Polypeptide Modulators of TRPV1 Produce Analgesia without Hyperthermia

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
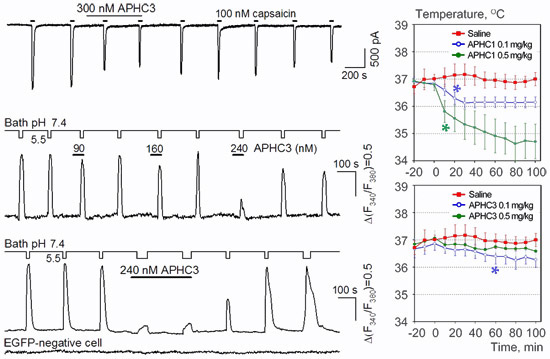
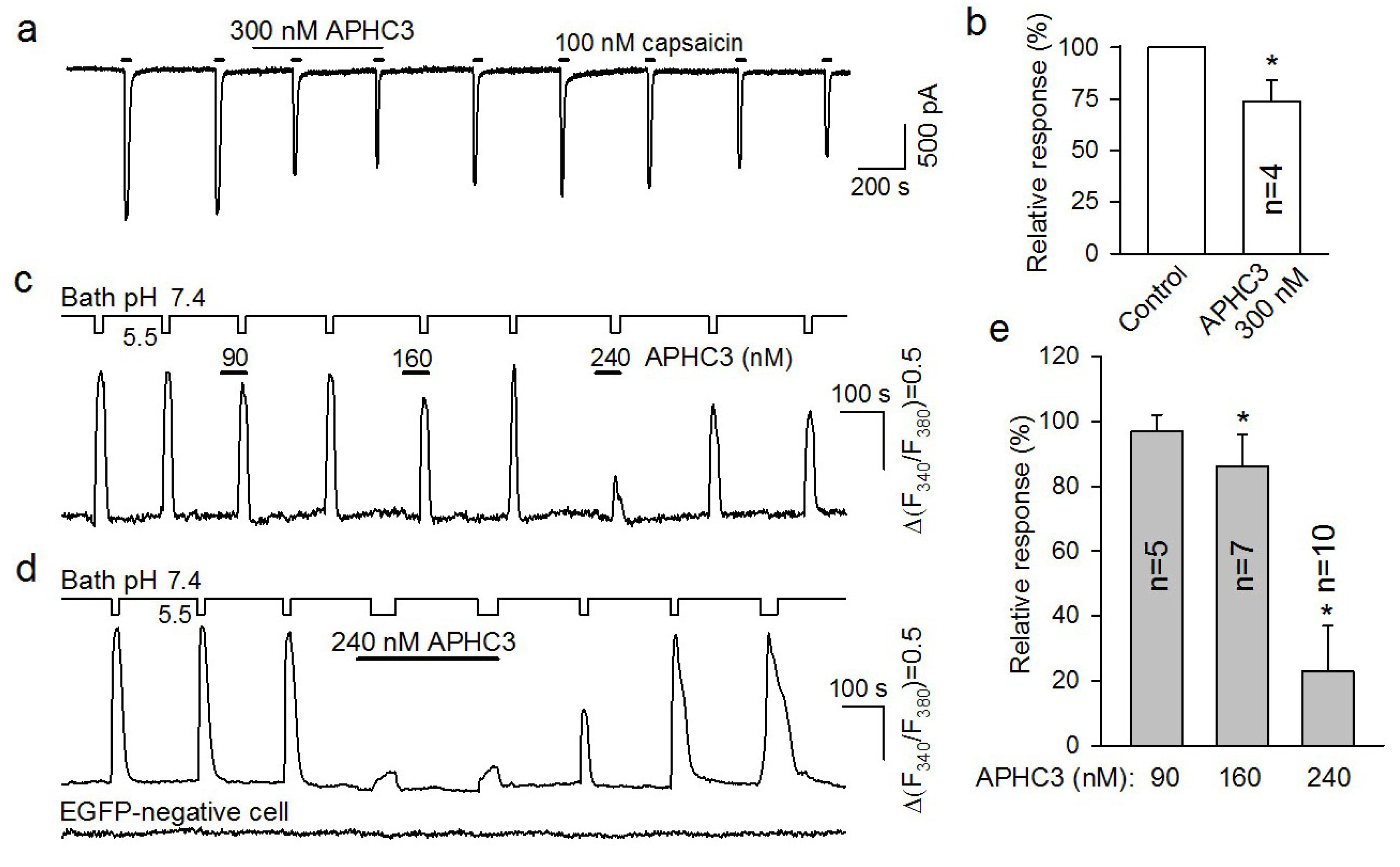
2.1. Pharmacology of APHC3


2.2. In Vivo Effects of APHC1 and APHC3 in Pain Models

2.2.1. Hot Plate Test
2.2.2. Capsaicin Test
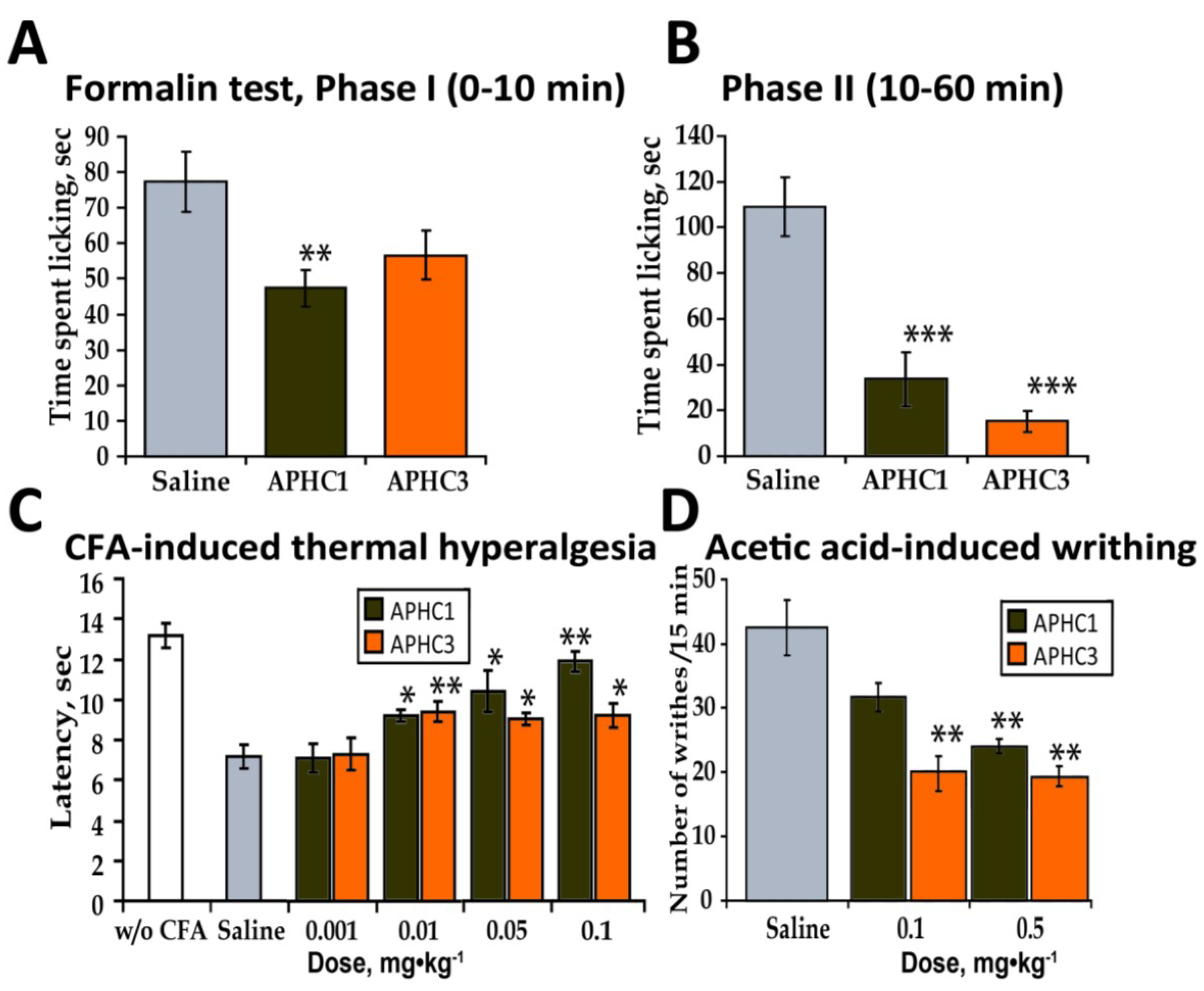
2.2.3. Formalin Test
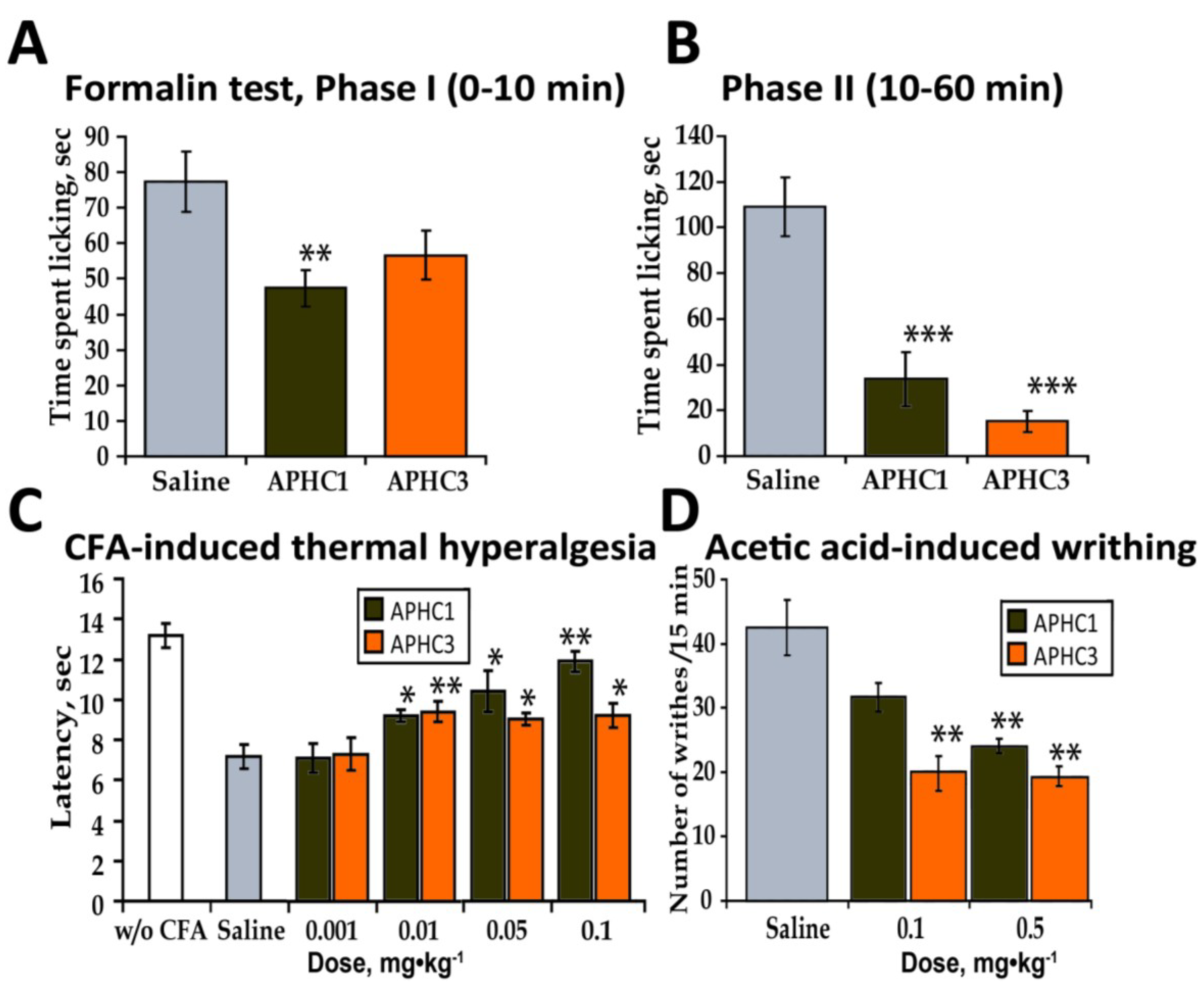
2.2.4. CFA-Induced Hyperalgesia
2.2.5. Abdominal Constriction Test of Visceral Pain (Acetic Acid-Induced Writhing)
2.3. APHC1 and APHC3 Decrease the Core Body Temperature
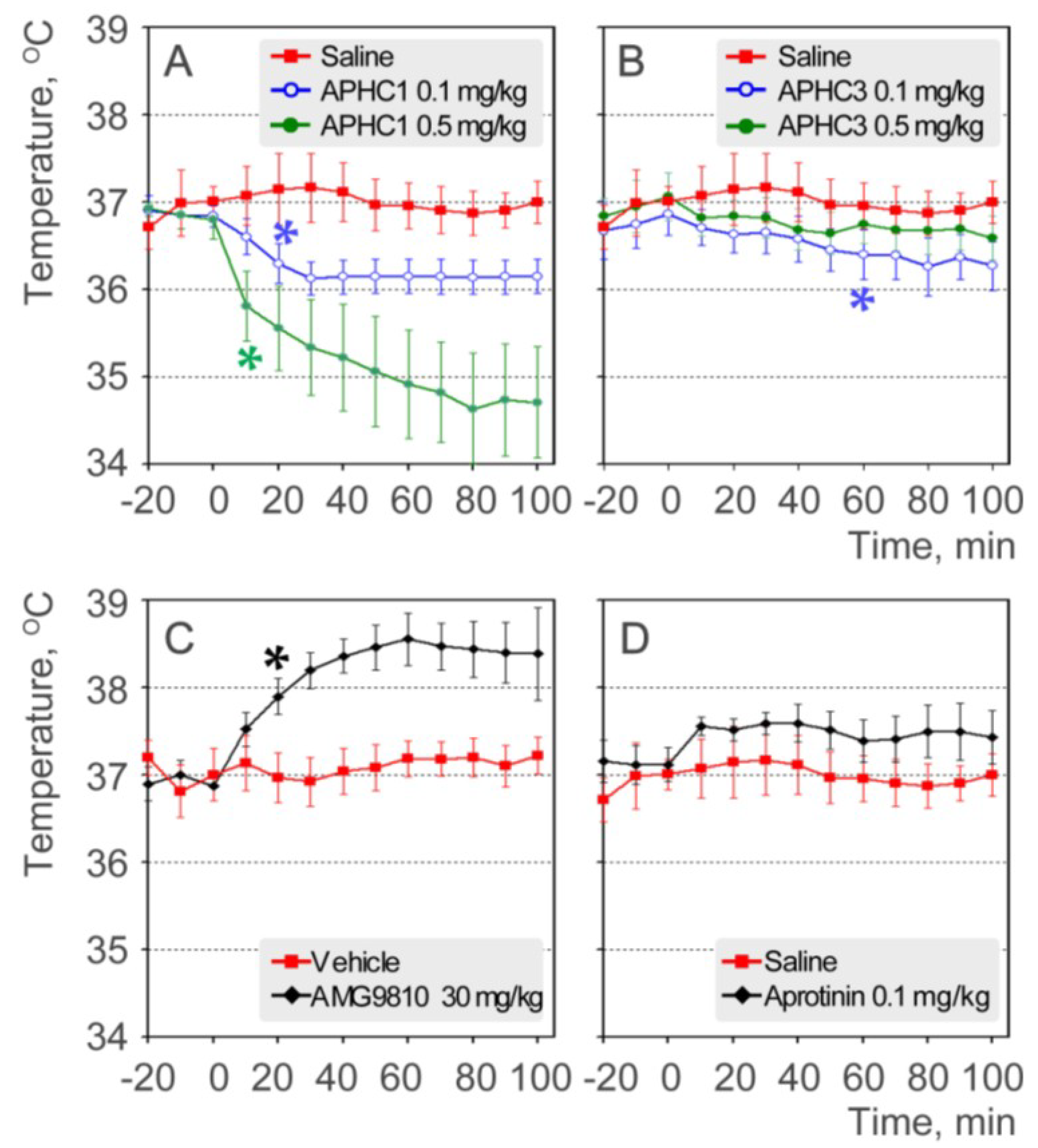
3. Experimental Section
3.1. Production of Recombinant Polypeptides
3.2. Cell Culture and Transient Transfection
3.3. Electrophysiology
3.4. Single Cell Ca2+ Imaging
3.5. Animals Experiments
3.5.1. Open-Field Activity in Mice
3.5.2. Hot-Plate Test
3.5.3. Capsaicin-Induced Acute Pain
3.5.4. Formalin Test
3.5.5. Complete Freund’s Adjuvant–Induced Thermal Hyperalgesia
3.5.6. Acetic Acid-Induced Writhing (Abdominal Constriction Test of Visceral Pain)
3.5.7. Body Temperature Measurements
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Szallasi, A.; Cortright, D.N.; Blum, C.A.; Eid, S.R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 2007, 6, 357–372. [Google Scholar] [CrossRef]
