.; Szpineta, A.; Dadlez, M. Structure-function relationship of serine protease-protein inhibitor interaction. Acta Biochim. Pol. 2001, 48, 419–428. [Google Scholar]
- Kawamura, K.; Yamada, T.; Kurihara, K.; Tamada, T.; Kuroki, R.; Tanaka, I.; Takahashi, H.; Niimura, N. X-ray and neutron protein crystallographic analysis of the trypsin-BPTI complex. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 140–148. [Google Scholar]
- Laskowski, M., Jr.; Kato, I. Protein inhibitors of proteinases. Annu. Rev. Biochem. 1980, 49, 593–626. [Google Scholar] [CrossRef]
- Scheidig, A.J.; Hynes, T.R.; Pelletier, L.A.; Wells, J.A.; Kossiakoff, A.A. Crystal structures of bovine chymotrypsin and trypsin complexed to the inhibitor domain of Alzheimer’s amyloid β-protein precursor (APPI) and basic pancreatic trypsin inhibitor (BPTI): Engineering of inhibitors with altered specificities. Protein Sci. 1997, 6, 1806–1824. [Google Scholar] [CrossRef]
- Navaneetham, D.; Sinha, D.; Walsh, P.N. Mechanisms and specificity of factor XIa and trypsin inhibition by protease nexin 2 and basic pancreatic trypsin inhibitor. J. Biochem. 2010, 148, 467–479. [Google Scholar]
- Fiedler, F. Effects of secondary interactions on the kinetics of peptide and peptide ester hydrolysis by tissue kallikrein and trypsin. Eur. J. Biochem. 1987, 163, 303–312. [Google Scholar] [CrossRef]
- Grzesiak, A.; Krokoszynska, I.; Krowarsch, D.; Buczek, O.; Dadlez, M.; Otlewski, J. Inhibition of six serine proteinases of the human coagulation system by mutants of bovine pancreatic trypsin inhibitor. J. Biol. Chem. 2000, 275, 33346–33352. [Google Scholar]
- Van Nostrand, W.E.; Schmaier, A.H.; Siegel, R.S.; Wagner, S.L.; Raschke, W.C. Enhanced plasmin inhibition by a reactive center lysine mutant of the Kunitz-type protease inhibitor domain of the amyloid β-protein precursor. J. Biol. Chem. 1995, 270, 22827–22830. [Google Scholar]
- Hanson, W.M.; Domek, G.J.; Horvath, M.P.; Goldenberg, D.P. Rigidification of a flexible protease inhibitor variant upon binding to trypsin. J. Mol. Biol. 2007, 366, 230–243. [Google Scholar] [CrossRef]
- Fritz, H.; Wunderer, G. Biochemistry and applications of aprotinin, the kallikrein inhibitor from bovine organs. Arzneimittelforschung 1983, 33, 479–494. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Vakorina, T.I.; Gladkikh, I.N.; Monastyrnaia, M.M.; Kozlovskaia, E.P. Conformational stability of serine proteinase inhibitor from the sea anemone Heteractis crispa (in Russian). Bioorg. Khim. 2011, 37, 310–318. [Google Scholar]
- Dy, C.Y.; Buczek, P.; Imperial, J.S.; Bulaj, G.; Horvath, M.P. Structure of conkunitzin-S1, a neurotoxin and Kunitz-fold disulfide variant from cone snail. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 980–990. [Google Scholar]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.; Rowan, E.G.; et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar] [CrossRef]
- Rodríguez de la Vega, R.C.; Possani, L.D. Current views on scorpion toxins specific for K+-channels. Toxicon 2004, 43, 865–875. [Google Scholar] [CrossRef]
- Berndt, K.D.; Guntert, P.; Wuthrich, K. Nuclear magnetic resonance solution structure of dendrotoxin K from the venom of Dendroaspis polylepis polylepis. J. Mol. Biol. 1993, 234, 735–750. [Google Scholar] [CrossRef]
