Bioactive Compounds from the Red Sea Marine Sponge Hyrtios Species



Abstract
:1. Introduction

2. Results and Discussion
2.1. Purification of Compounds 1–3
2.2. Structure Elucidation of Compounds 1–3

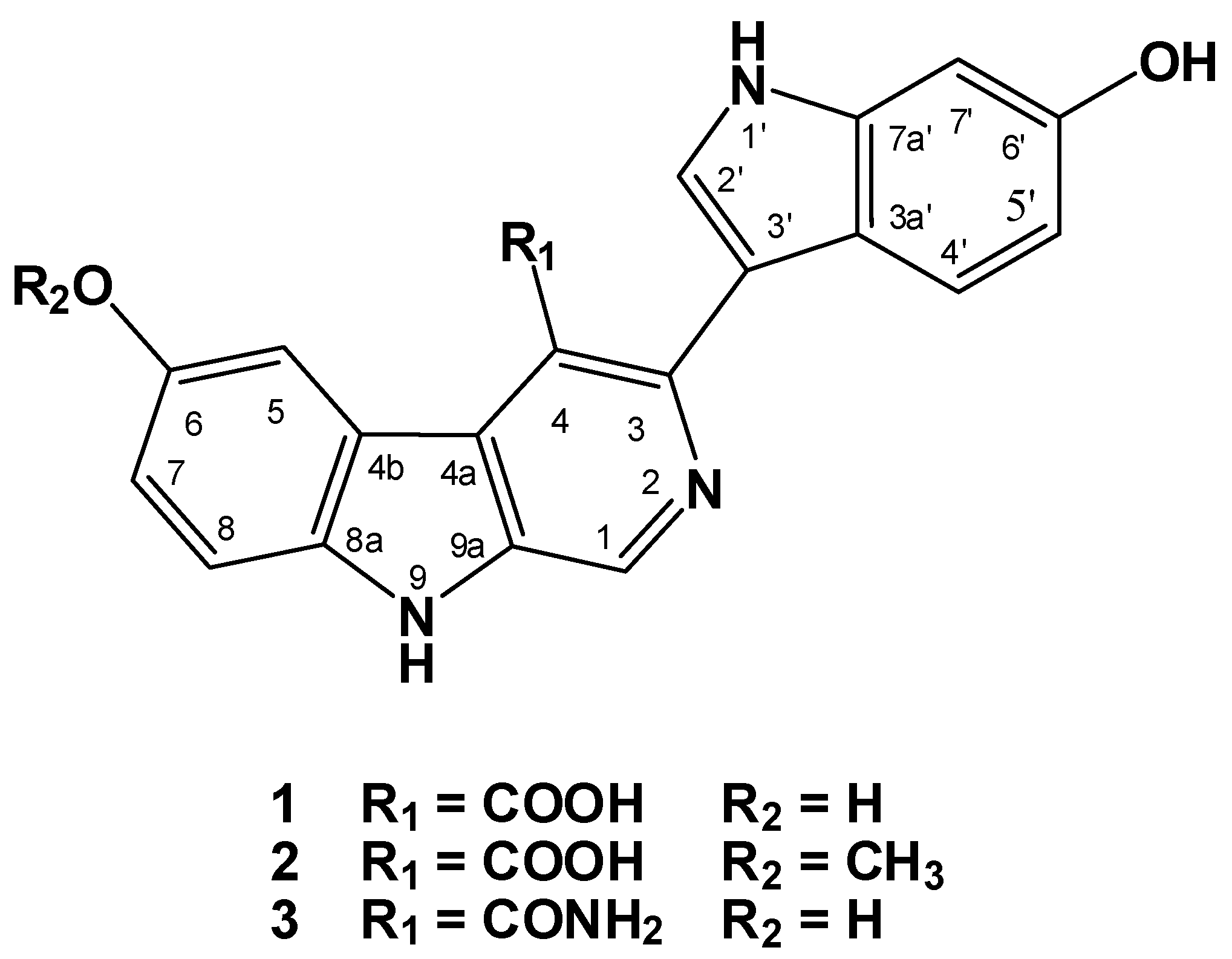{kind=link}
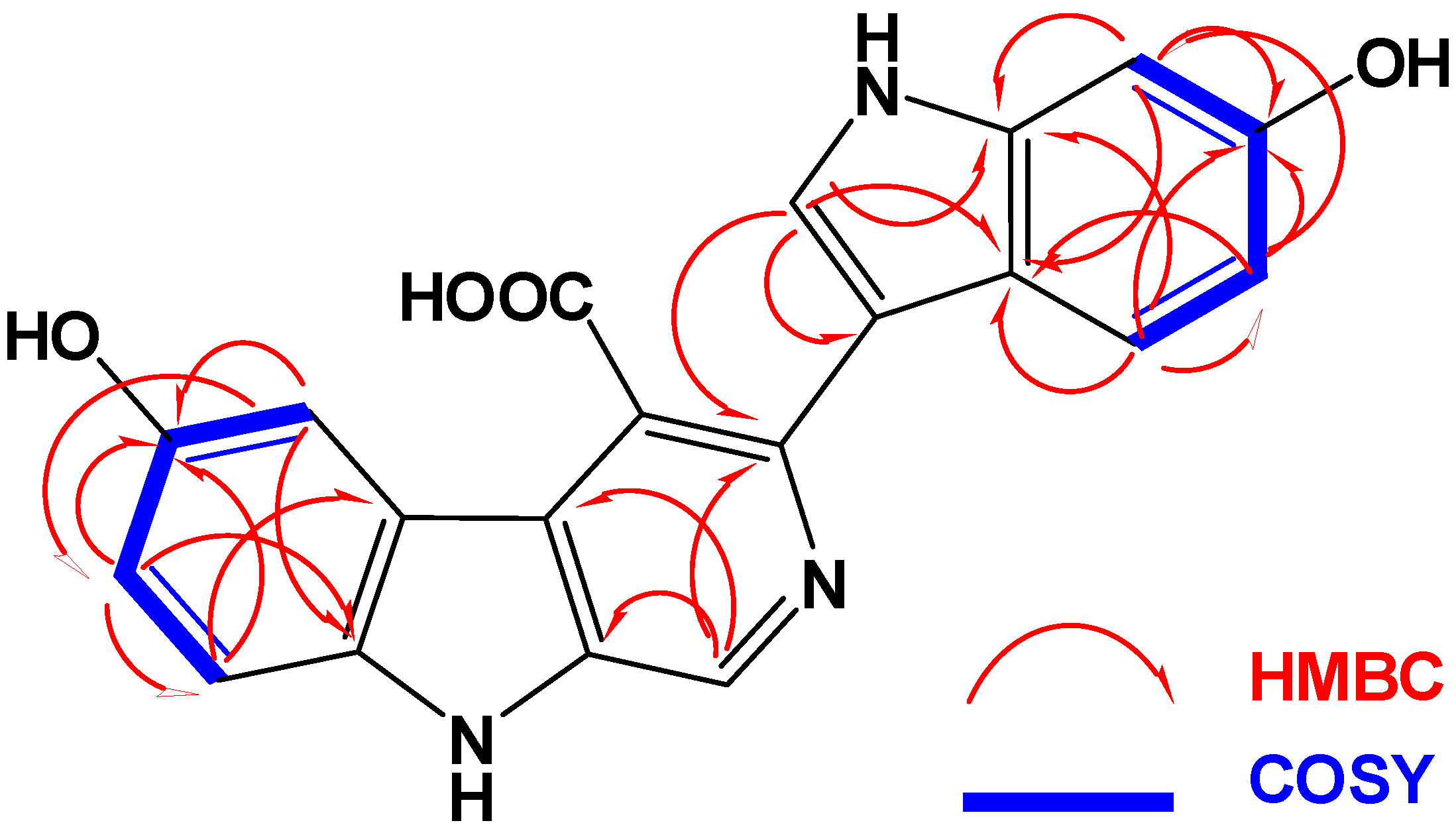{kind=link}
| Hyrtioerectine D | Hyrtioerectine E | Hyrtioerectine F | ||||
|---|---|---|---|---|---|---|
| Position | δC (mult.) a | δH, mult. (J in Hz) | δC (mult.) a | δH, mult. (J in Hz) | δC (mult.) a | δH, mult. (J in Hz) |
| 1 | 140.8 (CH) | 9.64, s | 141.2 (CH) | 9.66, s | 139.6 (CH) | 9.71, s |
| 3 | 138.1 (qC) | 138.2 (qC) | 138.3 (qC) | |||
| 4 | 138.8 (qC) | 138.8 (qC) | 138.8 (qC) | |||
| 4a | 115.9 (qC) | 115.8 (qC) | 115.9 (qC) | |||
| 4b | 130.1 (qC) | 130.3 (qC) | 130.4 (qC) | |||
| 5 | 108.2 (CH) | 8.07, d (2.0) | 110.1 (CH) | 8.10, d (2.2) | 107.2 (CH) | 8.09, d (2.2) |
| 6 | 154.3 (qC) | 156.2 (qC) | 154.2 (qC) | |||
| 7 | 113.5 (CH) | 6.80, dd (8.5, 2.0) | 113.5 (CH) | 6.78, dd (8.5, 2.2) | 113.7 (CH) | 6.76, dd (8.5, 2.2) |
| 8 | 113.1 (CH) | 7.30, d (8.5) | 113.1 (CH) | 7.27, d (8.5) | 113.5 (CH) | 7.32, d (8.5) |
| 8a | 132.1 (qC) | 132.2 (qC) | 132.4 (qC) | |||
| 9a | 133.2 (qC) | 133.1 (qC) | 133.2 (qC) | |||
| 2′ | 119.6 (CH) | 8.87, s | 119.5 (CH) | 8.88, s | 119.6 (CH) | 8.87, s |
| 3′ | 123.2 (qC) | 123.2 (qC) | 123.2 (qC) | |||
| 3a′ | 137.5 (qC) | 137.5 (qC) | 137.5 (qC) | |||
| 4′ | 114.1 (CH) | 7.57, d (8.7) | 114.1 (CH) | 7.61, d (8.7) | 114.1 (CH) | 7.64, d (8.0) |
| 5′ | 119.7 (CH) | 7.13, dd (8.7, 2.3) | 119.6 (CH) | 7.15, dd (8.7, 2.2) | 119.7 (CH) | 7.17, dd (8.7, 2.0) |
