Champacyclin, a New Cyclic Octapeptide from Streptomyces Strain C42 Isolated from the Baltic Sea




Abstract
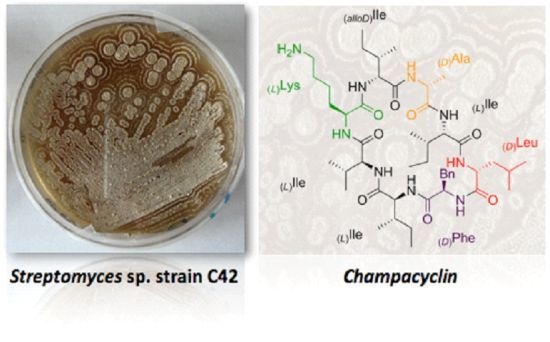
:
1. Introduction
2. Results and Discussion
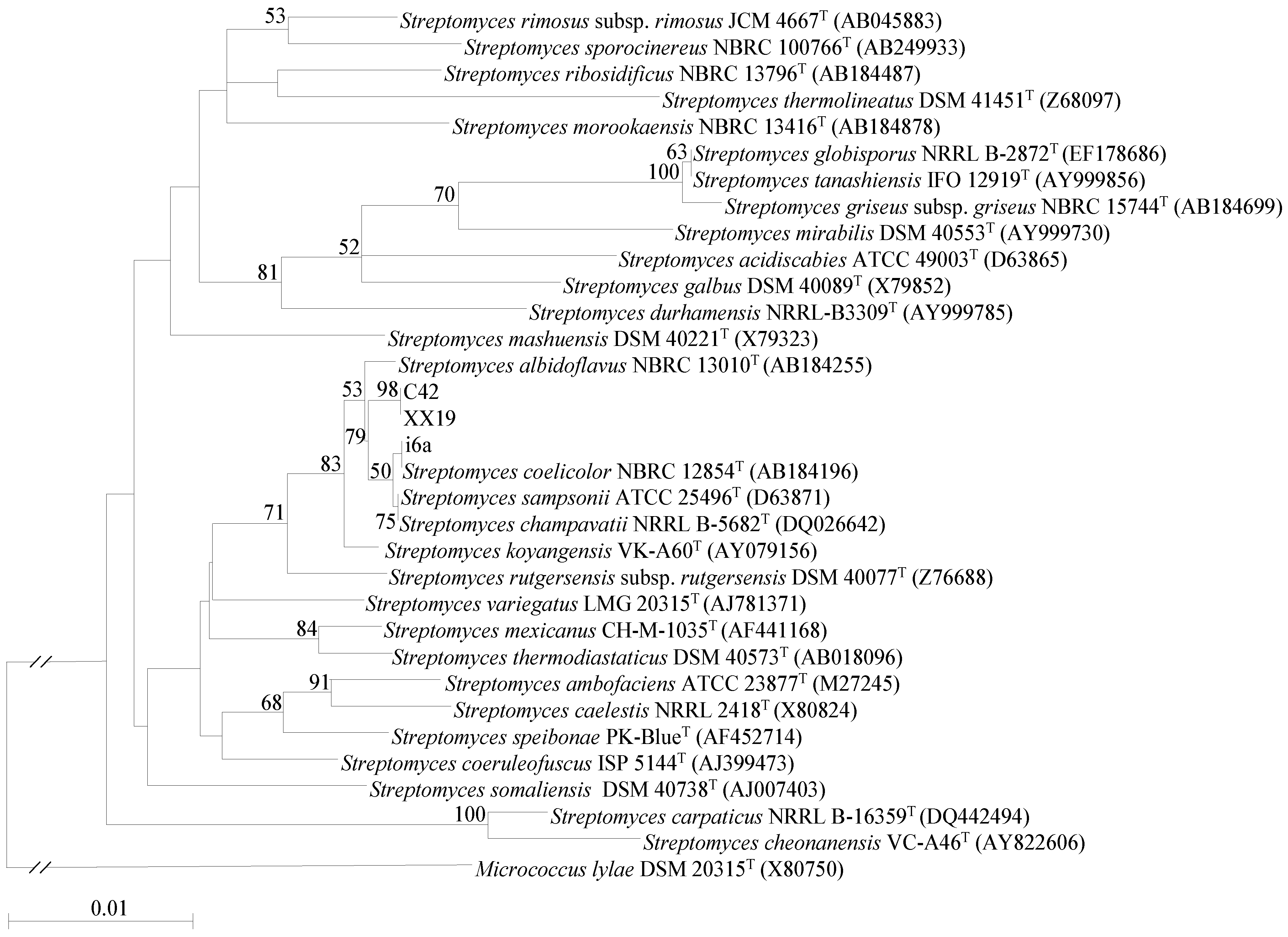
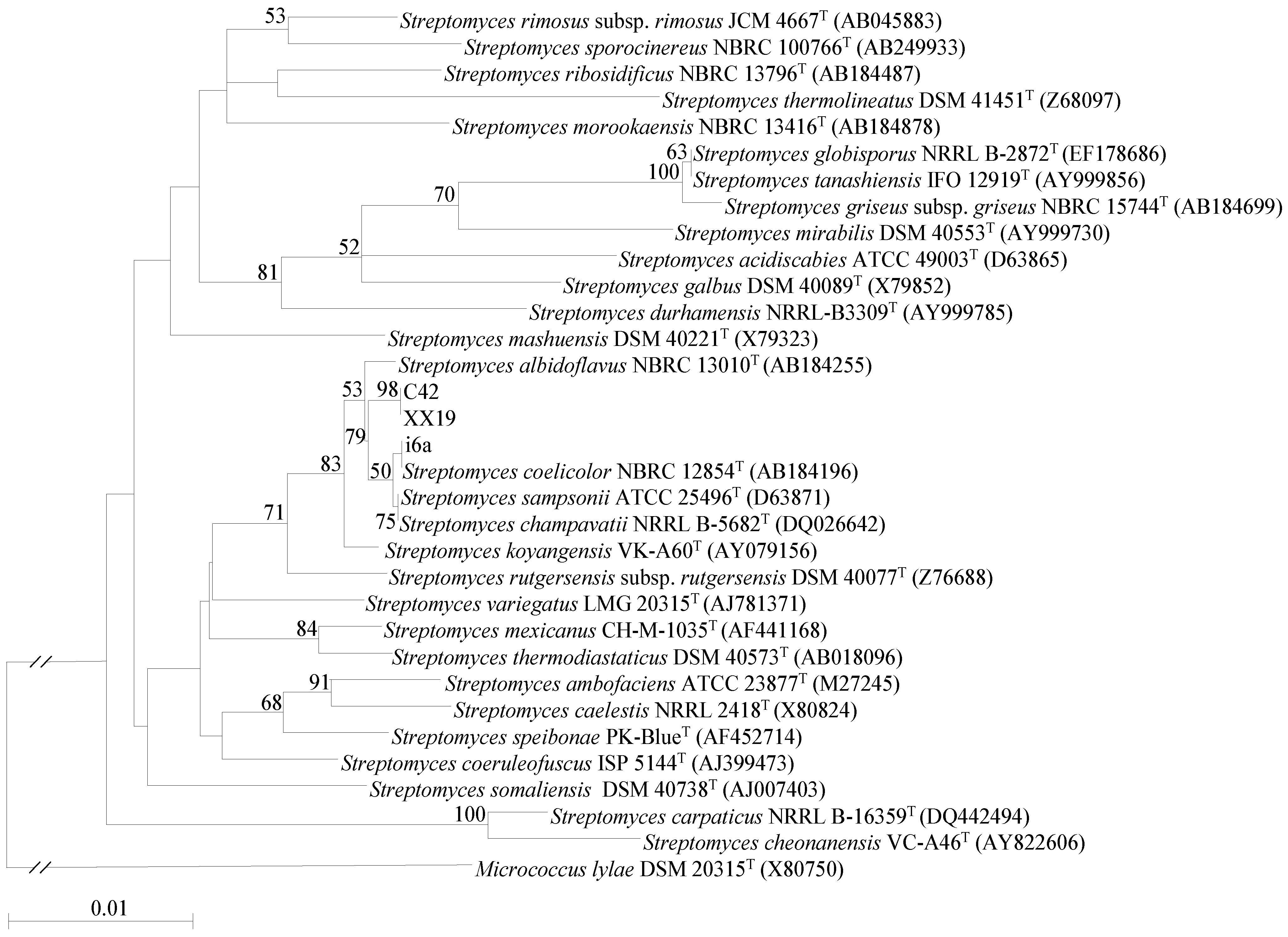
2.1. Isolation and Identification of Streptomyces Strains
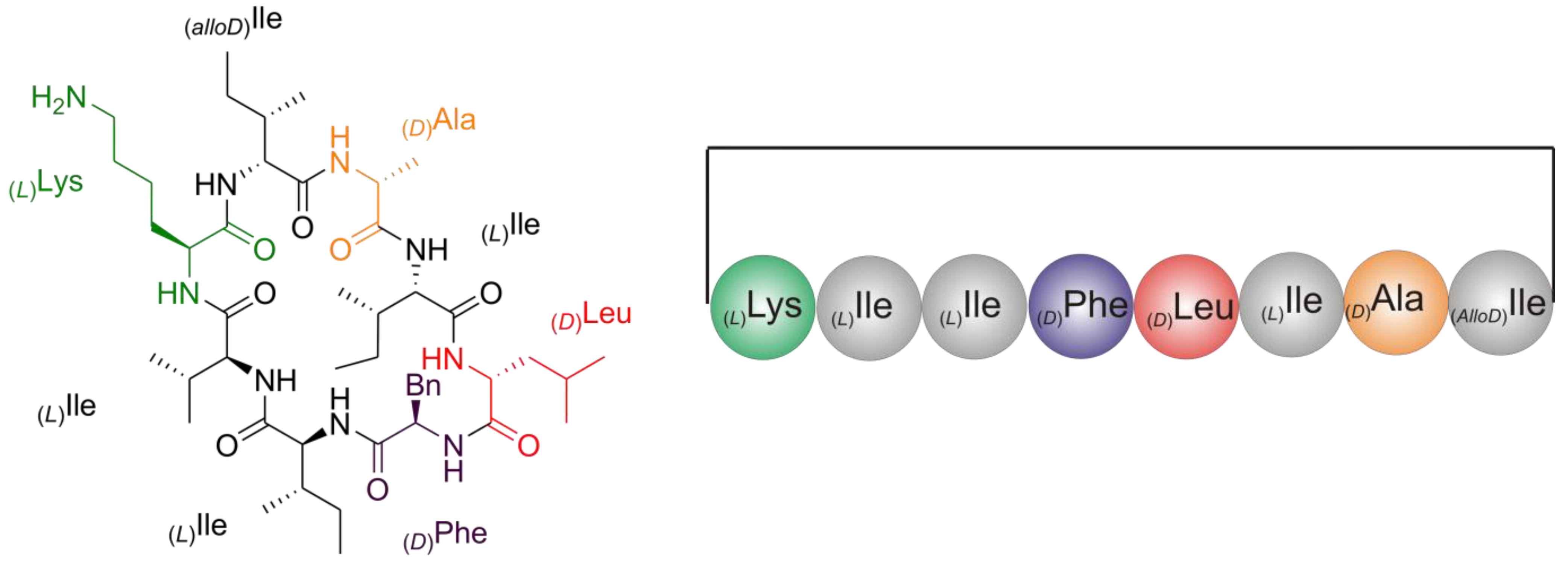
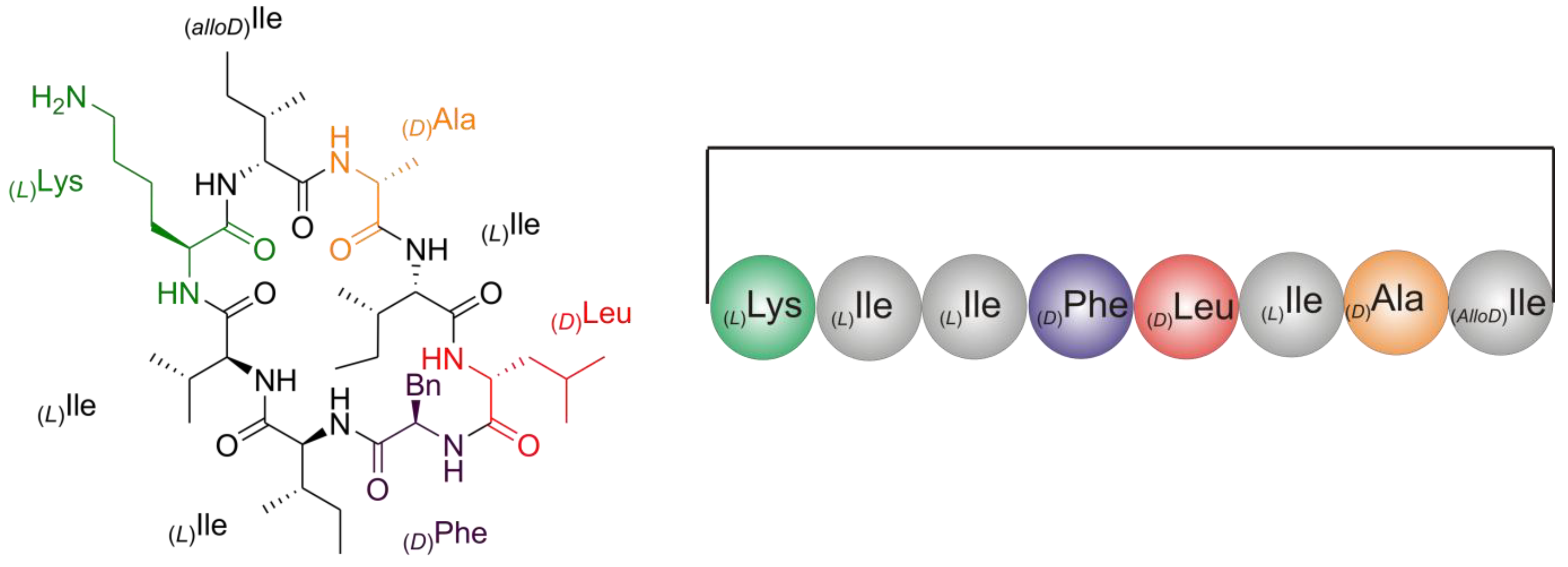
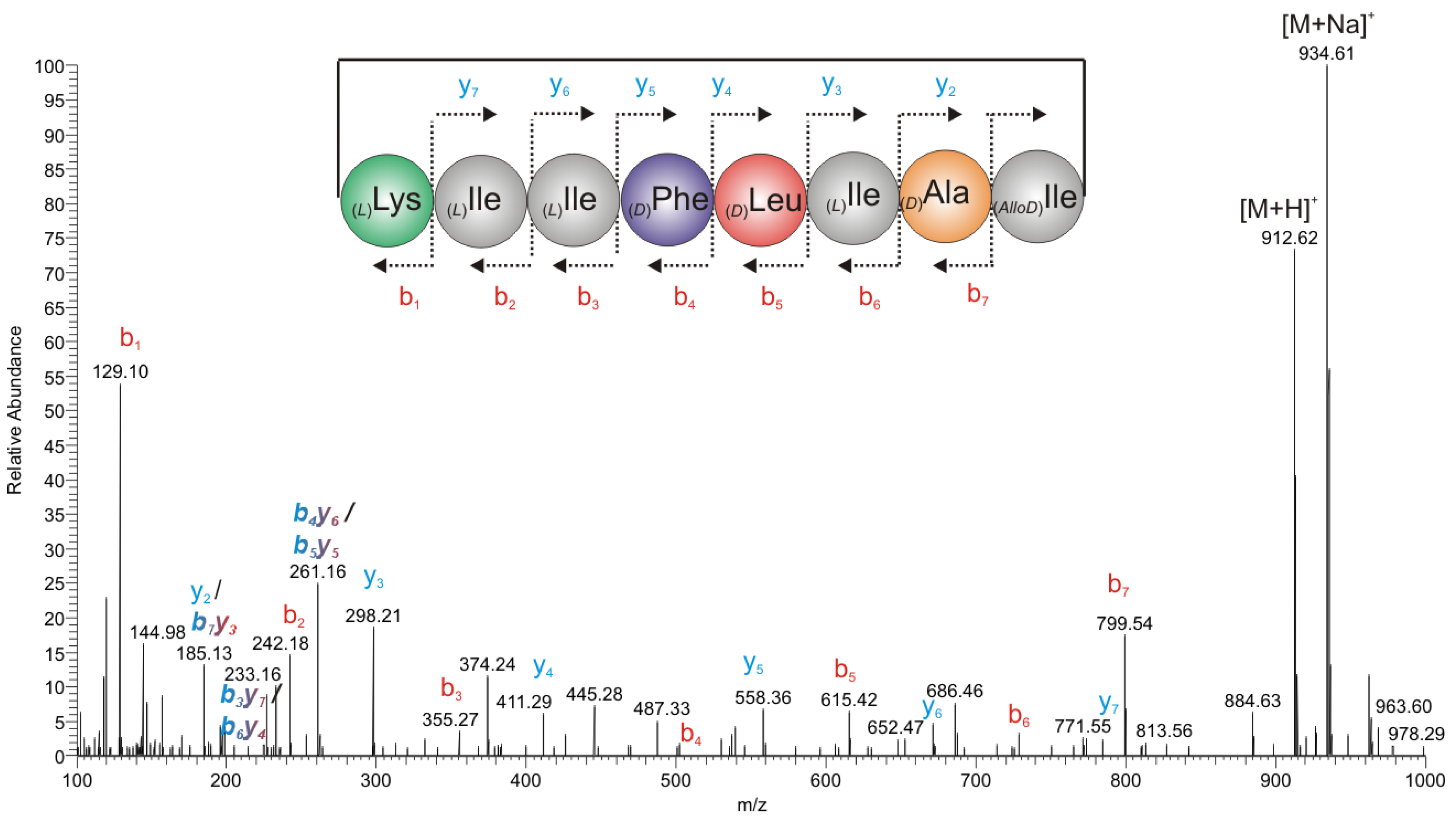
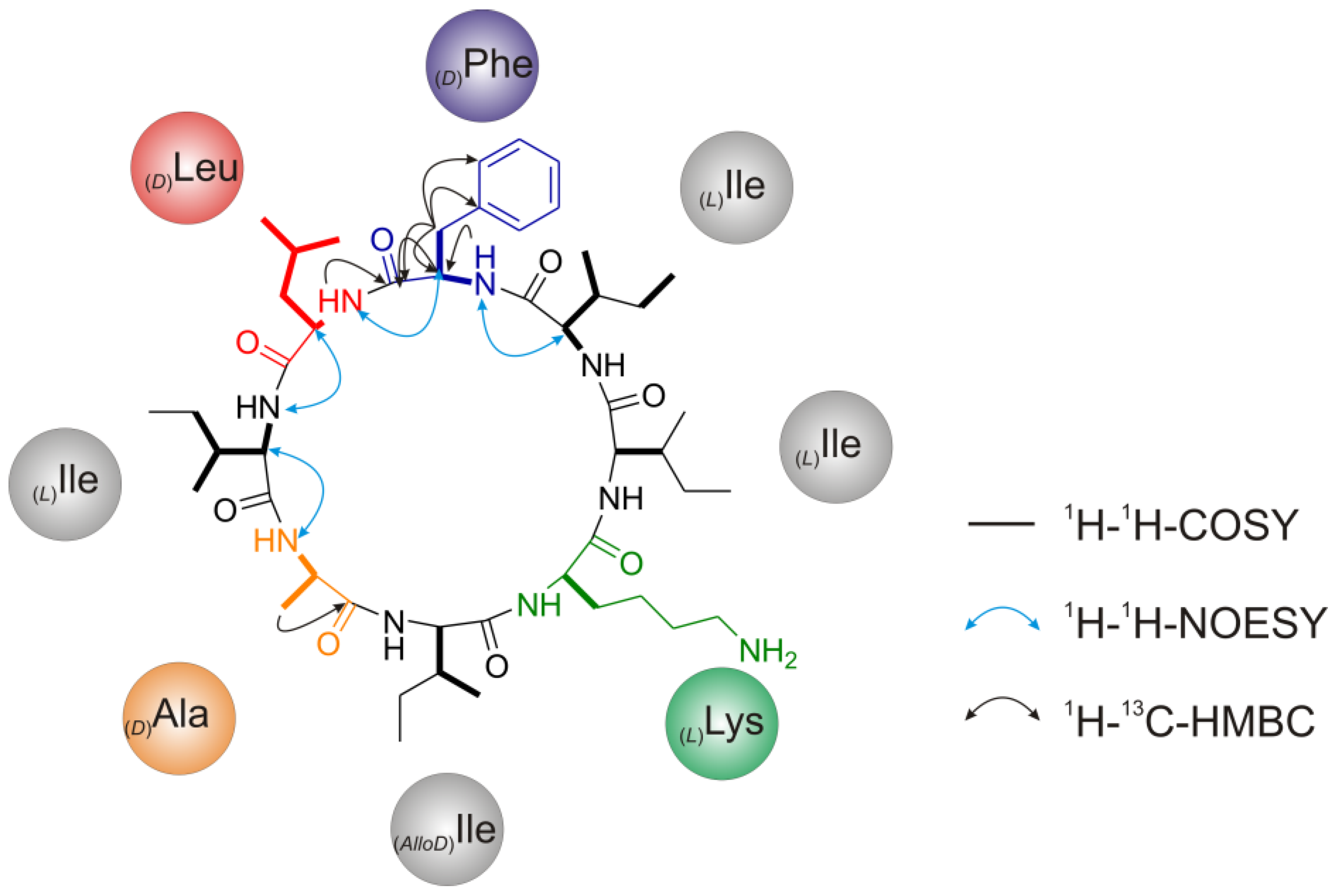
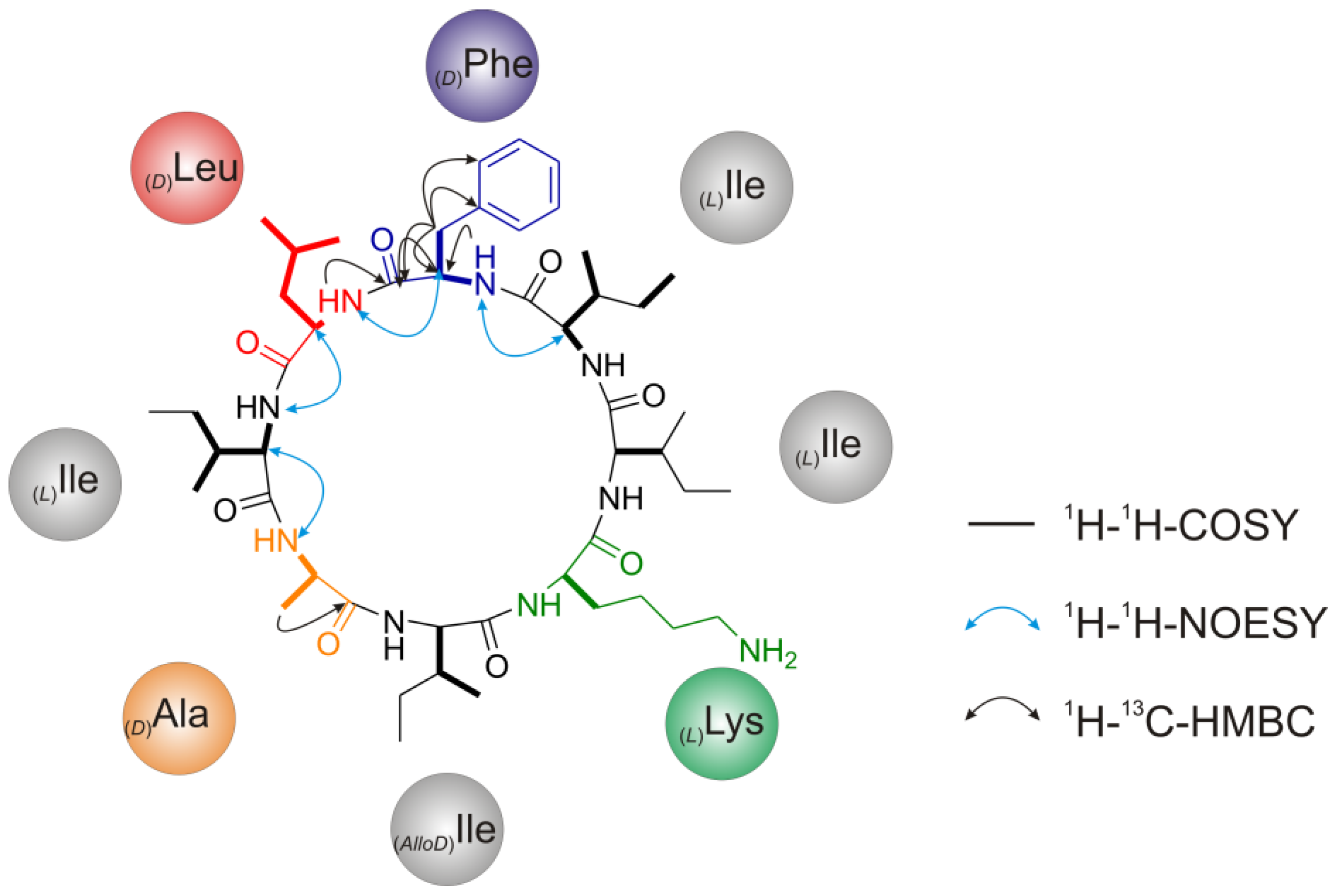
2.2. Structure Elucidation of Champacyclin (1a)
2.2.1. Quantitative Chiral Amino Acid Analysis
2.2.2. Mass Spectrometry
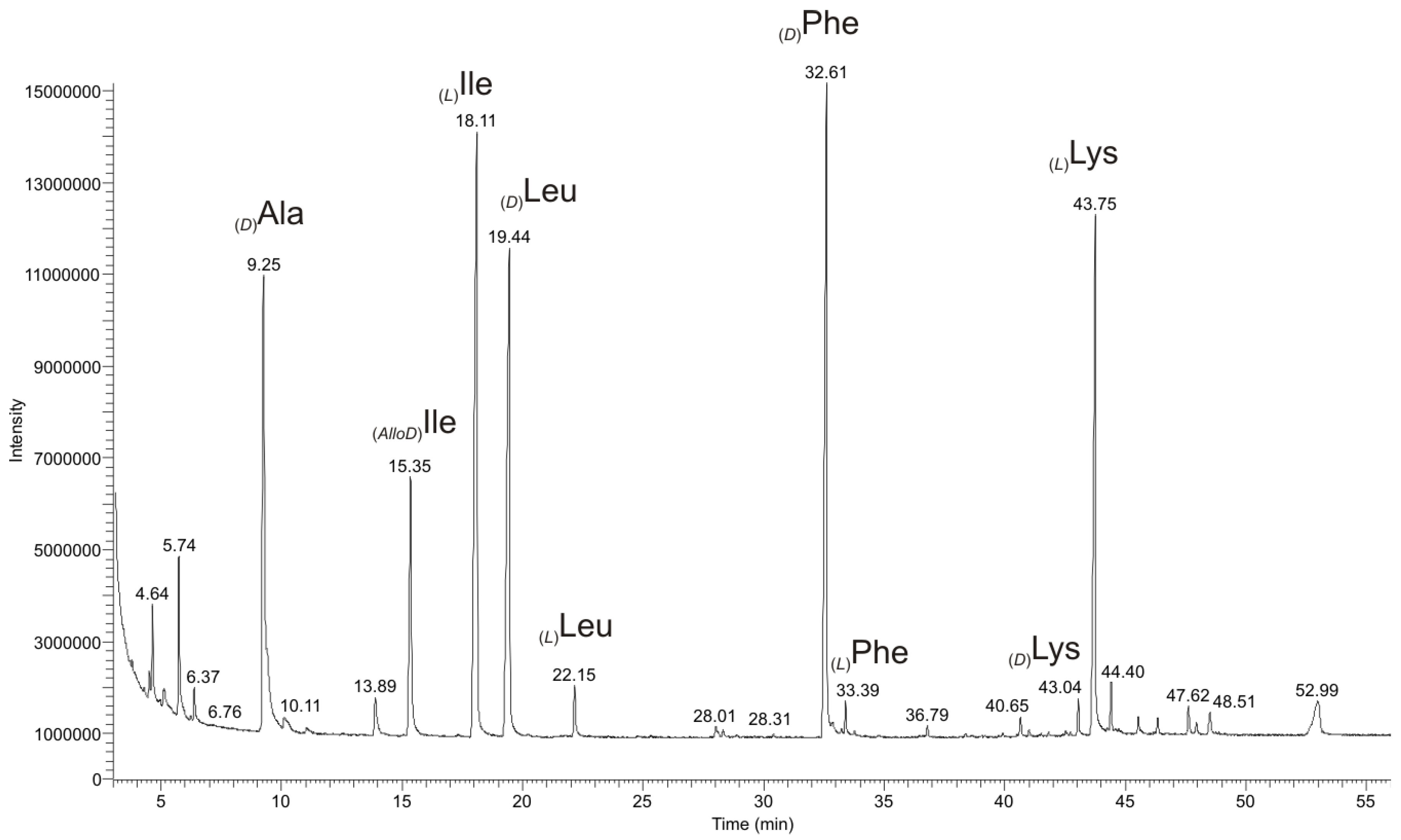

