c-Jun N-Terminal Kinase Phosphorylation Is a Biomarker of Plitidepsin Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
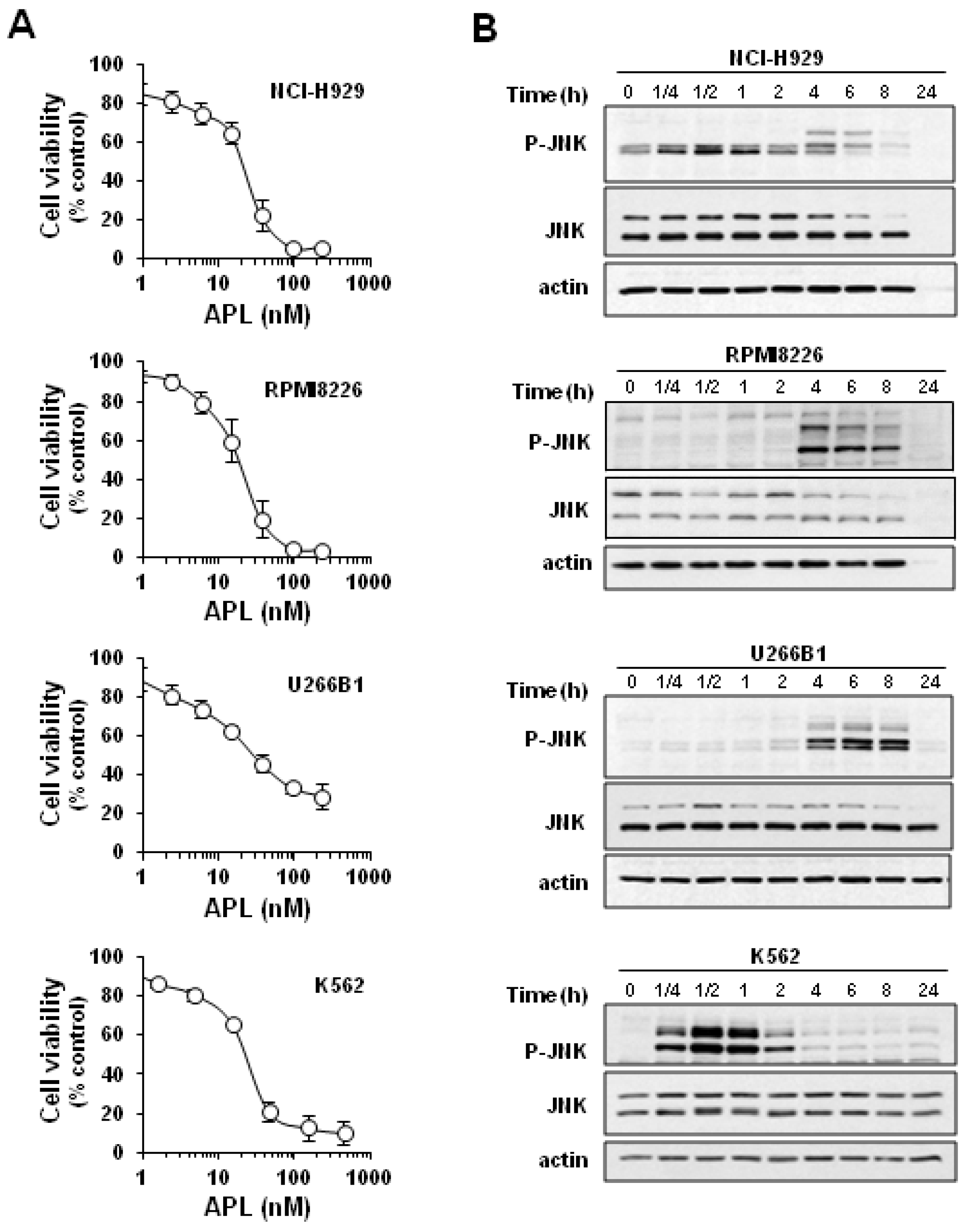
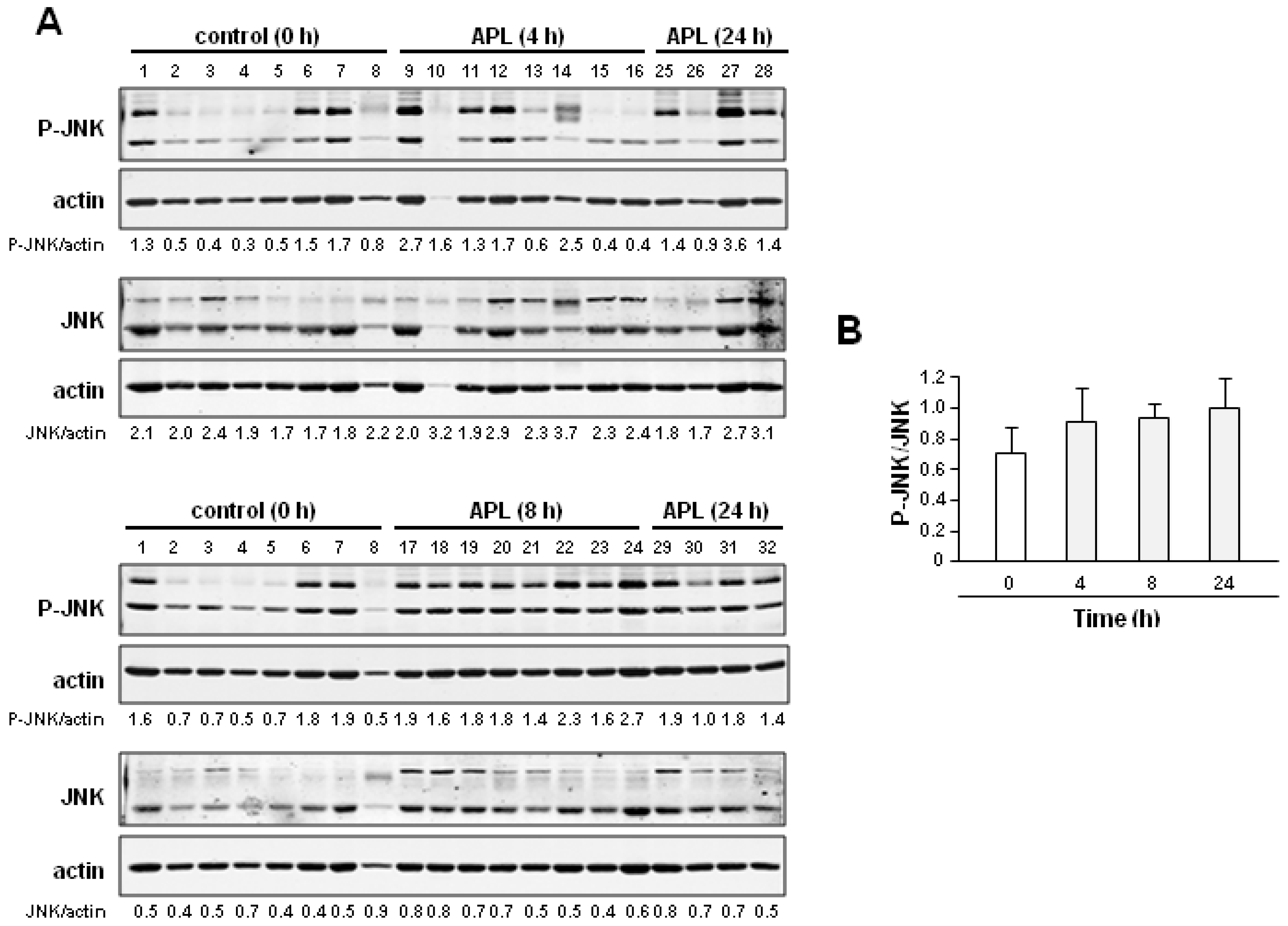
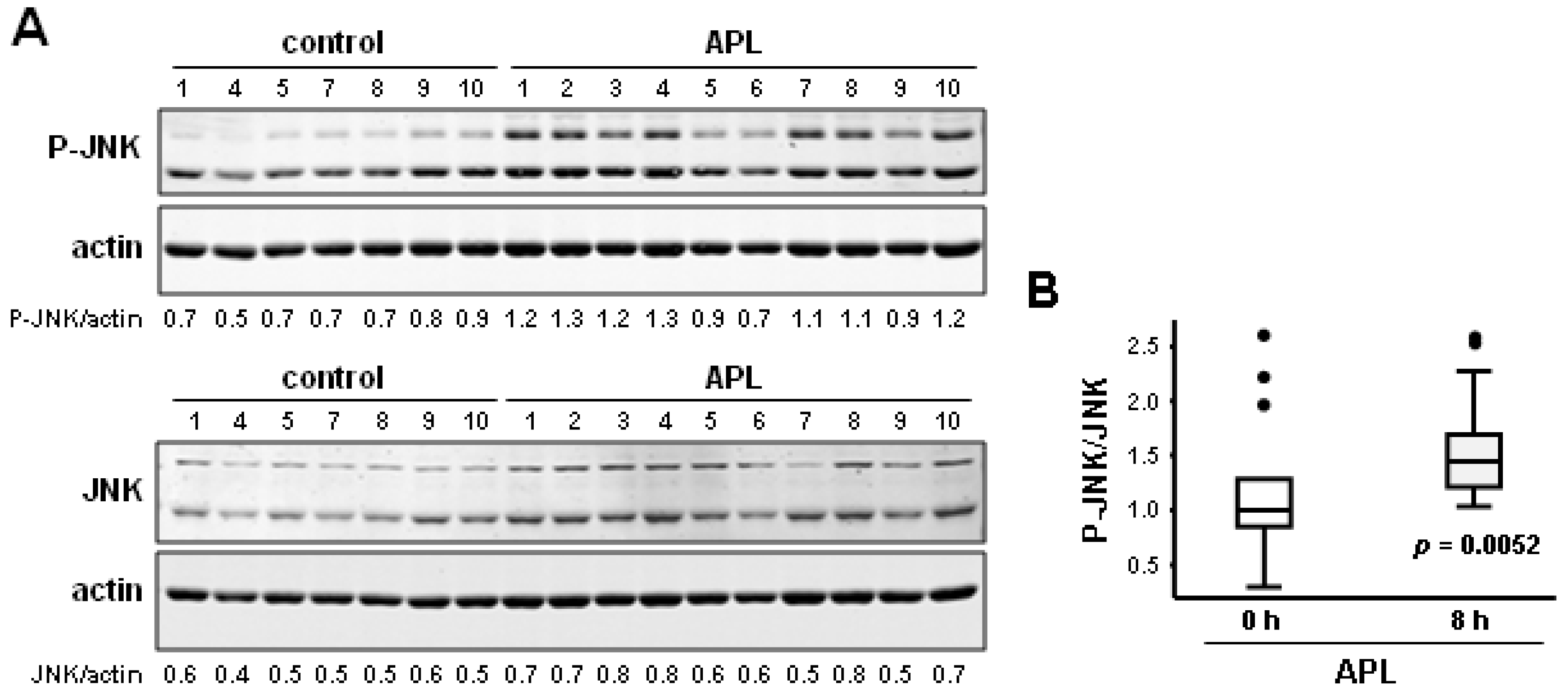
2.1. Plitidepsin Induces JNK Phosphorylation in Cultured K562 Leukemia and Other Hematological Cancer Cells

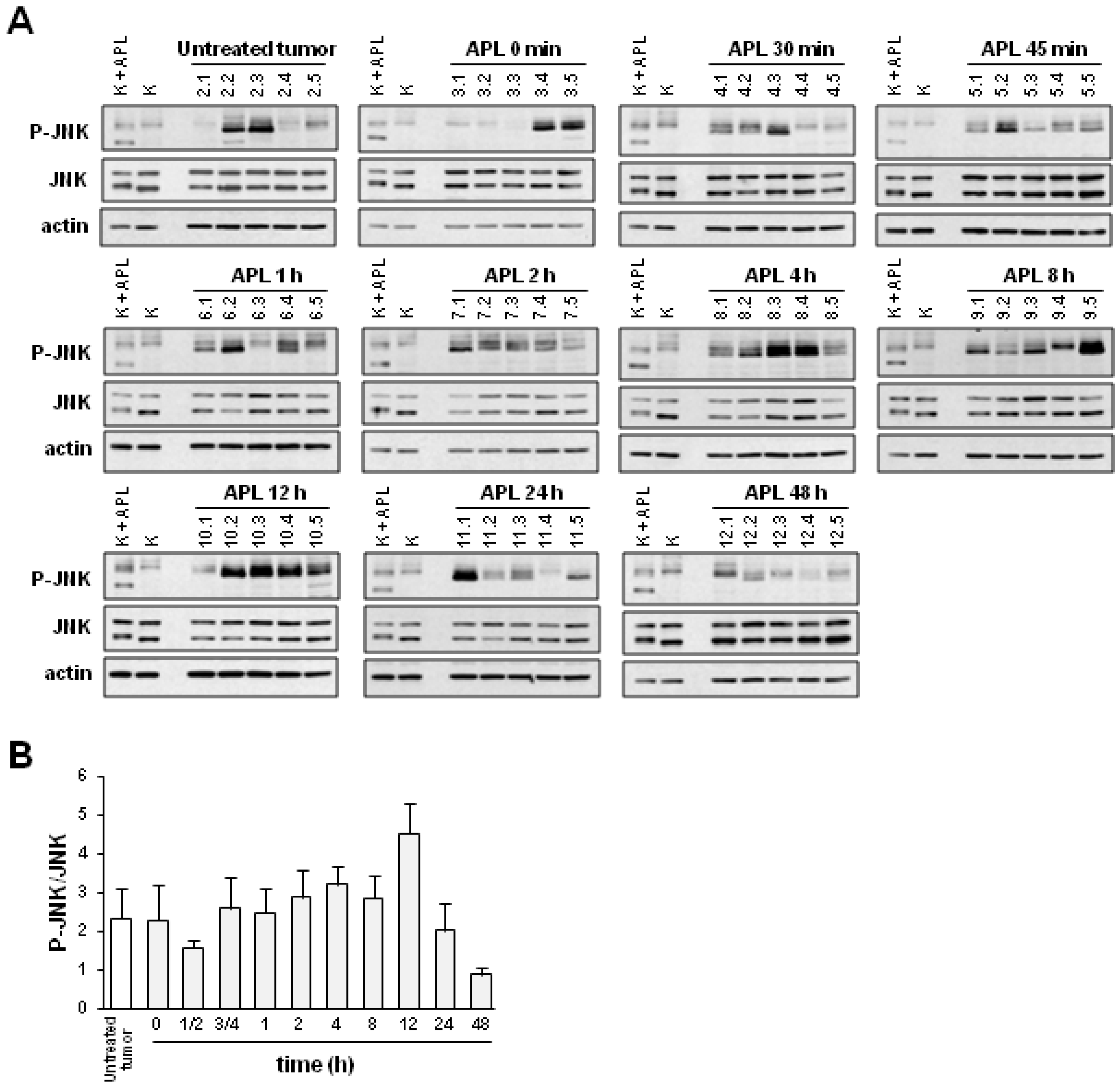
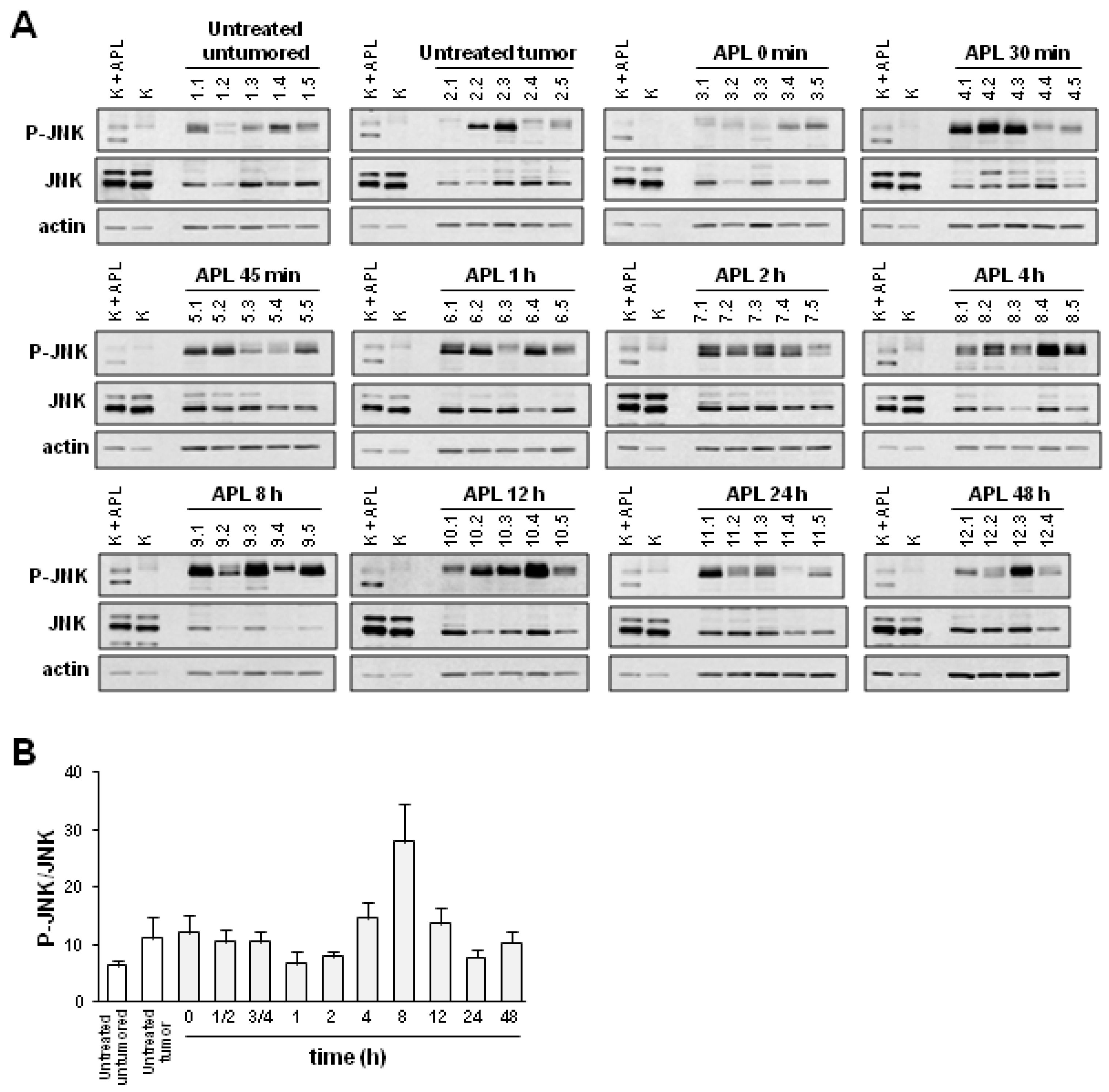
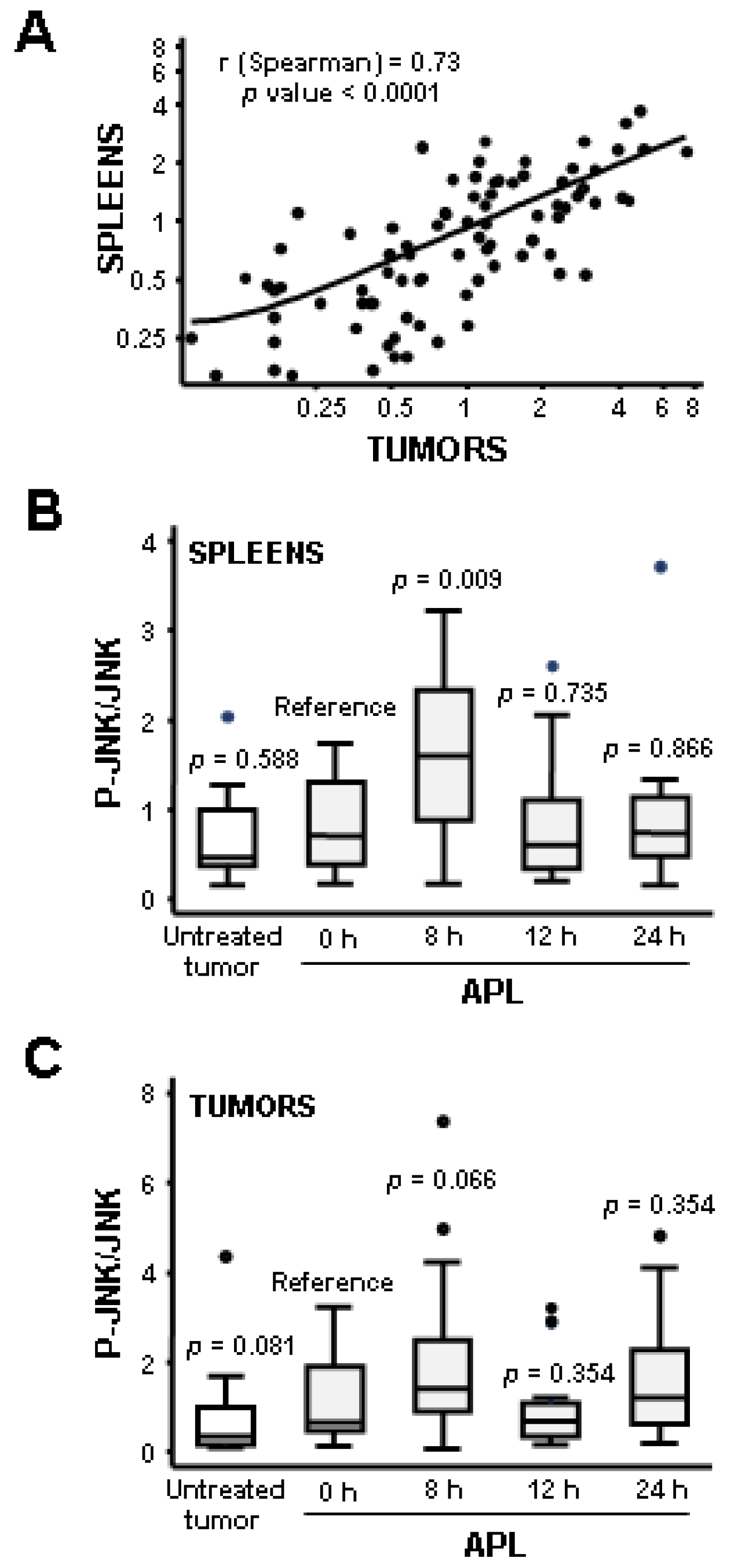
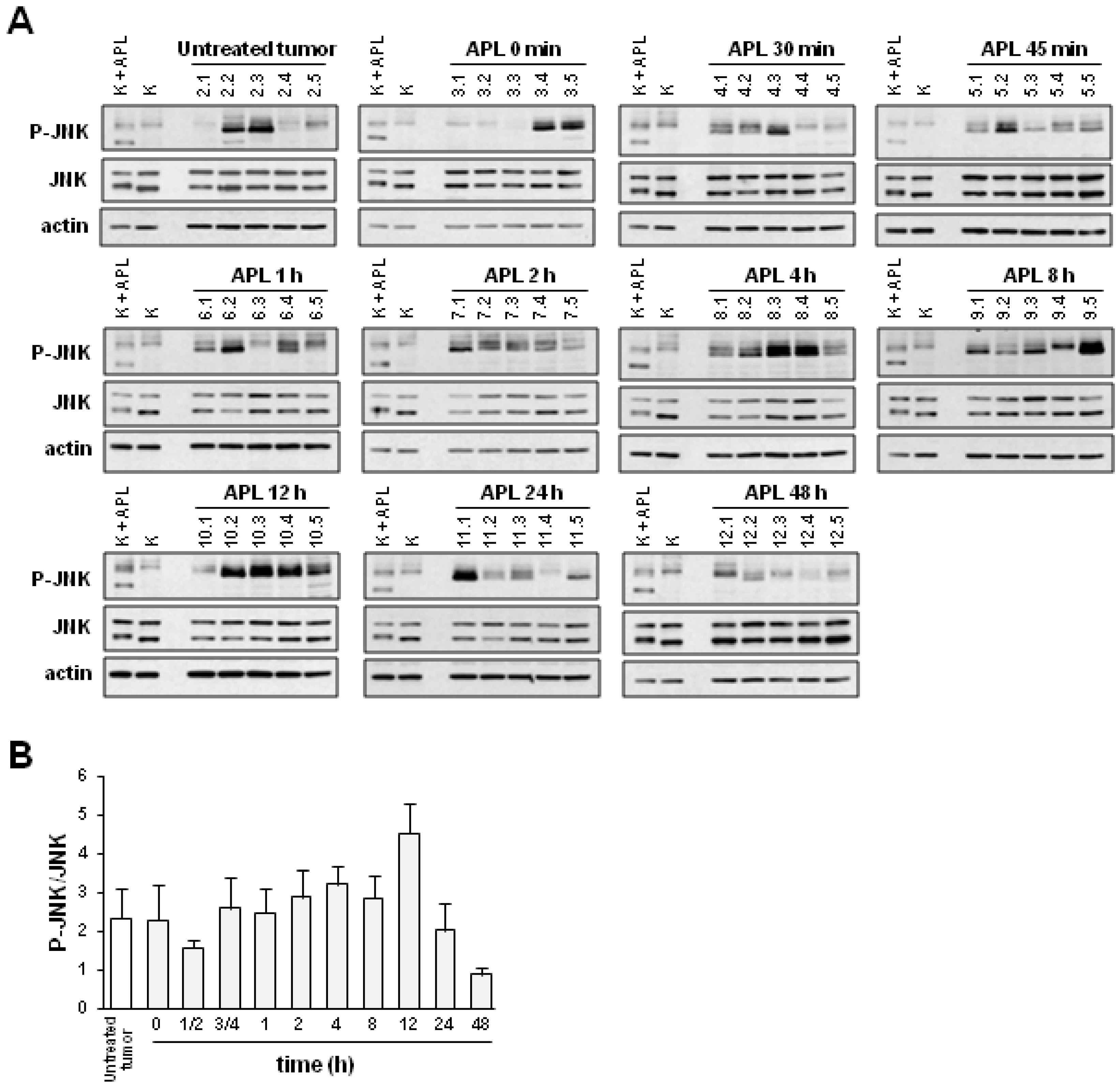
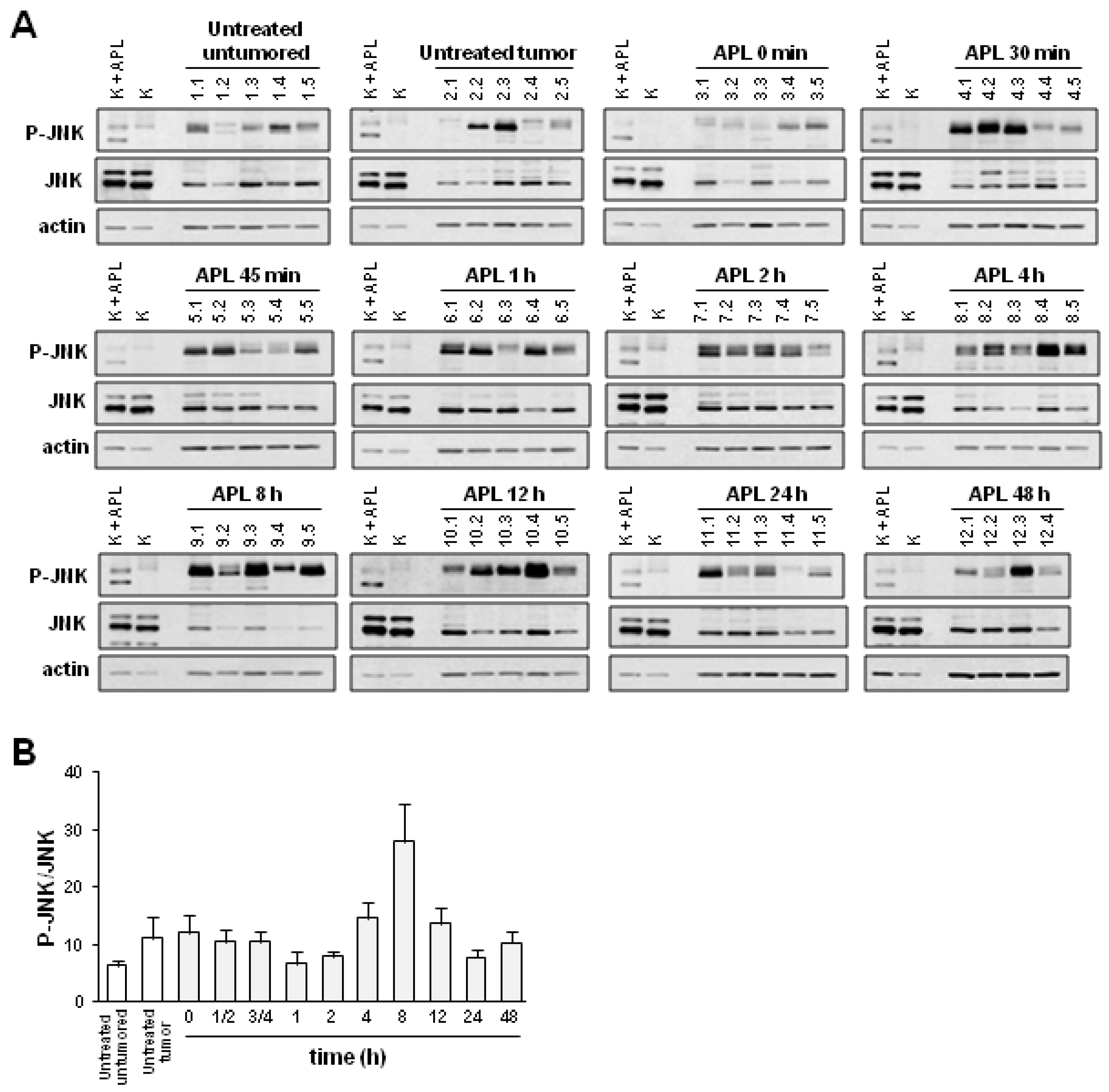
2.2. Plitidepsin Induces JNK Phosphorylation in Both Tumors and Spleens from K562 Xenografts

2.3. Plitidepsin Induces JNK Phosphorylation in Peripheral Mononuclear Blood Cells
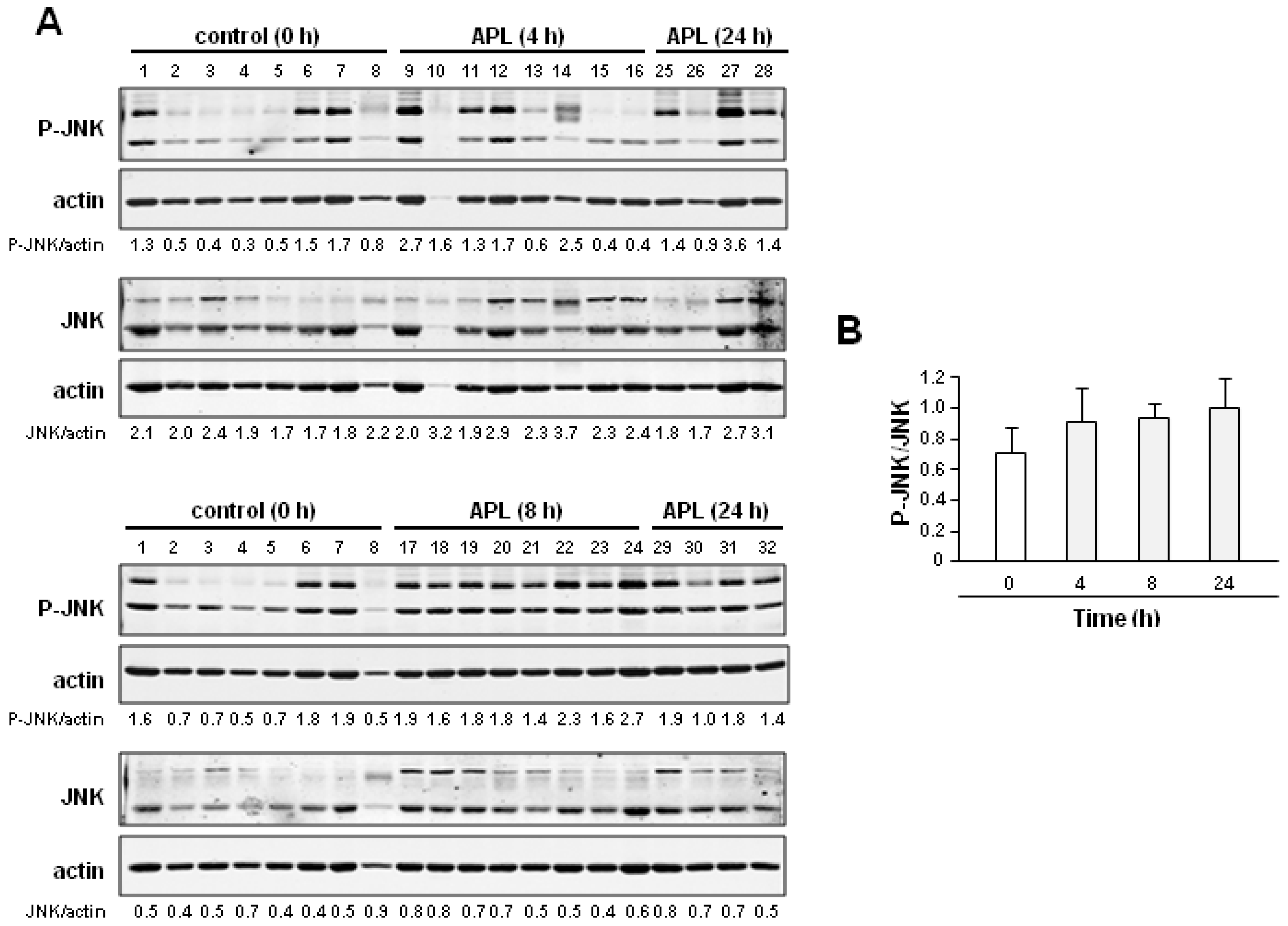
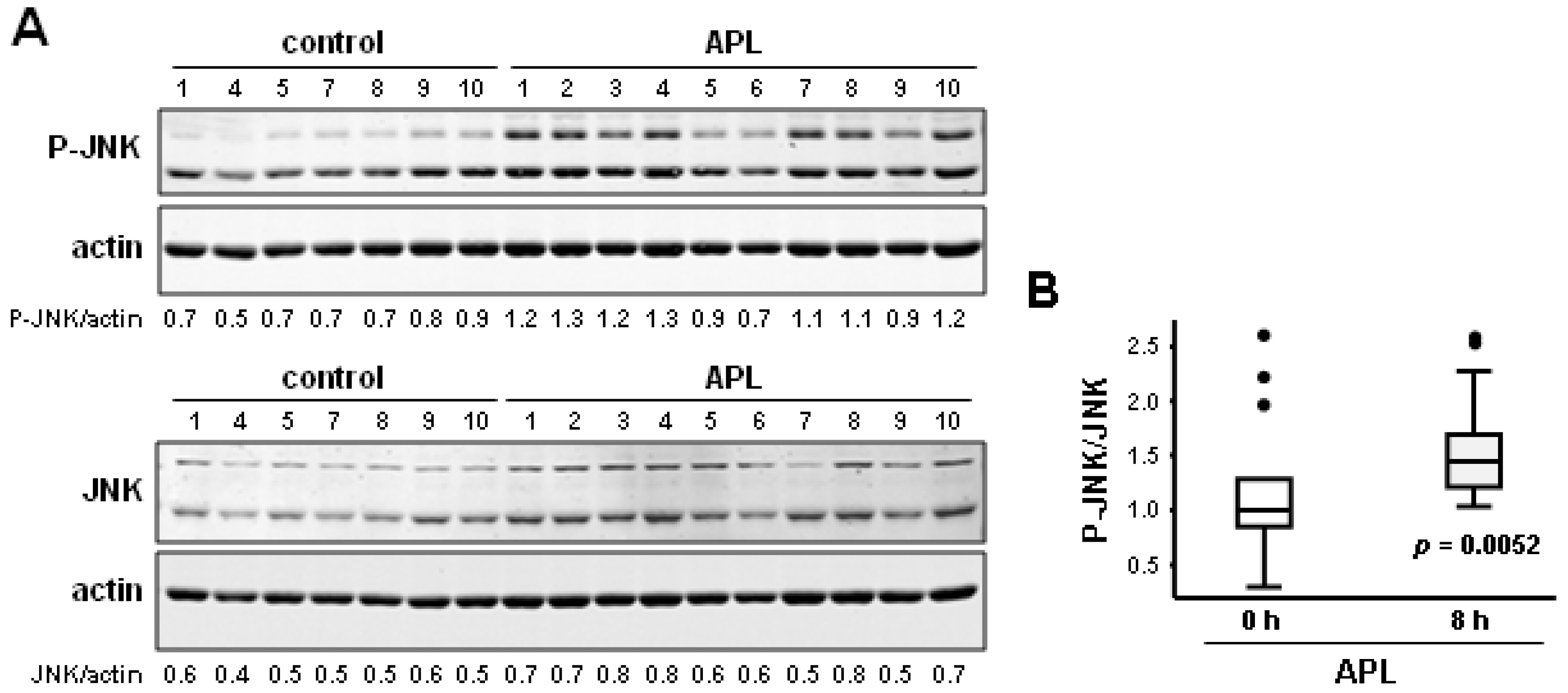
2.4. Discussion
3. Experimental Section
3.1. Cell Culture and Drug Solutions
3.2. Cell Proliferation Analysis
3.3. Xenograft Tissues Studies
3.4. Protein Lysates from Cells and in Vivo Tissues
3.5. Western Blot Analysis
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Munoz-Alonso, M.J.; Gonzalez-Santiago, L.; Martinez, T.; Losada, A.; Galmarini, C.M.; Munoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542. [Google Scholar]
- Bravo, S.B.; Garcia-Rendueles, M.E.; Seoane, R.; Dosil, V.; Cameselle-Teijeiro, J.; Lopez-Lazaro, L.; Zalvide, J.; Barreiro, F.; Pombo, C.M.; Alvarez, C.V. Plitidepsin has a cytostatic effect in human undifferentiated (anaplastic) thyroid carcinoma. Clin. Cancer Res. 2005, 11, 7664–7673. [Google Scholar] [CrossRef]
