Predictive Factors of Sensitivity to Elisidepsin, a Novel Kahalalide F-Derived Marine Compound

Abstract
:1. Introduction
2. Results
2.1. Antiproliferative Effects of Elisidepsin

{kind=link}
{kind=link}
{kind=link}
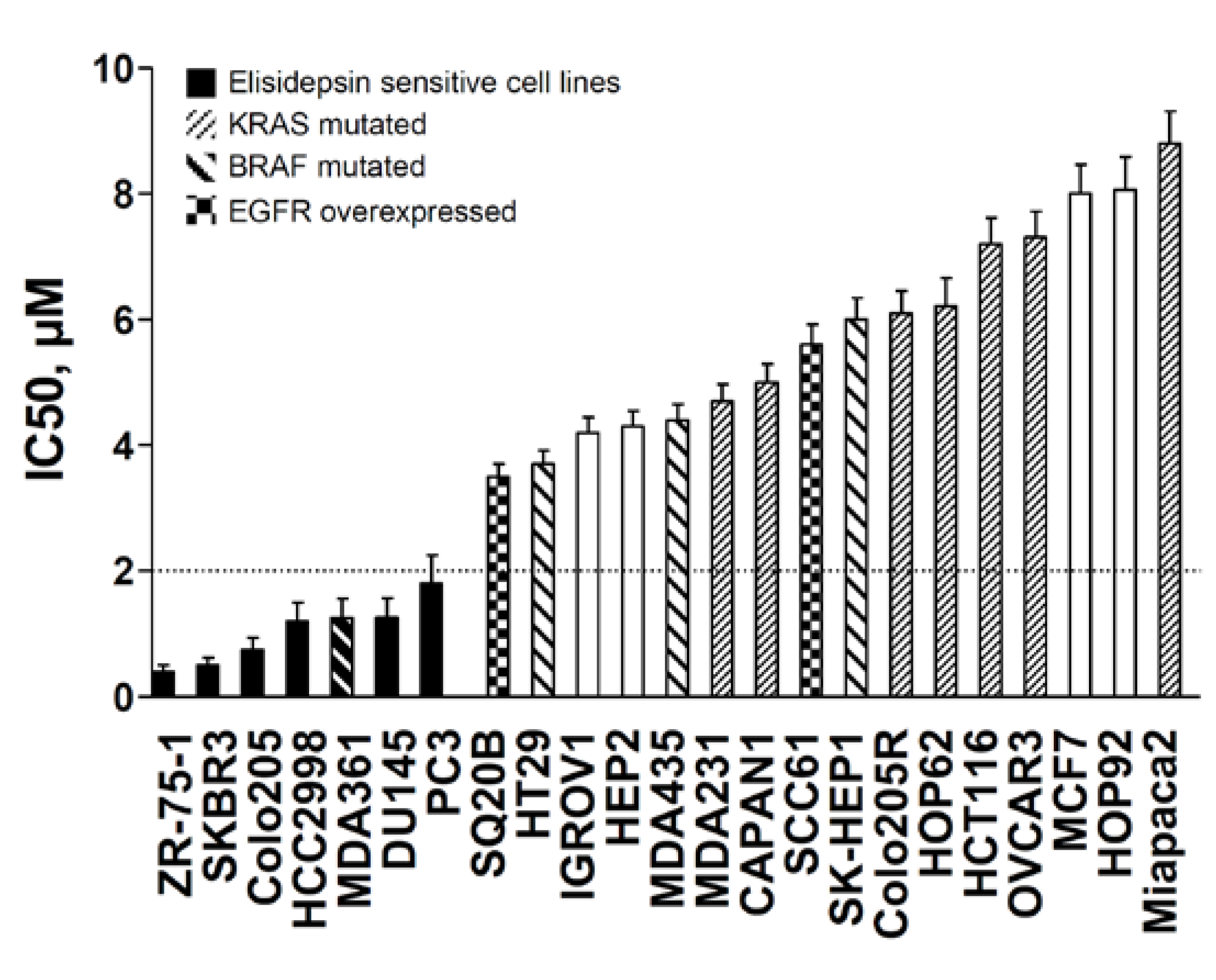
{kind=link}
| Cell line | Tumor type | IC50 (μM) |
|---|---|---|
| ZR-75-1 | Breast | 0.40 ± 0.1 |
| SKBR3 | Breast | 0.50 ± 0.1 |
| MDA-MB-361 | Breast | 1.25 ± 0.3 |
| MDA-MB-231 | Breast | 4.70 ± 1.2 |
| MCF7 | Breast | 8.00 ± 2.7 |
| Colo205 | Colon | 0.75 ± 0.2 |
| HCC2998 | Colon | 1.20 ± 0.4 |
| HT29 | Colon | 3.70 ± 0.8 |
| Colo205R | Colon | 6.10 ± 2.1 |
| HCT116 | Colon | 7.20 ± 2.2 |
| SQ20B | Head and Neck | 3.50 ± 1.1 |
| HEP2 | Head and Neck | 4.30 ± 1.2 |
| SCC61 | Head and Neck | 5.60 ± 1.8 |
| SK-HEP1 | Hepatocarcinoma | 6.00 ± 1.9 |
| HOP62 | Lung | 6.30 ± 1.9 |
| HOP92 | Lung | 8.00 ± 2.9 |
| MDA-MB-435 | Melanoma | 4.40 ± 0.9 |
| IGROV1 | Ovarian | 4.20 ± 0.8 |
| OVCAR3 | Ovarian | 7.30 ± 2.2 |
| CAPAN1 | Pancreas | 5.00 ± 1.6 |
| MiaPaCa2 | Pancreas | 8.80 ± 3.1 |
| DU145 | Prostate | 1.26 ± 0.4 |
| PC3 | Prostate | 1.80 ± 0.4 |
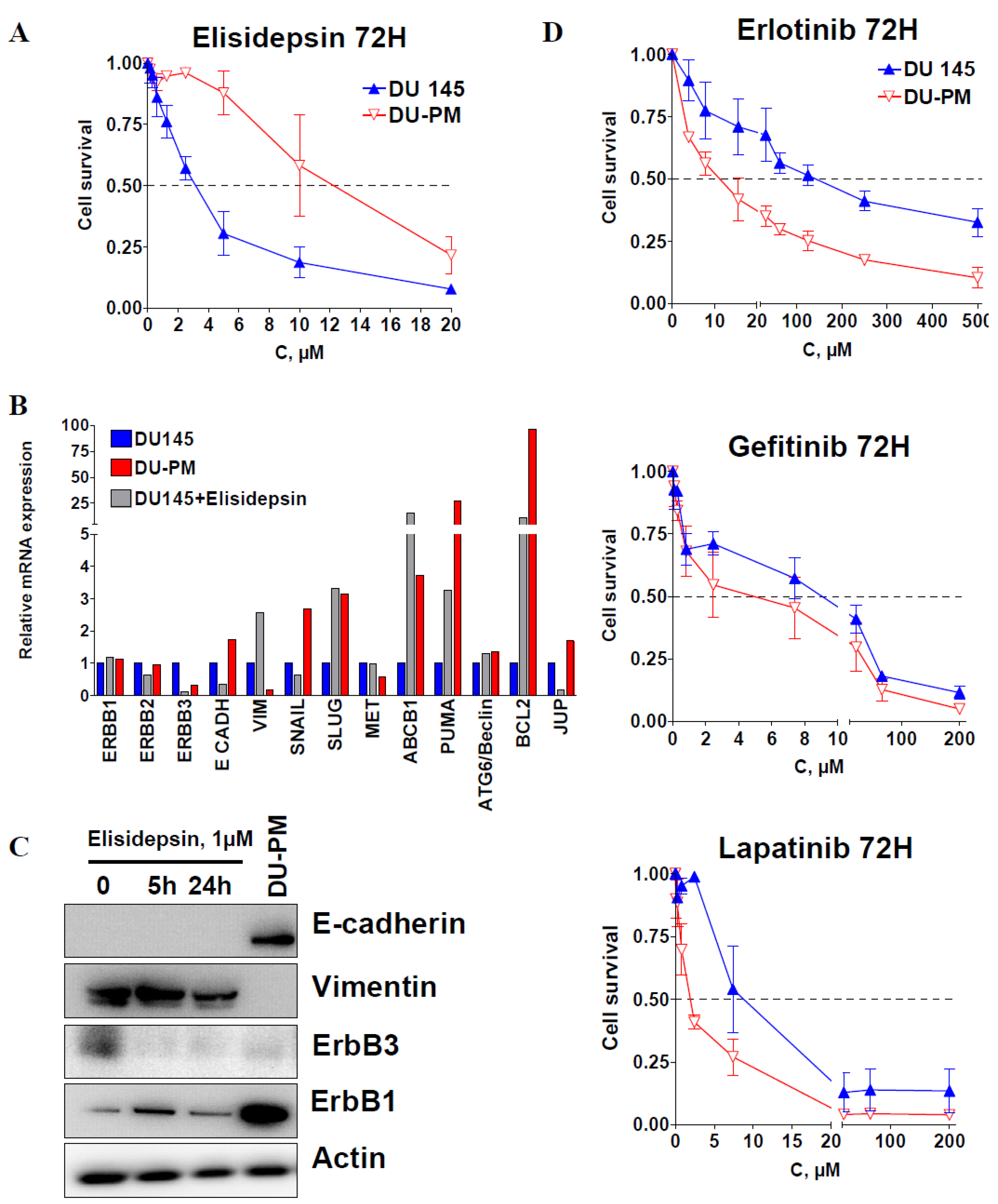
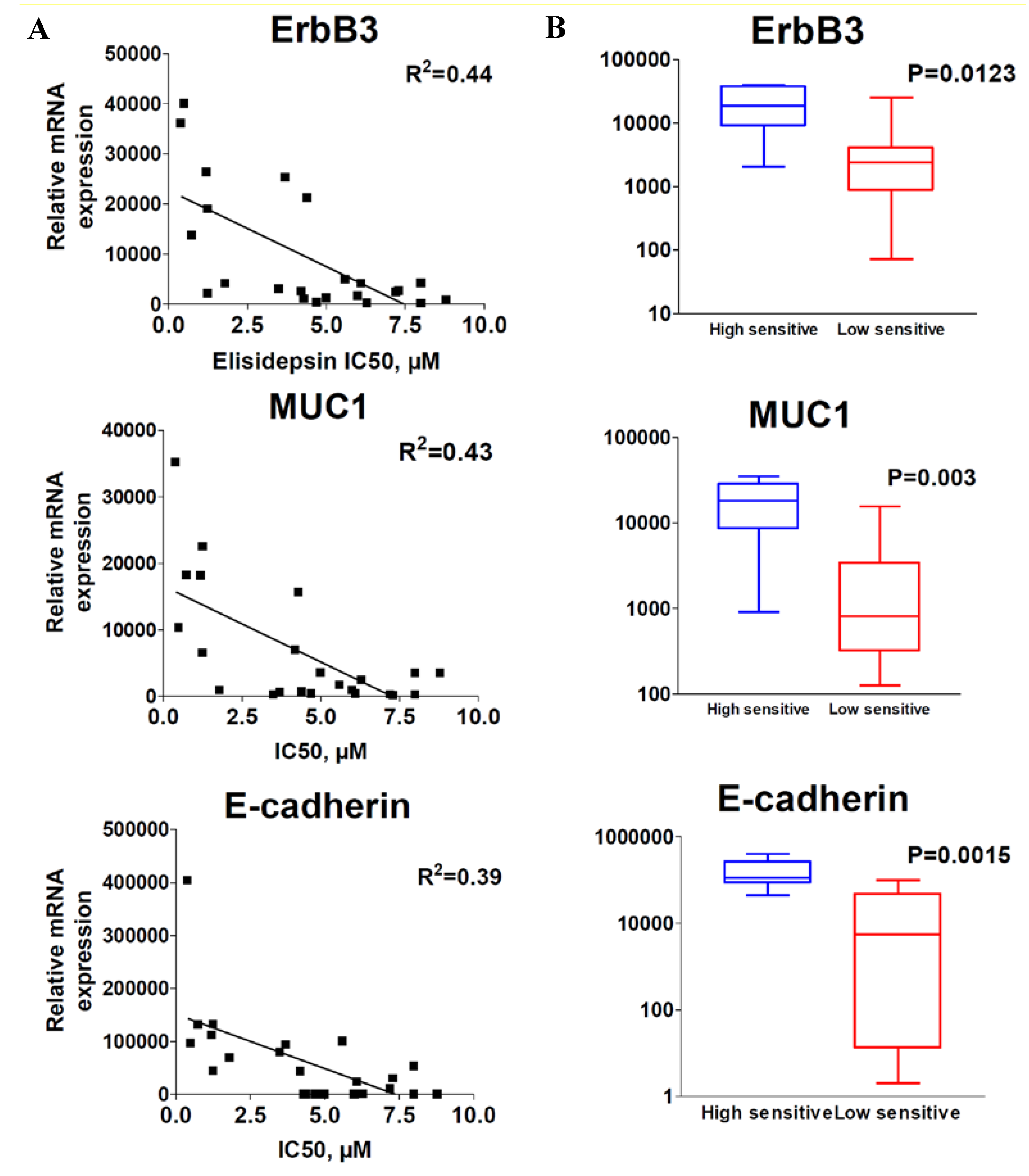
2.2. ErbB3, E-cadherin, Muc1 and the MAPK Activation Predict Elisidepsin Sensitivity and Resistance