2.2.3. Chiral GC-PCI/EI-MS Analysis of the Partial Hydrolyzate of (1a)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthetic Dipeptides | Retention Times Rt (min) of Synthetic Dipeptides | Dipeptides from the Partial Hydrolyzate (1a) | Retention Times Rt (min) of Dipeptides from the Partial Hydrolyzate (1a) |
|---|---|---|---|
| (L)Ile-(D)Ala | 20.00 | (L)Ile-(D)Ala | 19.97 |
| (AlloD)Ile-(D)Ala | 20.35 | ||
| (L)Ile-(L)Ala | 20.40 | ||
| (AlloD)Ile-(L)Ala | 20.45 | ||
| (L)Ile-(L)Ile | 22.55 | (L)Ile-(L)Ile | 22.53 |
| (AlloD)Ile-(L)Ile | 23.40 | ||
| (L)Ile-(AlloD)Ile | 22.65 | ||
| (AlloD)Ile-(AlloD)Ile | 22.90 | ||
| (L)Ile-(D)Leu | 24.30 | ||
| (L)Ile-(L)Leu | 23.75 | ||
| (AlloD)Ile-(D)Leu | 24.90 | ||
| (AlloD)Ile-(L)Leu | 25.05 | ||
| (D)Leu-(L)Ile | 24.75 | (D)Leu-(L)Ile | 24.61 |
| (D)Leu-(AlloD)Ile | 24.00 | ||
| (L)Leu-(AlloD)Ile | 23.30 | ||
| (L)Leu-(L)Ile | 22.05 | ||
| (D)Leu-(D)Leu | 25.90 | ||