- Munoz-Alonso, M.J.; Gonzalez-Santiago, L.; Zarich, N.; Martinez, T.; Alvarez, E.; Rojas, J.M.; Munoz, A. Plitidepsin has a dual effect inhibiting cell cycle and inducing apoptosis via rac1/c-jun nh2-terminal kinase activation in human melanoma cells. J. Pharmacol. Exp. Ther. 2008, 324, 1093–1101. [Google Scholar]
- Garcia-Fernandez, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernandez-Sousa, J.M.; Munoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated jnk and p38 activation and protein kinase c delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef]
- Cuadrado, A.; Garcia-Fernandez, L.F.; Gonzalez, L.; Suarez, Y.; Losada, A.; Alcaide, V.; Martinez, T.; Fernandez-Sousa, J.M.; Sanchez-Puelles, J.M.; Munoz, A. Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, src, jnk, and p38 mapk. J. Biol. Chem. 2003, 278, 241–250. [Google Scholar]
- Cuadrado, A.; Gonzalez, L.; Suarez, Y.; Martinez, T.; Munoz, A. Jnk activation is critical for aplidin-induced apoptosis. Oncogene 2004, 23, 4673–4680. [Google Scholar] [CrossRef]
- Marchini, S.; Chiorino, G.; Faircloth, G.T.; D’Incalci, M. Changes in gene expression profile induced by the anticancer agent aplidine in molt-4 leukemic cell lines. J. Biol. Regul. Homeost. Agents 2002, 16, 241–248. [Google Scholar]