2.3. Elisidepsin Sensitivity Is Dependent on Epithelial-to-Mesenchymal Transition (EMT)
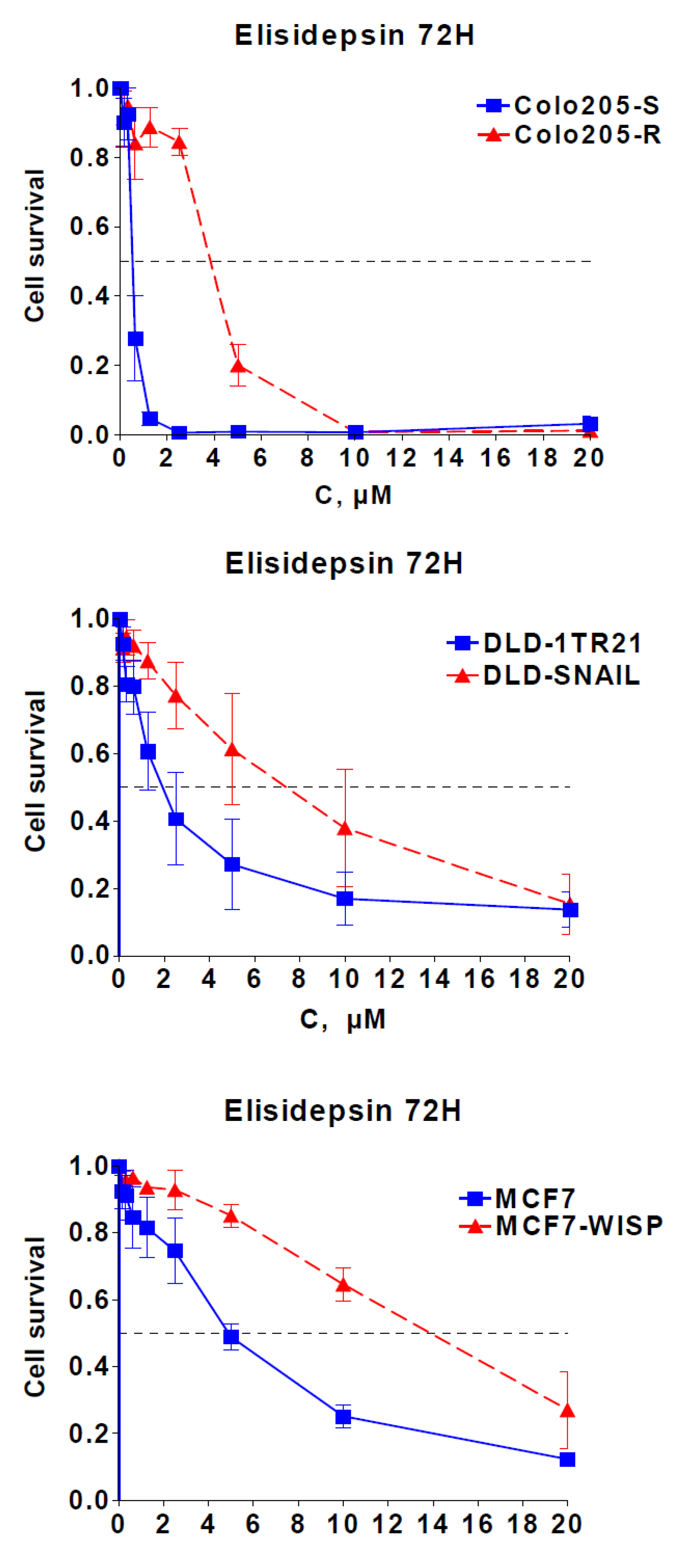
2.4. Characterization of Acquired Resistance to Elisidepsin
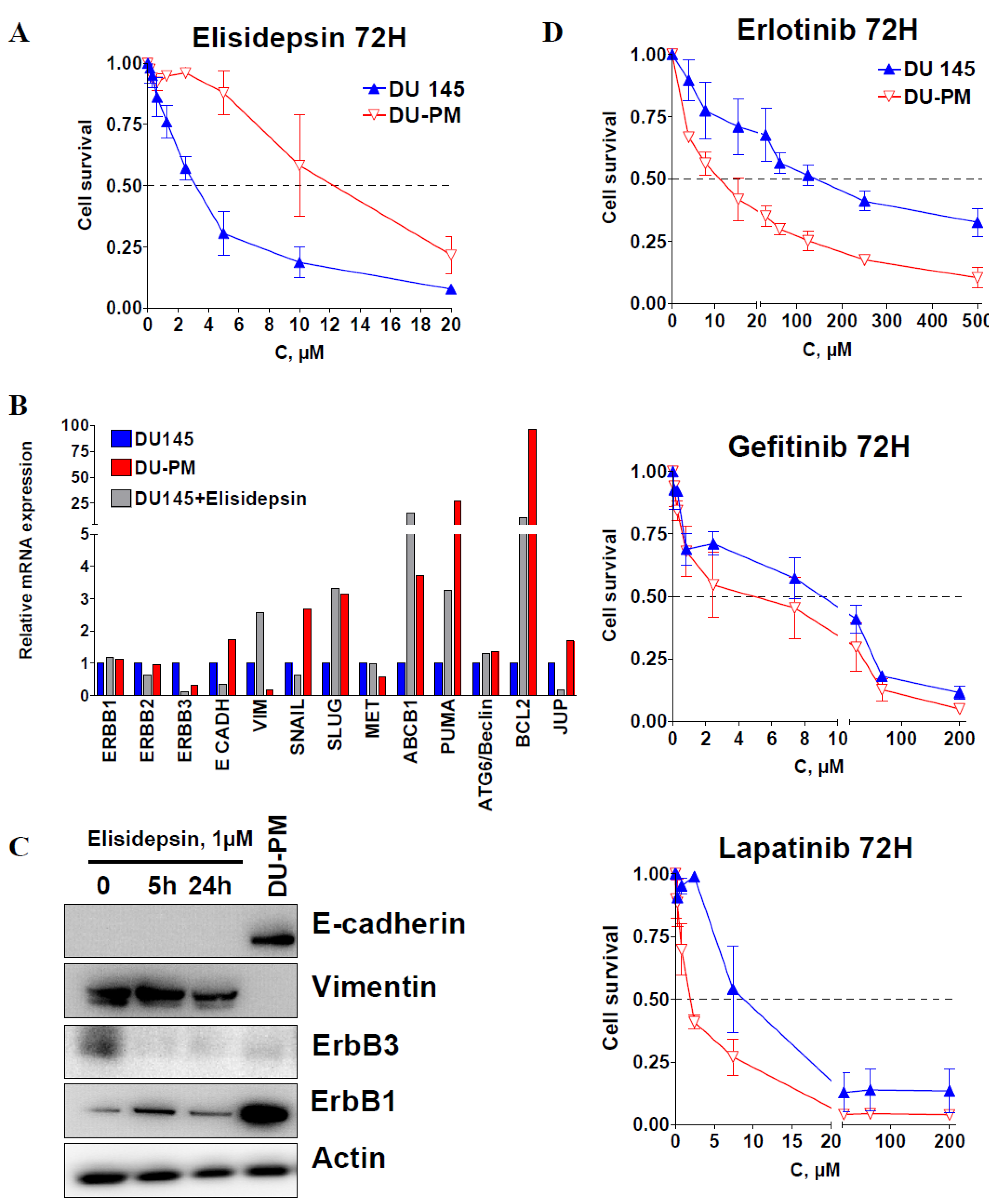
2.5. Elisidepsin Combination with Chemotherapies and Targeted Therapies
| Combination schedule | CI, median (min; max) | ||
|---|---|---|---|
| DU145 | Colo205 | ||
| 5-FU-based combinations | Elisidepsin → 5-FU | 0.83 (0.59; 1.14) | 1.36 (0.98; 1.52) |
| 5-FU → Elisidepsin | 0.65 (0.49; 0.88) | 0.93 (0.71; 1.24) | |
| Elisidepsin + 5-FU | 1.03 (0.59; 1.18) | 1.27 (1.14; 1.64) | |
| Gemcitabine-based combinations | Elisidepsin → Gemcitabine | 1.03 (0.93; 1.15) | - |
| Gemcitabine → Elisidepsin | 1.01 (0.96; 1.12) | - | |
| Elisidepsin + Gemcitabine | 1.07 (0.90; 1.22) | - | |
| Cisplatin-based combinations | Elisidepsin → Cisplatin | 1.18 (1.01; 1.38) | - |
| Cisplatin → Elisidepsin | 1.08 (0.92; 1.36) | - | |
| Elisidepsin + Cisplatin | 1.26 (1.09; 1.38) | - | |
| Oxaliplatin-based combinations | Elisidepsin → Oxaliplatin | 0.77 (0.75; 0.97) | 0.96 (0.92; 3.05) |
| Oxaliplatin → Elisidepsin | 0.69 (0.33; 0.94) | 0.22 (0.08; 1.85) | |
| Elisidepsin + Oxaliplatin | 0.82 (0.69; 0.89) | 0.62 (0.19; 1.67) | |