| (D)Leu-(L)Leu | 26.20 | ||
| (L)Leu-(L)Leu | 24.25 | ||
| (L)Leu-(D)Leu | 24.75 | ||
| (L)Ile-(D)Phe | 46.75 | (L)Ile-(D)Phe | 46.68 |
| (AlloD)Ile-(D)Phe | 44.80 | ||
| (L)Ile-(L)Phe | 40.90 | ||
| (AlloD)Ile-(L)Phe | 49.30 |
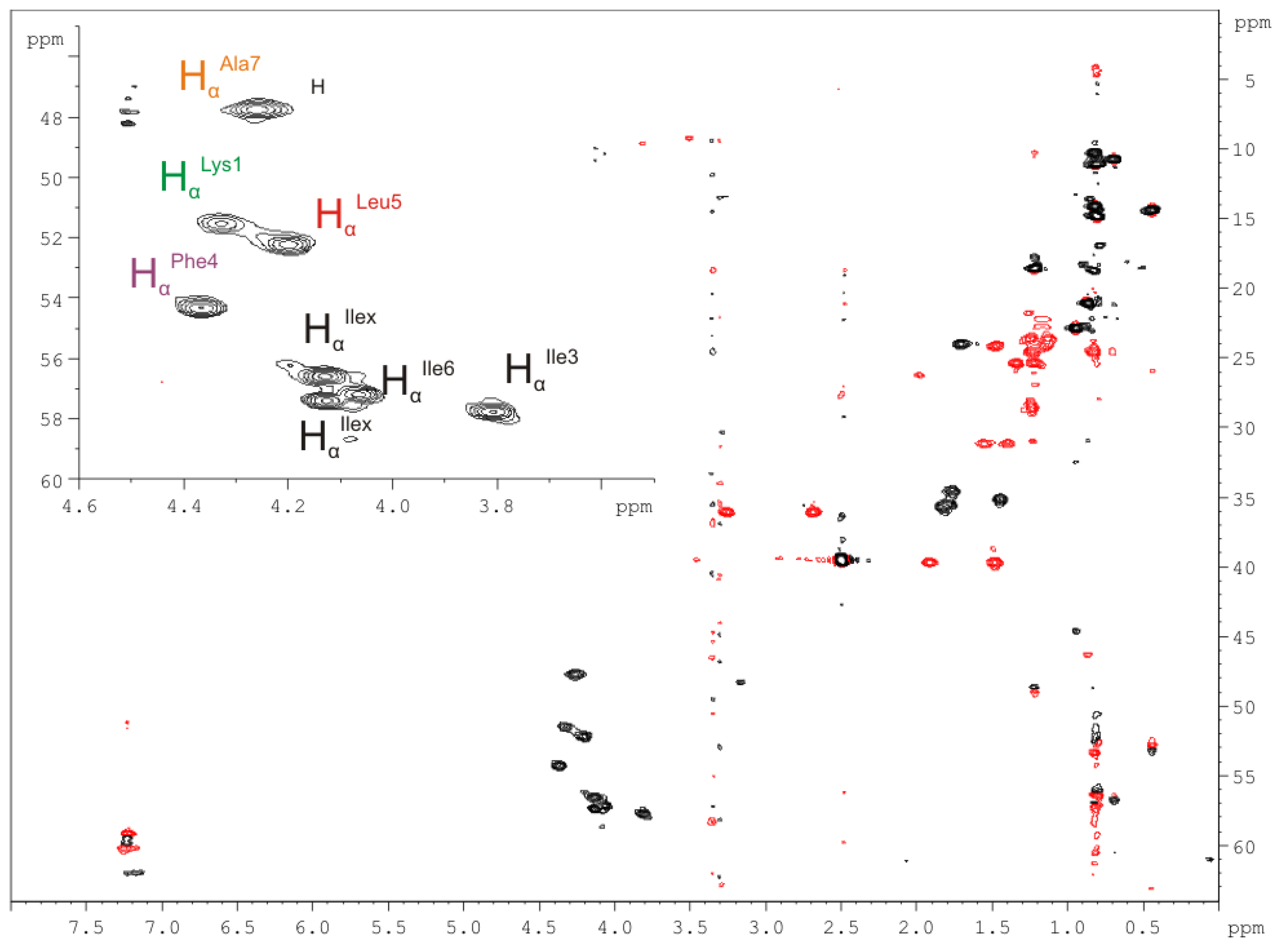
2.2.4. Nuclear Magnetic Resonance (NMR) Spectroscopy

| Amino Acid/Position | 1H NMR δ(1H) in ppm | 13C NMR δ(13C) in ppm (mult.) * | Amino Acid/Position | 1H NMR δ(1H) in ppm | 13C NMR δ(13C) in ppm (mult.) * |
|---|---|---|---|---|---|
| Lys(1) | Leu(5) | ||||
| C=O | − | n.d. | C=O | − | n.d. |
| NH | 7.40 | − | NH | 7.77 | − |
| Hα | 4.34 | 51.5, CH | Hα | 4.22 | 52.7, CH |
| Hβ/β′ | 1.55/1.40 | 31.6, CH2 | Hβ/β′ | 1.93/1.49 | 39.7, CH2 |
| Hγ/γ′ | n.d. | n.d. | Hγ | 1.72 | 24.1, CH |
| Hδ/δ′ | n.d. | n.d. | Hδ/δ′ | 0.96/0.89 | 22.9/21.4, CH2 |