- Malmberg, A.; Bley, K. Turning Up the Heat on Pain: TRPV1 Receptors in Pain and Inflammation; Birkhauser Verlag: Basel, Boston, Berlin, 2005. [Google Scholar]
- Szallasi, A.; Appendino, G. Vanilloid receptor TRPV1 antagonists as the next generation of painkillers. Are we putting the cart before the horse? J. Med. Chem. 2004, 47, 2717–2723. [Google Scholar] [CrossRef]
- Gunthorpe, M.J.; Chizh, B.A. Clinical development of TRPV1 antagonists: Targeting a pivotal point in the pain pathway. Drug Discov. Today 2009, 14, 56–67. [Google Scholar] [CrossRef]
- Gavva, N.R.; Bannon, A.W.; Surapaneni, S.; Hovland, D.N., Jr.; Lehto, S.G.; Gore, A.; Juan, T.; Deng, H.; Han, B.; Klionsky, L.; et al. The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J. Neurosci. 2007, 27, 3366–3374. [Google Scholar] [CrossRef]
- Gavva, N.R.; Treanor, J.J.; Garami, A.; Fang, L.; Surapaneni, S.; Akrami, A.; Alvarez, F.; Bak, A.; Darling, M.; Gore, A.; et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 2008, 136, 202–210. [Google Scholar] [CrossRef]
- Honore, P.; Chandran, P.; Hernandez, G.; Gauvin, D.M.; Mikusa, J.P.; Zhong, C.; Joshi, S.K.; Ghilardi, J.R.; Sevcik, M.A.; Fryer, R.M.; et al. Repeated dosing of ABT-102, a potent and selective TRPV1 antagonist, enhances TRPV1-mediated analgesic activity in rodents, but attenuates antagonist-induced hyperthermia. Pain 2009, 142, 27–35. [Google Scholar] [CrossRef]