| 6′ | 153.0 (qC) | 153.1 (qC) | 153.0 (qC) | |||
| 7′ | 106.7 (CH) | 7.60, d (2.3) | 106.9(CH) | 7.62, d (2.2) | 106.7 (CH) | 7.65, d (2.0) |
| 7a′ | 132.6 (qC) | 132.5 (qC) | 132.6 (qC) | |||
| 8′ | 173.0 (qC) | 173.0 (qC) | 164.2 (qC) | |||
| 9′ | 55.8 (CH3) | 3.84, s | ||||
| C# | Hyrtiooerectine D (1) | Hyrtioerectine E (2) | Hyrtioerectine F (3) |
|---|---|---|---|
| HMBC (H→C#) | HMBC (H→C#) | HMBC (H→C#) | |
| 3 | H-1, H-2′ | H-1, H-2′ | H-1, H-2′ |
| 4 | - | - | - |
| 4a | H-1, H-5 | H-1 | H-1, H-5 |
| 4b | H-8 | H-8 | H-8 |
| 5 | H-7 | H-7 | H-7 |
| 6 | H-5, H-7, H-8 | H-5, H-7, H-8, H3-9′ | H-5, H-8 |
| 7 | H-5 | H-5 | H-5 |
| 8 | H-7 | H-7 | H-7 |
| 8a | H-5, H-7 | H-5, H-7 | H-5, H-7 |
| 9a | H-1 | H-1 | H-1 |
| 2′ | - | - | - |
| 3′ | H-2′ | H-2′ | H-2′ |
| 3a′ | H-2′, H-4′, H-5′, H-7′ | H-2′, H-4′, H-7′ | H-2′, H-4′, H-7′ |
| 4′ | H-5′ | H-5′ | H-5′ |
| 5′ | H-4′, H-7′ | H-4′, H-7′ | H-4′, H-7′ |
| 6′ | H-4′, H-5′, H-7′ | H-4′, H-5′, H-7′ | H-4′, H-5′, H-7′ |
| 7′ | H-5′ | H-5′ | H-5′ |
| 7a′ | H-2′, H-4′, H-7′ | H-2′, H-4′, H-7′ | H-2′, H-4′, H-7′ |
| Compound | Cell line [GI50 (μM)] | ||
|---|---|---|---|
| MDA-MB-231 | A549 | HT-29 | |
| Compound 1 | 25 | 30 | 28 |
| Compound 2 | 90 | 100 | 85 |
| Compound 3 | 42 | 35 | 45 |
| Doxorubicin a | 0.30 | 0.35 | 0.40 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Specimen
3.3. Extraction and Purifications of Compounds 1–3
3.4. Biological Evaluation of the Compounds
3.4.1. Determination of the Antimicrobial Activity
3.4.2. Determination of the Free Radical Scavenging Activity Using DPPH
3.4.3. Cancer Cell Growth Inhibition Assay
4. Conclusions
Acknowledgments
References