| Hε/ε′ | n.d. | n.d. | |||
| NH2 | n.d. | n.d. | |||
| Ile(x) | Ile(6) | ||||
| C=O | − | n.d. | C=O | − | n.d. |
| NH | n.d. | − | NH | 6.91 | − |
| Hα | 4.15 | 57.1, CH | Hα | 4.05 | 57.7, CH |
| Hβ | 1.85 | 35.6, CH | Hβ | 1.82 | 35.5, CH |
| Hγ | 0.84 | 14.7, CH3 | Hγ | 0.80 | n.d. |
| Hγ′ | n.d. | n.d. | Hγ′ | 1.23/1.13 | 23.5, CH2 |
| Hδ | n.d. | n.d. | Hδ | n.d. | n.d. |
| Ile(3) | Ala(7) | ||||
| C=O | − | 171.4 | C=O | − | 172.4 |
| NH | 7.93 | − | NH | 7.78 | − |
| Hα | 3.78 | 57.7, CH | Hα | 4.28 | 47.7, CH |
| Hβ | 1.47 | 35.5, CH | Hβ/β′/β′′ | 1.25 | 18.4, CH3 |
| Hγ | 0.45 | 14.9, CH | |||
| Hγ′ | 1.22/0.83 | 24.1, CH2 | |||
| Hδ | 0.72 | 10.7, CH3 | |||
| Phe(4) | Ile(x) | ||||
| C=O | − | 171.0 | C=O | − | n.d. |
| NH | 8.40 | − | NH | n.d. | − |