- Immke, D.C.; Gavva, N.R. The TRPV1 receptor and nociception. Semin. Cell Dev. Biol. 2006, 17, 582–591. [Google Scholar] [CrossRef]
- Lehto, S.G.; Tamir, R.; Deng, H.; Klionsky, L.; Kuang, R.; Le, A.; Lee, D.; Louis, J.C.; Magal, E.; Manning, B.H.; et al. Antihyperalgesic effects of (R,E)-N-(2-hydroxy-2,3-dihydro-1H-inden-4-yl)-3-(2-(piperidin-1-yl)-4-(trifluorom ethyl)phenyl)-acrylamide (AMG8562), a novel transient receptor potential vanilloid type 1 modulator that does not cause hyperthermia in rats. J. Pharmacol. Exp. Ther. 2008, 326, 218–229. [Google Scholar] [CrossRef]
- Garami, A.; Shimansky, Y.P.; Pakai, E.; Oliveira, D.L.; Gavva, N.R.; Romanovsky, A.A. Contributions of different modes of TRPV1 activation to TRPV1 antagonist-induced hyperthermia. J. Neurosci. 2010, 30, 1435–1440. [Google Scholar] [CrossRef]
- Andreev, Y.A.; Kozlov, S.A.; Koshelev, S.G.; Ivanova, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E.V. Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 2008, 283, 23914–23921. [Google Scholar] [CrossRef]
- Andreev, Y.A.; Kozlov, S.A.; Kozlovskaya, E.P.; Grishin, E.V. Analgesic effect of a polypeptide inhibitor of the TRPV1 receptor in noxious heat pain models. Dok. Biochem. Biophys. 2009, 424, 46–48. [Google Scholar] [CrossRef]
- Kozlov, S.A.; Andreev, Y.A.; Murashev, A.N.; Skobtsov, D.I.; D'yachenko, I.A.; Grishin, E.V. New polypeptide components from the Heteractis crispa sea anemone with analgesic activity. Russ. J. Bioorgan. Chem. 2009, 35, 711–719. [Google Scholar] [CrossRef]