- Gonzalez-Santiago, L.; Alfonso, P.; Suarez, Y.; Nunez, A.; Garcia-Fernandez, L.F.; Alvarez, E.; Munoz, A.; Casal, J.I. Proteomic analysis of the resistance to aplidin in human cancer cells. J. Proteome Res. 2007, 6, 1286–1294. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Ocio, E.M.; Pandiella, A.; Maiso, P.; Gajate, C.; Garayoa, M.; Vilanova, D.; Montero, J.C.; Mitsiades, N.; McMullan, C.J.; et al. Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res. 2008, 68, 5216–5225. [Google Scholar] [CrossRef]
- Moneo, V.; Serelde, B.G.; Leal, J.F.; Blanco-Aparicio, C.; Diaz-Uriarte, R.; Aracil, M.; Tercero, J.C.; Jimeno, J.; Carnero, A. Levels of p27(kip1) determine aplidin sensitivity. Mol. Cancer Ther. 2007, 6, 1310–1316. [Google Scholar] [CrossRef]
- Gajate, C.; An, F.; Mollinedo, F. Rapid and selective apoptosis in human leukemic cells induced by aplidine through a fas/cd95- and mitochondrial-mediated mechanism. Clin. Cancer Res. 2003, 9, 1535–1545. [Google Scholar]
- Caers, J.; Menu, E.; de Raeve, H.; Lepage, D.; van Valckenborgh, E.; van Camp, B.; Alvarez, E.; Vanderkerken, K. Antitumour and antiangiogenic effects of aplidin in the 5tmm syngeneic models of multiple myeloma. Br. J. Cancer 2008, 98, 1966–1974. [Google Scholar] [CrossRef]