- Landis, R.C.; Asimakopoulos, G.; Poullis, M.; Haskard, D.O.; Taylor, K.M. The antithrombotic and antiinflammatory mechanisms of action of aprotinin. Ann. Thorac. Surg. 2001, 72, 2169–2175. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Bolognesi, M.; Spallarossa, A.; Coletta, M.; de Cristofaro, R.; Menegatti, E. The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): A milestone protein. Curr. Protein Pept. Sci. 2003, 4, 231–251. [Google Scholar] [CrossRef]
- Bidstrup, B.P.; Royston, D.; Sapsford, R.N.; Taylor, K.M. Reduction in blood loss and blood use after cardiopulmonary bypass with high dose aprotinin (Trasylol). J. Thorac. Cardiovasc. Surg. 1989, 97, 364–372. [Google Scholar]
- Lemmer, J.H., Jr.; Stanford, W.; Bonney, S.L.; Breen, J.F.; Chomka, E.V.; Eldredge, W.J.; Holt, W.W.; Karp, R.B.; Laub, G.W.; Lipton, M.J.; et al. Aprotinin for coronary bypass operations: efficacy, safety, and influence on early saphenous vein graft patency. A multicenter, randomized, double-blind, placebo-controlled study. J. Thorac. Cardiovasc. Surg. 1994, 107, 543–551; discussion 551–553. [Google Scholar]
- Millers, E.K.; Trabi, M.; Masci, P.P.; Lavin, M.F.; de Jersey, J.; Guddat, L.W. Crystal structure of textilinin-1, a Kunitz-type serine protease inhibitor from the venom of the Australian common brown snake (Pseudonaja textilis). FEBS J. 2009, 276, 3163–3175. [Google Scholar] [CrossRef]
- Puente, X.S.; Sanchez, L.M.; Overall, C.M.; Lopez-Otin, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef]
- Pandey, K.C. Centenary celebrations article: Cysteine proteases of human malaria parasites. J. Parasit. Dis. 2011, 35, 94–103. [Google Scholar]
- Ghany, M.G.; Nelson, D.R.; Strader, D.B.; Thomas, D.L.; Seeff, L.B. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 1433–1444. [Google Scholar] [CrossRef]
- Randolph, J.T.; Huang, P.P.; Flosi, W.J.; DeGoey, D.; Klein, L.L.; Yeung, C.M.; Flentge, C.; Sun, M.; Zhao, C.; Dekhtyar, T.; et al. Synthesis, antiviral activity, and pharmacokinetic evaluation of P3 pyridylmethyl analogs of oximinoarylsulfonyl HIV-1 protease inhibitors. Bioorg. Med. Chem. 2006, 14, 4035–4046. [Google Scholar] [CrossRef]
- Lu, D.; Sham, Y.Y.; Vince, R. Design, asymmetric synthesis, and evaluation of pseudosymmetric sulfoximine inhibitors against HIV-1 protease. Bioorg. Med. Chem. 2010, 18, 2037–2048. [Google Scholar] [CrossRef]
- Wensing, A.M.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antiviral Res. 2010, 85, 59–74. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mourão, C.B.F.; Schwartz, E.F. Protease Inhibitors from Marine Venomous Animals and Their Counterparts in Terrestrial Venomous Animals. Mar. Drugs 2013, 11, 2069-2112. https://doi.org/10.3390/md11062069
Mourão CBF, Schwartz EF. Protease Inhibitors from Marine Venomous Animals and Their Counterparts in Terrestrial Venomous Animals. Marine Drugs. 2013; 11(6):2069-2112. https://doi.org/10.3390/md11062069
Chicago/Turabian StyleMourão, Caroline B. F., and Elisabeth F. Schwartz. 2013. "Protease Inhibitors from Marine Venomous Animals and Their Counterparts in Terrestrial Venomous Animals" Marine Drugs 11, no. 6: 2069-2112. https://doi.org/10.3390/md11062069