- Philyppov, I.B.; Paduraru, O.N.; Andreev, Y.A.; Grishin, E.V.; Shuba, Y.M. Modulation of TRPV1-dependent contractility of normal and diabetic bladder smooth muscle by analgesic toxins from sea anemone Heteractis crispa. Life Sci. 2012, 91, 912–920. [Google Scholar] [CrossRef]
- Kozlov, S.; Grishin, E. Convenient nomenclature of cysteine-rich polypeptide toxins from sea anemones. Peptides 2012, 33, 240–244. [Google Scholar] [CrossRef]
- Antuch, W.; Berndt, K.D.; Chavez, M.A.; Delfin, J.; Wuthrich, K. The NMR solution structure of a Kunitz-type proteinase inhibitor from the sea anemone Stichodactyla helianthus. Eur. J. Biochem. 1993, 212, 675–684. [Google Scholar] [CrossRef]
- Gebhard, W.; Tschesche, H.; Fritz, H. Biochemistry of aprotinin and aprotinin-like inhibitors. In Proteinase Inhibitors; Barrett, A.J., Salvesen, G., Eds.; Elsevier: Amsterdam, The Netherlands, 1986; pp. 375–388. [Google Scholar]
- Creighton, T.E.; Charles, I.G. Sequences of the genes and polypeptide precursors for two bovine protease inhibitors. J. Mol. Biol. 1987, 194, 11–22. [Google Scholar] [CrossRef]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef]