- Hooper, J.N.A.; van Soest, R.W.M. Kluwer Academic/Plenum Publishers; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2002; Volume 1, pp. 1028–1050. [Google Scholar]
- Pettit, G.R.; Tan, R.; Cichacz, Z.A. Antineoplastic agents. 542. Isolation and structure of sesterstatin 6 from the Indian Ocean sponge Hyrtios erecta. J. Nat. Prod. 2005, 68, 1253–1255. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Shaala, L.A.; Emara, S. Antimycobacterial scalarane-based sesterterpenes from the Red Sea sponge Hyrtios erecta. J. Nat. Prod. 2005, 68, 1782–1784. [Google Scholar] [CrossRef]
- Qiu, Y.; Deng, Z.; Pei, Y.; Fu, H.; Li, J.; Proksh, P.; Lin, W. Sesterterpenoids from the marine sponge Hyrtios erectus. J. Nat. Prod. 2004, 67, 921–924. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Yamaki, R.K.; Kelly, M.; Scheuer, P.J. Salmahyrtisol A, a novel cytotoxic sesterterpene from a Red Sea sponge Hyrtios erecta. J. Nat. Prod. 2002, 65, 2–6. [Google Scholar] [CrossRef]
- Miyaoka, H.; Nishijima, S.; Mitome, H.; Yamada, Y. Three new scalarane sesterterpenoids from the Okinawan sponge Hyrtios erectus. J. Nat. Prod. 2000, 63, 1369–1372. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Singab, A.B.; van Soest, R.W.M.; Fusetani, N. Hyrtiosenolides A and B, two new sesquiterpene γ-methoxybutenolides and a new sterol from a Red Sea sponge Hyrtios species. J. Nat. Prod. 2004, 67, 1736–1739. [Google Scholar] [CrossRef]
- Pina, I.C.; Sanders, M.L.; Crews, P. Puupehenone congeners from an Indo-Pacific Hyrtios sponge. J. Nat. Prod. 2003, 66, 2–6. [Google Scholar] [CrossRef]