- Morande, P.E.; Zanetti, S.R.; Borge, M.; Nannini, P.; Jancic, C.; Bezares, R.F.; Bitsmans, A.; Gonzalez, M.; Rodriguez, A.L.; Galmarini, C.M.; et al. The cytotoxic activity of aplidin in chronic lymphocytic leukemia (cll) is mediated by a direct effect on leukemic cells and an indirect effect on monocyte-derived cells. Invest. New Drugs 2012, 30, 1830–1840. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Cytoskeleton-mediated death receptor and ligand concentration in lipid rafts forms apoptosis-promoting clusters in cancer chemotherapy. J. Biol. Chem. 2005, 280, 11641–11647. [Google Scholar] [CrossRef]
- Gonzalez-Santiago, L.; Suarez, Y.; Zarich, N.; Munoz-Alonso, M.J.; Cuadrado, A.; Martinez, T.; Goya, L.; Iradi, A.; Saez-Tormo, G.; Maier, J.V.; et al. Aplidin induces jnk-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, rac1 gtpase activation, and mkp-1 phosphatase downregulation. Cell Death Differ. 2006, 13, 1968–1981. [Google Scholar] [CrossRef]
- Albella, B.; Faircloth, G.; Lopez-Lazaro, L.; Guzman, C.; Jimeno, J.; Bueren, J.A. In vitro toxicity of et-743 and aplidine, two marine-derived antineoplastics, on human bone marrow haematopoietic progenitors. Comparison with the clinical results. Eur. J. Cancer 2002, 38, 1395–1404. [Google Scholar]
- Faivre, S.; Chieze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.P.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef]
- Weston, C.R.; Davis, R.J. The jnk signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by jnk and p38 mapk pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by map kinase: Physiological roles of ask1-map kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef]