| Lapatinib-based combinations | Elisidepsin → Lapatinib | 0.98 (0.71; 1.18) | 0.98 (0.89; 1.6) |
| Lapatinib → Elisidepsin | 0.71 (0.34; 1.16) | 0.71 (0.67; 0,89) | |
| Elisidepsin + Lapatinib | 1.04 (0.69; 1.12) | 0.77 (0.60; 1,21) | |
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Lines
4.3. Cell Cytotoxicity Assay
4.4. Western Blot Analysis
4.5. Real-Time RT-PCR
4.6. DNA Extraction and Mutation Screening
4.7. Determination of Synergistic Activity
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Shilabin, A.G.; Kasanah, N.; Wedge, D.E.; Hamann, M.T. Lysosome and HER3 (ErbB3) selective anticancer agent kahalalide F: Semisynthetic modifications and antifungal lead-exploration studies. J. Med. Chem. 2007, 50, 4340–4350. [Google Scholar] [CrossRef]
- Garcia-Rocha, M.; Bonay, P.; Avila, J. The antitumoral compound kahalalide facts on cell lysosomes. Cancer Lett. 1996, 99, 43–50. [Google Scholar] [CrossRef]
- Suarez, Y.; Gonzalez, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Munoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer. Ther. 2003, 2, 863–872. [Google Scholar]
- Faircloth, G.T.; Smith, B.; Grant, W.; Jimeno, J.M.; Garcia-Gravalos, L.; Scotto, K.; Shtil, A. Selective antitumor activity of Kahalalide F, a marine-derived cyclic depsipeptide. Proc. Am. Assoc. Cancer Res. 2001, 42, 213. [Google Scholar]