- Klionsky, L.; Tamir, R.; Holzinger, B.; Bi, X.; Talvenheimo, J.; Kim, H.; Martin, F.; Louis, J.C.; Treanor, J.J.; Gavva, N.R. A polyclonal antibody to the prepore loop of transient receptor potential vanilloid type 1 blocks channel activation. J. Pharmacol. Exp. Ther. 2006, 319, 192–198. [Google Scholar] [CrossRef]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Tang, L.; Chen, Y.; Chen, Z.; Blumberg, P.M.; Kozikowski, A.P.; Wang, Z.J. Antinociceptive pharmacology of N-(4-chlorobenzyl)-N′-(4-hydroxy-3-iodo-5-methoxybenzyl) thiourea, a high-affinity competitive antagonist of the transient receptor potential vanilloid 1 receptor. J. Pharmacol. Exp. Ther. 2007, 321, 791–798. [Google Scholar] [CrossRef]
- Staruschenko, A.; Jeske, N.A.; Akopian, A.N. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J. Biol. Chem. 2010, 285, 15167–15177. [Google Scholar] [CrossRef]
- Davis, J.B.; Gray, J.; Gunthorpe, M.J.; Hatcher, J.P.; Davey, P.T.; Overend, P.; Harries, M.H.; Latcham, J.; Clapham, C.; Atkinson, K.; et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000, 405, 183–187. [Google Scholar] [CrossRef]
- Jara-Oseguera, A.; Simon, S.A.; Rosenbaum, T. TRPV1: On the road to pain relief. Curr. Mol. Pharmacol. 2008, 1, 255–269. [Google Scholar]
- Ikeda, Y.; Ueno, A.; Naraba, H.; Oh-ishi, S. Involvement of vanilloid receptor VR1 and prostanoids in the acid-induced writhing responses of mice. Life Sci. 2001, 69, 2911–2919. [Google Scholar] [CrossRef]