- Karin, M.; Gallagher, E. From jnk to pay dirt: Jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 2005, 57, 283–295. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the jnk group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Chen, Z.; Seimiya, H.; Naito, M.; Mashima, T.; Kizaki, A.; Dan, S.; Imaizumi, M.; Ichijo, H.; Miyazono, K.; Tsuruo, T. Ask1 mediates apoptotic cell death induced by genotoxic stress. Oncogene 1999, 18, 173–180. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. Jnk signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Sanchez-Perez, I.; Perona, R. Lack of c-jun activity increases survival to cisplatin. FEBS Lett. 1999, 453, 151–158. [Google Scholar] [CrossRef]
- Eichhorst, S.T.; Muller, M.; Li-Weber, M.; Schulze-Bergkamen, H.; Angel, P.; Krammer, P.H. A novel ap-1 element in the cd95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol. Cell Biol. 2000, 20, 7826–7837. [Google Scholar] [CrossRef]
- Seimiya, H.; Mashima, T.; Toho, M.; Tsuruo, T. c-Jun nh2-terminal kinase-mediated activation of interleukin-1beta converting enzyme/ced-3-like protease during anticancer drug-induced apoptosis. J. Biol. Chem. 1997, 272, 4631–4636. [Google Scholar] [CrossRef]