- Janmaat, M.; Rodriguez, J.; Jimeno, J.; Kruyt, F.; Giaccone, G. Kahalalide F induces necrosis-like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol. Pharmacol. 2005, 68, 502–510. [Google Scholar]
- Ling, Y.H.; Aracil, M.; Jimeno, J.; Perez-Solera, R.; Zoua, Y. Molecular pharmacodynamics of PM02734 (elisidepsin) as single agent and in combination with erlotinib; synergistic activity in human non-small cell lung cancer cell lines and xenograft models. Eur. J. Cancer 2009, 45, 1855–1864. [Google Scholar] [CrossRef]
- Teixidó, C.; Arguelaguet, E.; Pons, B.; Aracil, M.; Jimeno, J.; Somoza, R.; Marés, R.; Ramón, Y.; Cajal, S.; Hernández-Losa, J. ErbB3 expression predicts sensitivity to elisidepsin treatment: In vitro synergism with cisplatin, paclitaxel and gemcitabine in lung, breast and colon cancer cell lines. Int. J. Oncol. 2012, 41, 317–324. [Google Scholar]
- Varadi, T.; Rosnik, J.; Lisboa, D.; Vereb, G.; Molina-Guijarro, J.M.; Galmarini, C.M.; Szöllosi, J.; Nagy, P. ErbB protein modifications are secondary to severe cell membrane alterations induced by elisidepsin treatment. Eur. J. Pharmacol. 2011, 667, 91–99. [Google Scholar]