| Hα | 4.33 | 54.8, CH | Hα | 4.14 | 57.9, CH |
| Hβ/β′ | 3.29/2.70 | 36.6, CH2 | Hβ | 1.85 | 35.6, CH |
| Hγ | − | 138.3, CH | Hγ | n.d. | n.d. |
| Hδ/δ′ | 7.20 | 129.4, CH | Hγ′ | 1.34/1.22 | 25.3, CH2 |
| Hε/ε′ | 7.26 | 128.1, CH | Hδ | n.d. | n.d. |
| Hζ | 7.36 | 126.3, CH |
2.2.5. Solid Phase Peptide Synthesis as a Proof of Head-to-Tail Cyclization of (1a)
3. Experimental Section
3.1. Sampling Sites
3.2. Isolation Procedure
3.3. Phylogenetic Classification
3.4. PCR for Non-Ribosomal Peptide Synthetases (NRPS)
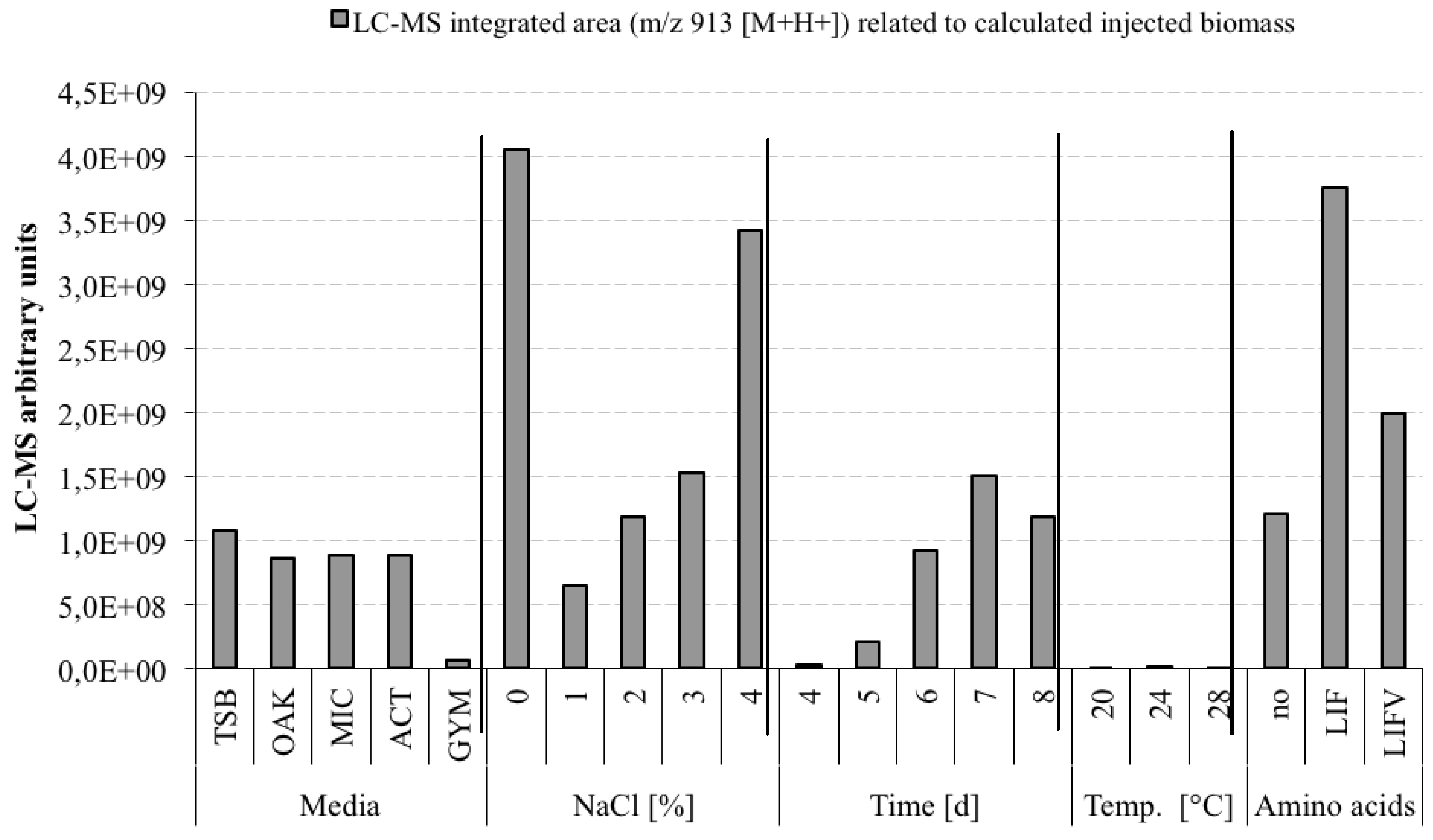
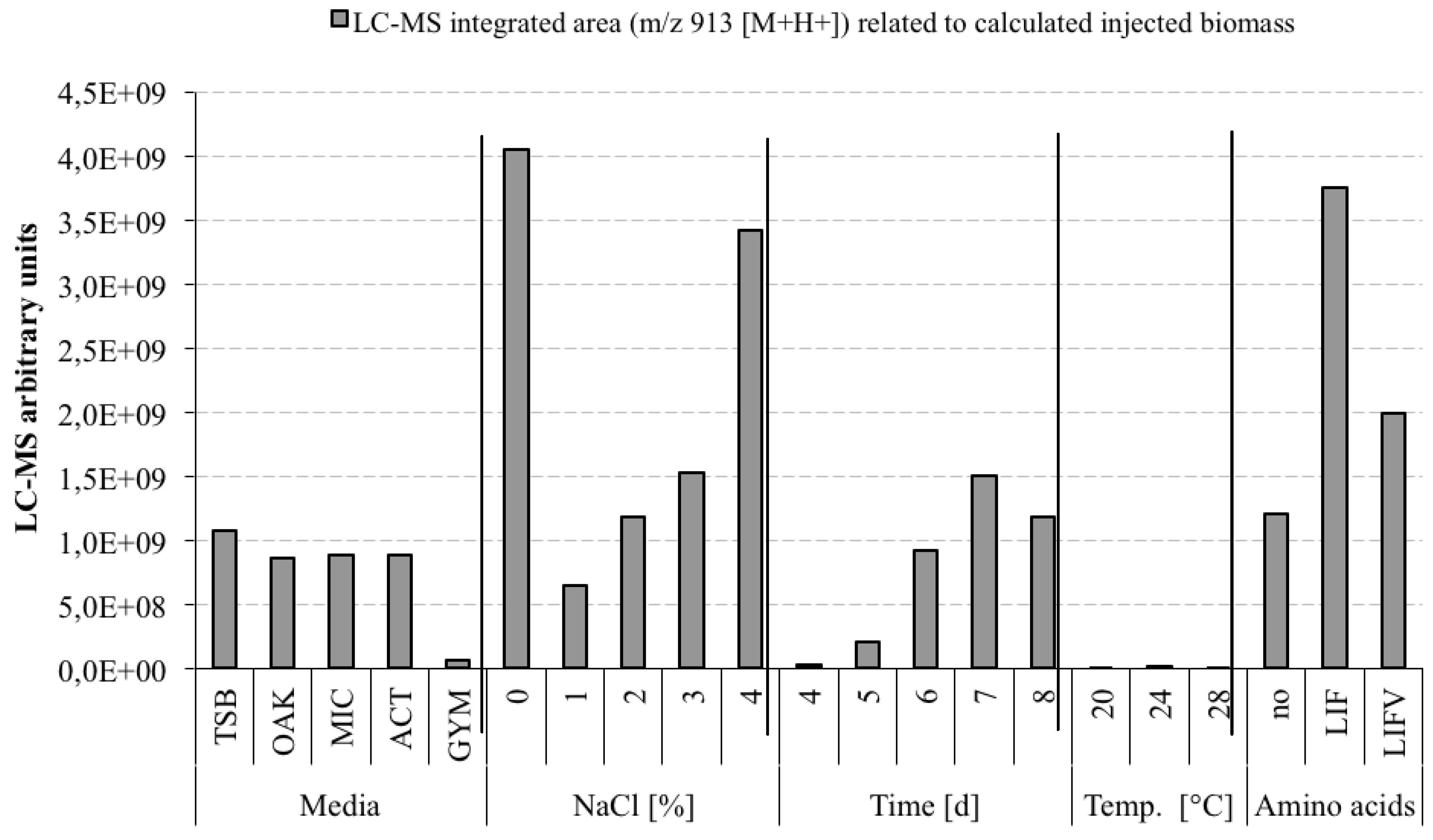
3.5. Optimization Experiments to Increase Champacyclin (1a) Synthesis
- TSB—Trypticase Soy Broth medium contained 3 g soybean casein digest broth (BD-BBL™ Trypticase™ Soy Broth, Dickinson and Company, Sparks, MD, USA), 10 g tropic marine salt in 1 L deionized water.