- Fan, M.; Goodwin, M.E.; Birrer, M.J.; Chambers, T.C. The c-jun nh(2)-terminal protein kinase/ap-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 2001, 61, 4450–4458. [Google Scholar]
- Srivastava, R.K.; Mi, Q.S.; Hardwick, J.M.; Longo, D.L. Deletion of the loop region of bcl-2 completely blocks paclitaxel-induced apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3775–3780. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the bcr-abl tyrosine kinase in chronic myeloid leukemia. New Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Baselga, J.; Rischin, D.; Ranson, M.; Calvert, H.; Raymond, E.; Kieback, D.G.; Kaye, S.B.; Gianni, L.; Harris, A.; Bjork, T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of zd1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J. Clin. Oncol. 2002, 20, 4292–4302. [Google Scholar] [CrossRef]
- Baselga, J.; Tripathy, D.; Mendelsohn, J.; Baughman, S.; Benz, C.C.; Dantis, L.; Sklarin, N.T.; Seidman, A.D.; Hudis, C.A.; Moore, J.; et al. Phase ii study of weekly intravenous recombinant humanized anti-p185her2 monoclonal antibody in patients with her2/neu-overexpressing metastatic breast cancer. J. Clin. Oncol. 1996, 14, 737–744. [Google Scholar]
- Lipton, A.; Ali, S.M.; Leitzel, K.; Demers, L.; Chinchilli, V.; Engle, L.; Harvey, H.A.; Brady, C.; Nalin, C.M.; Dugan, M.; et al. Elevated serum her-2/neu level predicts decreased response to hormone therapy in metastatic breast cancer. J. Clin. Oncol. 