- Molina-Guijarro, J.M.; Macías, Á.; García, C.; Muñoz, E.; García-Fernández, L.F.; David, M.; Núñez, L.; Martínez-Leal, J.F.; Moneo, V.; Cuevas, C.; et al. Irvalec inserts into the plasma membrane causing rapid loss of integrity and necrotic cell death in tumor cells. PLoS One 2011, 6, e19042. [Google Scholar]
- Herrero, A.B.; Astudillo, A.M.; Balboa, M.A.; Cuevas, C.; Balsinde, J.; Moreno, S. Levels of SCS7/FA2H-Mediated Fatty Acid 2-Hydroxylation determine the sensitivity of cells to antitumor PM02734. Cancer Res. 2008, 68, 9779–9787. [Google Scholar] [CrossRef]
- Bruce, J.Y.; Geary, D.; de las Heras, B.; Soto, A.; Garcia Paramio, P.; Yovine, A.; Schilsky, R.L.; Undevia, S.D.; Ratain, M.J. Phase I study of PM02734: Association of dose-limiting hepatotoxicity with plasma concentrations. J. Clin. Oncol. 2008, 26, 2513. [Google Scholar]
- Faivre, S.J.; Ropert, S.; Coronado, C.; Alcantara, J.; de Miguel, B.; Soto, A.; Szyldergemajn, S.A.; Loussilaho, G.; Mir, O.; Goldwasser, F.; et al. Phase I study of elisidepsin (E) in combination with carboplatin (C) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2011, 29, e13097. [Google Scholar]
- Sewell, J.M.; Mayer, I.; Langdon, S.P.; Smyth, J.F.; Jodrell, D.I.; Guichard, S.M. The mechanism of action of Kahalalide F: Variable cell permeability in human hepatoma cell lines. Eur. J. Cancer 2005, 41, 1637–1644. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Kono, K.; Takahashi, A.; Amemiya, H.; Ichihara, F.; Sugai, H.; Iizuka, H.; Fujii, H.; Matsumoto, Y. Frequencies of HER-2/neu overexpression relating to HLA haplotype in patients with gastric cancer. Int. J. Cancer 2002, 98, 216–220. [Google Scholar] [CrossRef]
- Ghoul, A.; Serova, M.; Astorgues-Xerri, L.; Bieche, I.; Bousquet, G.; Varna, M.; Vidaud, M.; Phillips, E.; Weill, S.; Benhadji, K.A.; et al. Epithelial-to-mesenchymal transition and resistance to ingenol 3-angelate, a novel protein kinase C modulator, in colon cancer cells. Cancer Res. 2009, 69, 4260–4269. [Google Scholar] [CrossRef]
- Serova, M.; Astorgues-Xerri, L.; Bieche, I.; Albert, S.; Vidaud, M.; Benhadji, K.A.; Emami, S.; Vidaud, D.; Hammel, P.; Theou-Anton, N.; et al. Epithelial-to-mesenchymal transition and oncogenic Ras expression in resistance to the protein kinase Cbeta inhibitor enzastaurin in colon cancer cells. Mol. Cancer Ther. 2010, 9, 1308–1317. [Google Scholar] [CrossRef]
- De Craene, B.; Gilbert, B.; Stove, C.; Bruyneel, E.; van Roy, F.; Berx, G. The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res. 2005, 65, 6237–6244. [Google Scholar] [CrossRef]