- Romanovsky, A.A.; Almeida, M.C.; Garami, A.; Steiner, A.A.; Norman, M.H.; Morrison, S.F.; Nakamura, K.; Burmeister, J.J.; Nucci, T.B. The transient receptor potential vanilloid-1 channel in thermoregulation: A thermosensor it is not. Pharmacol. Rev. 2009, 61, 228–261. [Google Scholar] [CrossRef]
- Steiner, A.A.; Turek, V.F.; Almeida, M.C.; Burmeister, J.J.; Oliveira, D.L.; Roberts, J.L.; Bannon, A.W.; Norman, M.H.; Louis, J.C.; Treanor, J.J.; et al. Nonthermal activation of transient receptor potential vanilloid-1 channels in abdominal viscera tonically inhibits autonomic cold-defense effectors. J. Neurosci. 2007, 27, 7459–7468. [Google Scholar] [CrossRef]
- Fosgerau, K.; Weber, U.J.; Gotfredsen, J.W.; Jayatissa, M.; Buus, C.; Kristensen, N.B.; Vestergaard, M.; Teschendorf, P.; Schneider, A.; Hansen, P.; et al. Drug-induced mild therapeutic hypothermia obtained by administration of a transient receptor potential vanilloid type 1 agonist. BMC Cardiovasc. Disord. 2010, 10, 51. [Google Scholar] [CrossRef]
- Andreev, Y.A.; Kozlov, S.A.; Vassilevski, A.A.; Grishin, E.V. Cyanogen bromide cleavage of proteins in salt and buffer solutions. Anal. Biochem. 2010, 407, 144–146. [Google Scholar] [CrossRef]
- Sakurada, T.; Katsumata, K.; Tan-No, K.; Sakurada, S.; Kisara, K. The capsaicin test in mice for evaluating tachykinin antagonists in the spinal cord. Neuropharmacology 1992, 31, 1279–1285. [Google Scholar] [CrossRef]