2002, 20, 1467–1472. [Google Scholar] [CrossRef]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of su11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Luo, F.R.; Barrett, Y.C.; Yang, Z.; Camuso, A.; McGlinchey, K.; Wen, M.L.; Smykla, R.; Fager, K.; Wild, R.; Palme, H.; et al. Identification and validation of phospho-src, a novel and potential pharmacodynamic biomarker for dasatinib (sprycel), a multi-targeted kinase inhibitor. Cancer Chemother. Pharmacol. 2008, 62, 1065–1074. [Google Scholar] [CrossRef]
- Luo, F.R.; Yang, Z.; Camuso, A.; Smykla, R.; McGlinchey, K.; Fager, K.; Flefleh, C.; Castaneda, S.; Inigo, I.; Kan, D.; et al. Dasatinib (bms-354825) pharmacokinetics and pharmacodynamic biomarkers in animal models predict optimal clinical exposure. Clin. Cancer Res. 2006, 12, 7180–7186. [Google Scholar] [CrossRef]
- Boulay, A.; Zumstein-Mecker, S.; Stephan, C.; Beuvink, I.; Zilbermann, F.; Haller, R.; Tobler, S.; Heusser, C.; O’Reilly, T.; Stolz, B.; et al. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative rad001 correlates with prolonged inactivation of ribosomal protein s6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 2004, 64, 252–261. [Google Scholar] [CrossRef]
- Mateos, M.V.; Cibeira, M.T.; Richardson, P.G.; Prosper, F.; Oriol, A.; de la Rubia, J.; Lahuerta, J.J.; Garcia-Sanz, R.; Extremera, S.; Szyldergemajn, S.; et al. Phase ii clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. 2010, 16, 3260–3269. [Google Scholar] [CrossRef]