- Fritah, A.; Saucier, C.; de Wever, O.; Bracke, M.; Bièche, I.; Lidereau, R.; Gespach, C.; Drouot, S.; Redeuilh, G.; Sabbah, M. Role of WISP-2/CCN5 in the maintenance of a differentiated and noninvasive phenotype in human breast cancer cells. Mol. Cell Biol. 2008, 28, 1114–1123. [Google Scholar] [CrossRef]
- Ling, Y.H.; Aracil, M.; Zou, Y.; Yuan, Z.; Lu, B.; Jimeno, J.; Cuervo, A.M.; Perez-Soler, R. PM02734 (elisidepsin) induces caspase-independent cell death associated with features of autophagy, inhibition of the Akt/mTOR signaling pathway, and activation of death-associated protein kinase. Clin. Cancer Res. 2011, 17, 5353–5366. [Google Scholar] [CrossRef]
- Sabbah, M.; Emami, S.; Redeuilh, G.; Julien, S.; Prévost, G.; Zimber, A.; Ouelaa, R.; Bracke, M.; de Wever, O.; Gespach, C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist. Updat. 2008, 11, 123–151. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Teixidó, C.; Marés, R.; Aracil, M.; Ramón, Y.; Cajal, S.; Hernández-Losa, J. Epithelial-mesenchymal transition markers and HER3 expression are predictors of elisidepsin treatment response in breast and pancreatic cancer cell lines. PLoS One 2013, 8, e53645. [Google Scholar]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Bièche, I.; Parfait, B.; Laurendeau, I.; Girault, I.; Vidaud, M.; Lidereau, R. Quantification of estrogen receptor alpha and beta expression in sporadic breast cancer. Oncogene 