- Salmoun, M.; Devijver, C.; Daloze, D.; Braekman, J.-C.; Gomez, R.; de Kluijver, M.; van Soest, R.W.M. New sesquiterpenes/quinones from two sponges of the genus Hyrtios. J. Nat. Prod. 2000, 63, 452–456. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Sakai, H.; Kawazoe, K.; Kihara, N.; Sasaki, T.; Kitagawa, I. Altohyrtin A, a potent anti-tumor macrolide from the Okinawan marine sponge Hyrtios altum. Tetrahedron Lett. 1993, 34, 2795–2798. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Sakai, H.; Kawazoe, K.; Kihara, N.; Sasaki, T.; Kitagawa, I. Altohyrtins B and C and 5-desacetylalthyrtin A, potent cytotoxic macrolide congeners of altohyrtin A, from the Okinawan marine sponge Hyrtios altum. Chem. Pharm. Bull. 1993, 41, 989–991. [Google Scholar] [CrossRef]
- Youssef, D.T.A. Hyrtioerectines A–C, cytotoxic alkaloids from the Red Sea sponge Hyrtios erectus. J. Nat. Prod. 2005, 68, 1416–1419. [Google Scholar] [CrossRef]
- Sauleau, P.; Martin, M.; Dau, M.T.H.; Youssef, D.T.A.; Bourguet-Kondracki, M. Hyrtiazepine, an azepino-indole-type alkaloid from the Red Sea marine sponge Hyrtios erectus. J. Nat. Prod. 2006, 69, 1676–1679. [Google Scholar] [CrossRef]
- Salmoun, M.; Devijver, C.; Daloze, D.; Braekman, J.-C.; van Soest, R.W.M. 5-Hydroxytryptamine-derived alkaloids from two marine sponges of the genus Hyrtios. J. Nat. Prod. 2002, 65, 1173–1176. [Google Scholar] [CrossRef]
- Aoki, S.; Ye, Y.; Higuchi, K.; Takashima, A.; Tanaka, Y.; Kitagawa, I.; Kobayashi, M. Novel neuronal nitric oxide synthase (nNOS) selective inhibitor, aplysinopsin-type indole alkaloid, from marine sponge Hyrtios erecta. Chem. Pharm. Bull. 2001, 49, 1372–1374. [Google Scholar] [CrossRef]