- OAK—Oak Flakes medium contained 20 g oak flakes and 3 mL trace element solution SL12 (3 g EDTA sodium salt, 42 mg ZnCl2, 50 mg MnCl2-tetrahydrate, 300 mg H3BO3, 190 mg CoCl2-hexahydrate, 1100 mg FeSO4 heptahydrate, 2 mg CuCl2-dihydrate, 24 mg NiCl2-hexahydrate, and 18 mg Na2Mo4-dihydrate in 1 L bidestilled water). The trace element solution was sterile filtered and added to the medium after autoclaving. The pH was 6.3 after autoclaving without adjustment.
- MIC—Micromonospora medium contained 10 g starch (VWR, Darmstadt, Germany), 5 g d-(+)-glucose-monohydrate (VWR), 4 g casein sodium salt from bovine milk (Sigma-Aldrich Chemie GmbH, Munich, Germany), 4 g yeast extract (BD-Bacto™, Dickinson and Company, Sparks, MD, USA), and 1 g calcium carbonate (Sigma-Aldrich Chemie GmbH) in 1 L bidestilled water. The pH was adjusted to 7.2 before autoclaving.
- ACT—Actinomycetes medium: contained 2 g casein sodium salt from bovine milk (Sigma-Aldrich Chemie GmbH, Munich, Germany), 4 g propionate acid sodium salt (Sigma-Aldrich Chemie GmbH), 0.1 g magnesium sulphate heptahydrate (VWR, Darmstadt, Germany), l-asparagine monohydrate (Roth, Karlsruhe, Germany), 0.5 g di-potassium hydrogenphosphate (VWR), 0.001 g ferric(III)-sulphate hydrate (Sigma-Aldrich Chemie GmbH), and 5 g glycerol (VWR) in 1 L sterile filtered Baltic Sea water (1.5% salt content). The pH was adjusted to 8.1 prior to autoclaving.
- GYM—GYM medium contained 4 g glucose, 4 g yeast extract, 4 g malt extract, 2 g CaCO3, 30 g tropic marine salt in 1 L bidestilled water. The pH of 7.2 was measured after autoclaving.