- Tjolsen, A.; Berge, O.G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The formalin test: An evaluation of the method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Andreev, Y.A.; Kozlov, S.A.; Korolkova, Y.V.; Dyachenko, I.A.; Bondarenko, D.A.; Skobtsov, D.I.; Murashev, A.N.; Kotova, P.D.; Rogachevskaja, O.A.; Kabanova, N.V.; et al. Polypeptide Modulators of TRPV1 Produce Analgesia without Hyperthermia. Mar. Drugs 2013, 11, 5100-5115. https://doi.org/10.3390/md11125100
Andreev YA, Kozlov SA, Korolkova YV, Dyachenko IA, Bondarenko DA, Skobtsov DI, Murashev AN, Kotova PD, Rogachevskaja OA, Kabanova NV, et al. Polypeptide Modulators of TRPV1 Produce Analgesia without Hyperthermia. Marine Drugs. 2013; 11(12):5100-5115. https://doi.org/10.3390/md11125100
Chicago/Turabian StyleAndreev, Yaroslav A., Sergey A. Kozlov, Yuliya V. Korolkova, Igor A. Dyachenko, Dmitrii A. Bondarenko, Denis I. Skobtsov, Arkadii N. Murashev, Polina D. Kotova, Olga A. Rogachevskaja, Natalia V. Kabanova, and et al. 2013. "Polypeptide Modulators of TRPV1 Produce Analgesia without Hyperthermia" Marine Drugs 11, no. 12: 5100-5115. https://doi.org/10.3390/md11125100
APA StyleAndreev, Y. A., Kozlov, S. A., Korolkova, Y. V., Dyachenko, I. A., Bondarenko, D. A., Skobtsov, D. I., Murashev, A. N., Kotova, P. D., Rogachevskaja, O. A., Kabanova, N. V., Kolesnikov, S. S., & Grishin, E. V. (2013). Polypeptide Modulators of TRPV1 Produce Analgesia without Hyperthermia. Marine Drugs, 11(12), 5100-5115. https://doi.org/10.3390/md11125100