- Kobayashi, J.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Sasaki, T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Ito, Y.; Suzuki, M.; Hirota, A. Indole derivatives from a marine sponge-derived yeast as DPPH radical scavengers. J. Nat. Prod. 2009, 72, 2069–2071. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooper, J.N.A. Antineoplastic agents. 257. Isolation and structure of spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] [CrossRef]
- Badre, A.; Boulanger, A.; Abou-Mansour, E.; Banaigs, B.; Combaut, G.; Francisco, C. Eudistomin U and isoeudistomin U, new alkaloids from the Carribean ascidian Lissoclinum fragile. J. Nat. Prod. 1994, 57, 528–533. [Google Scholar] [CrossRef]
- Williams, B.W.; Cuverlier, M.E.; Berset, C. Use of free radical method to evaluate antioxidant activity. Food. Sci. Technol. 1995, 28, 25–30. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Youssef, D.T.A.; Shaala, L.A.; Asfour, H.Z. Bioactive Compounds from the Red Sea Marine Sponge Hyrtios Species. Mar. Drugs 2013, 11, 1061-1070. https://doi.org/10.3390/md11041061
Youssef DTA, Shaala LA, Asfour HZ. Bioactive Compounds from the Red Sea Marine Sponge Hyrtios Species. Marine Drugs. 2013; 11(4):1061-1070. https://doi.org/10.3390/md11041061
Chicago/Turabian StyleYoussef, Diaa T. A., Lamiaa A. Shaala, and Hani Z. Asfour. 2013. "Bioactive Compounds from the Red Sea Marine Sponge Hyrtios Species" Marine Drugs 11, no. 4: 1061-1070. https://doi.org/10.3390/md11041061
APA StyleYoussef, D. T. A., Shaala, L. A., & Asfour, H. Z. (2013). Bioactive Compounds from the Red Sea Marine Sponge Hyrtios Species. Marine Drugs, 11(4), 1061-1070. https://doi.org/10.3390/md11041061