- Yin, J.; Aviles, P.; Lee, W.; Ly, C.; Floriano, P.; Ignacio, M.; Faircloth, G. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of aplidin, a novel marine-derived antineoplastic agent, in human plasma. Rapid Commun. Mass Spectrom. 2003, 17, 1909–1914. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Muñoz-Alonso, M.J.; Álvarez, E.; Guillén-Navarro, M.J.; Pollán, M.; Avilés, P.; Galmarini, C.M.; Muñoz, A. c-Jun N-Terminal Kinase Phosphorylation Is a Biomarker of Plitidepsin Activity. Mar. Drugs 2013, 11, 1677-1692. https://doi.org/10.3390/md11051677
Muñoz-Alonso MJ, Álvarez E, Guillén-Navarro MJ, Pollán M, Avilés P, Galmarini CM, Muñoz A. c-Jun N-Terminal Kinase Phosphorylation Is a Biomarker of Plitidepsin Activity. Marine Drugs. 2013; 11(5):1677-1692. https://doi.org/10.3390/md11051677
Chicago/Turabian StyleMuñoz-Alonso, María J., Enrique Álvarez, María José Guillén-Navarro, Marina Pollán, Pablo Avilés, Carlos M. Galmarini, and Alberto Muñoz. 2013. "c-Jun N-Terminal Kinase Phosphorylation Is a Biomarker of Plitidepsin Activity" Marine Drugs 11, no. 5: 1677-1692. https://doi.org/10.3390/md11051677
APA StyleMuñoz-Alonso, M. J., Álvarez, E., Guillén-Navarro, M. J., Pollán, M., Avilés, P., Galmarini, C. M., & Muñoz, A. (2013). c-Jun N-Terminal Kinase Phosphorylation Is a Biomarker of Plitidepsin Activity. Marine Drugs, 11(5), 1677-1692. https://doi.org/10.3390/md11051677