3.6. Determination of the Molar Absorption Coefficient
3.7. Antimicrobial Activity
3.8. Mass Spectrometry (MS)
3.9. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.10. Hydrolysis of (1a)
3.10.1. Partial Hydrolysis
3.10.2. Total Hydrolysis
3.11. Chiral GC/EI-PCI-MS Analysis
3.11.1. Amino Acid Analysis as N-Pentafluoropropionic 2-Propyl Amino Acid Derivatives
3.11.2. Dipeptide Analysis as N-Trifluoroacetyl Methyl Dipeptide Derivatives
3.12. Solid Phase Peptide Synthesis (SPPS)
3.12.1. Synthesis of Dipeptides
3.12.2. Synthesis of Head-to-Tail (1b) between Nα(L)Lys1-CO-(AlloD)Ile8 and Head-to-Side-Chain (2) between Nζ-(L)Lys-CO-(AlloD)Ile8 Cyclized Peptides for Mass Spectrometry
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Ribosomal peptide natural products: bridging the ribosomal and non-ribosomal worlds. Nat. Prod. Rep. 2009, 26, 537–559. [Google Scholar] [CrossRef]
- Bockhus, A.T.; McEwen, C.M.; Lokey, R.S. Form and function in cyclic peptide natural products: a pharmacokinetic perspective. Curr. Top. Med. Chem. 2013, 13, 821–836. [Google Scholar]
- Harrison, A.G.; Young, A.B.; Bleiholder, C.; Suhai, S.; Paizs, B. Scrambling of sequence information in collision-induced dissociation of peptides. J. Am. Chem. Soc. 2006, 128, 10364–10365. [Google Scholar]
- Kjeldsen, F.; Haselmann, K.F.; Sørensen, E.S.; Zubarev, R.A. Distinguishing of Ile/Leu amino acid residues in the PP3 protein by (hot) electron capture dissociation in Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2003, 75, 1267–1274. [Google Scholar] [CrossRef]
- Armirotti, A.; Milo, E.; Damonte, G. How to discriminate between leucine and isoleucine by low energy ESI-TRAP MSn. J. Am. Soc. Mass Spectrom. 2007, 18, 57–63. [Google Scholar] [CrossRef]
- Weygand, F.; Prox, A.; Fessel, H.H.; Kun Sun, K. Sequenzanalyse von Peptiden als N-Trifluoracetyl-peptid-ester. Z. Naturforschg. 1965, 20, 1169–1182. [Google Scholar]
- Pätzold, R.; Theis, C.; Brückner, H. Gas-chromatographic separation of steroisomers of dipeptides. Chirality 2006, 18, 551–557. [Google Scholar]
- Narasimha Rao, P.L.; Uma, B.N. Formation of antifungal antibiotic by a new Streptomyces species. Nature 1958, 182, 115–116. [Google Scholar]
- Rao, U.K.; Narasimha Rao, P.L. Actinomycetes. II. Purification and pharmacological properties of champamycin-B from Streptomyces champavatii. Indian J. Exp. Biol. 1967, 5, 39–42. [Google Scholar]
- Rong, X.; Guo, Y.; Huand, Y. Proposal to reclassify the Streptomyces albidoflavus clade on the basis of multilocus sequence analysis and DNA-DNA hybridization, and taxonomic elucidation of Streptomyces griseus subsp. solvifaciens. Syst. Appl. Microbiol. 2009, 32, 314–322. [Google Scholar] [CrossRef]
- Roepstorff, P.; Fohlman, J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 1984, 11, 601. [Google Scholar] [CrossRef]
- Rosenbaum, C.; Waldmann, H. Solid phase synthesis of cyclic peptides by oxidative cyclative cleavage of an aryl hydrazide linker—synthesis of stylostatin 1. Tetrahedron Lett. 2001, 42, 5677–5680. [Google Scholar] [CrossRef]
- Takada, K.; Ninomiya, A.; Naruse, M.; Sun, Y.; Miyazaki, M.; Nogi, Y.; Okada, S.; Matsunaga, S. Surugamides A–E, Cyclic Octapeptides with Four d-Amino Acid Residues, from a Marine Streptomyces sp.: LC-MS-Aided Inspection of Partial Hydrolysates for the Distinction of d- and l-Amino Acid Residues in the Sequence. J. Org. Chem. 2013, 78, 6746–6750. [Google Scholar] [CrossRef]
- Fan, L.; Liu, Y.; Li, Z.; Baumann, H.I.; Kleinschmidt, K.; Ye, W.; Imhoff, J.F.; Kleine, M.; Cai, D. Draft genome sequence of the marine Streptomyces sp. strain PP-C42, isolated from the Baltic Sea. J. Bacteriol. 2011, 193, 3691–3692. [Google Scholar]
- Pätzold, J.; Halbach, P.E.; Hempel, G.; Weikert, H. METEOR-Berichte 00-3. Östliches Mittelmeer—Nördliches Rotes Meer 1999. Cruise No. 44. 22 January–16 May 1999; Leitstelle Meteor: Hamburg, Germay, 1999. [Google Scholar]
- Noda, S.; Inoue, T.; Hongoh, Y.; Kawai, M.; Nalepa, C.A.; Vongkaluang, C.; Kudo, T.; Ohkuma, M. Identification and characterization of ectosymbionts of distinct lineages in Bacteroidales attached to flagellated protists in the gut of termites and a wood-feeding cockroach. Environ. Microbiol. 2006, 8, 11–20. [Google Scholar] [CrossRef]
- Reysenbach, A.L.; Longnecker, K.; Kirshtein, J. Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a Mid-Atlantic Ridge hydrothermal vent. Appl. Environ. Microbiol. 2000, 66, 3798–3806. [Google Scholar] [CrossRef]
- Wiese, J.; Thiel, V.; Nagel, K.; Staufenberger, T.; Imhoff, J.F. Diversity of antibiotic-active bacteria associated with the brown alga Laminaria saccharina from the Baltic Sea. Mar. Biotechnol. 2009, 11, 287–300. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Klam-Syed-Mohideen, A.S.; McCarell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef]
- Tatsuova, T.A.; Madden, T.L. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999, 174, 247–250. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Perrière, G.; Goug, M. WWW-Query: An on-line retrieval system for biological sequence banks. Biochemie 1996, 78, 364–369. [Google Scholar] [CrossRef]
- Doekel, S.; Marahiel, M.A. Biosynthesis of natural products on modular peptide synthetases. Metab. Eng. 2001, 3, 64–77. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pesic, A.; Baumann, H.I.; Kleinschmidt, K.; Ensle, P.; Wiese, J.; Süssmuth, R.D.; Imhoff, J.F. Champacyclin, a New Cyclic Octapeptide from Streptomyces Strain C42 Isolated from the Baltic Sea. Mar. Drugs 2013, 11, 4834-4857. https://doi.org/10.3390/md11124834
Pesic A, Baumann HI, Kleinschmidt K, Ensle P, Wiese J, Süssmuth RD, Imhoff JF. Champacyclin, a New Cyclic Octapeptide from Streptomyces Strain C42 Isolated from the Baltic Sea. Marine Drugs. 2013; 11(12):4834-4857. https://doi.org/10.3390/md11124834
Chicago/Turabian StylePesic, Alexander, Heike I. Baumann, Katrin Kleinschmidt, Paul Ensle, Jutta Wiese, Roderich D. Süssmuth, and Johannes F. Imhoff. 2013. "Champacyclin, a New Cyclic Octapeptide from Streptomyces Strain C42 Isolated from the Baltic Sea" Marine Drugs 11, no. 12: 4834-4857. https://doi.org/10.3390/md11124834
APA StylePesic, A., Baumann, H. I., Kleinschmidt, K., Ensle, P., Wiese, J., Süssmuth, R. D., & Imhoff, J. F. (2013). Champacyclin, a New Cyclic Octapeptide from Streptomyces Strain C42 Isolated from the Baltic Sea. Marine Drugs, 11(12), 4834-4857. https://doi.org/10.3390/md11124834