2001, 56, 8109–8115. [Google Scholar]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Serova, M.; De Gramont, A.; Bieche, I.; Riveiro, M.E.; Galmarini, C.M.; Aracil, M.; Jimeno, J.; Faivre, S.; Raymond, E. Predictive Factors of Sensitivity to Elisidepsin, a Novel Kahalalide F-Derived Marine Compound. Mar. Drugs 2013, 11, 944-959. https://doi.org/10.3390/md11030944
Serova M, De Gramont A, Bieche I, Riveiro ME, Galmarini CM, Aracil M, Jimeno J, Faivre S, Raymond E. Predictive Factors of Sensitivity to Elisidepsin, a Novel Kahalalide F-Derived Marine Compound. Marine Drugs. 2013; 11(3):944-959. https://doi.org/10.3390/md11030944
Chicago/Turabian StyleSerova, Maria, Armand De Gramont, Ivan Bieche, Maria Eugenia Riveiro, Carlos Maria Galmarini, Miguel Aracil, José Jimeno, Sandrine Faivre, and Eric Raymond. 2013. "Predictive Factors of Sensitivity to Elisidepsin, a Novel Kahalalide F-Derived Marine Compound" Marine Drugs 11, no. 3: 944-959. https://doi.org/10.3390/md11030944
APA StyleSerova, M., De Gramont, A., Bieche, I., Riveiro, M. E., Galmarini, C. M., Aracil, M., Jimeno, J., Faivre, S., & Raymond, E. (2013). Predictive Factors of Sensitivity to Elisidepsin, a Novel Kahalalide F-Derived Marine Compound. Marine Drugs, 11(3), 944-959. https://doi.org/10.3390/md11030944