Molecular Docking Studies of Marine Diterpenes as Inhibitors of Wild-Type and Mutants HIV-1 Reverse Transcriptase

 ,
,
Abstract
:1. Introduction
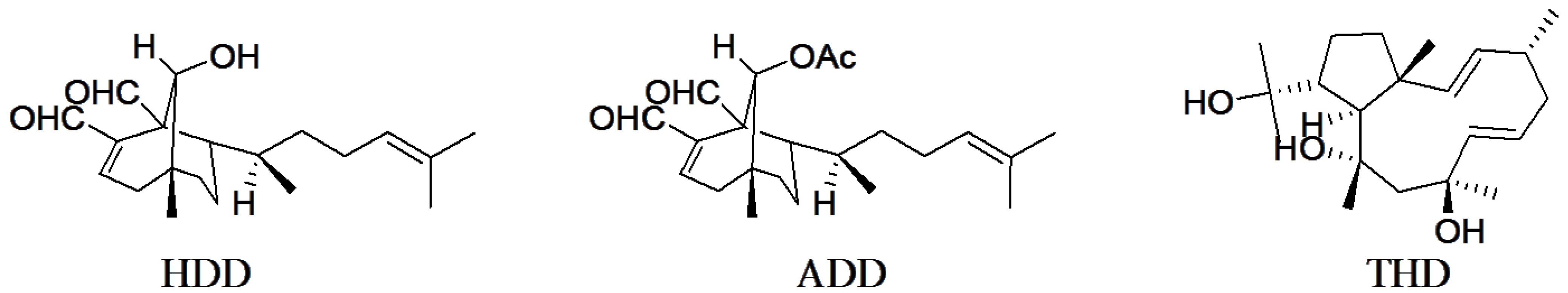
2. Results and Discussion
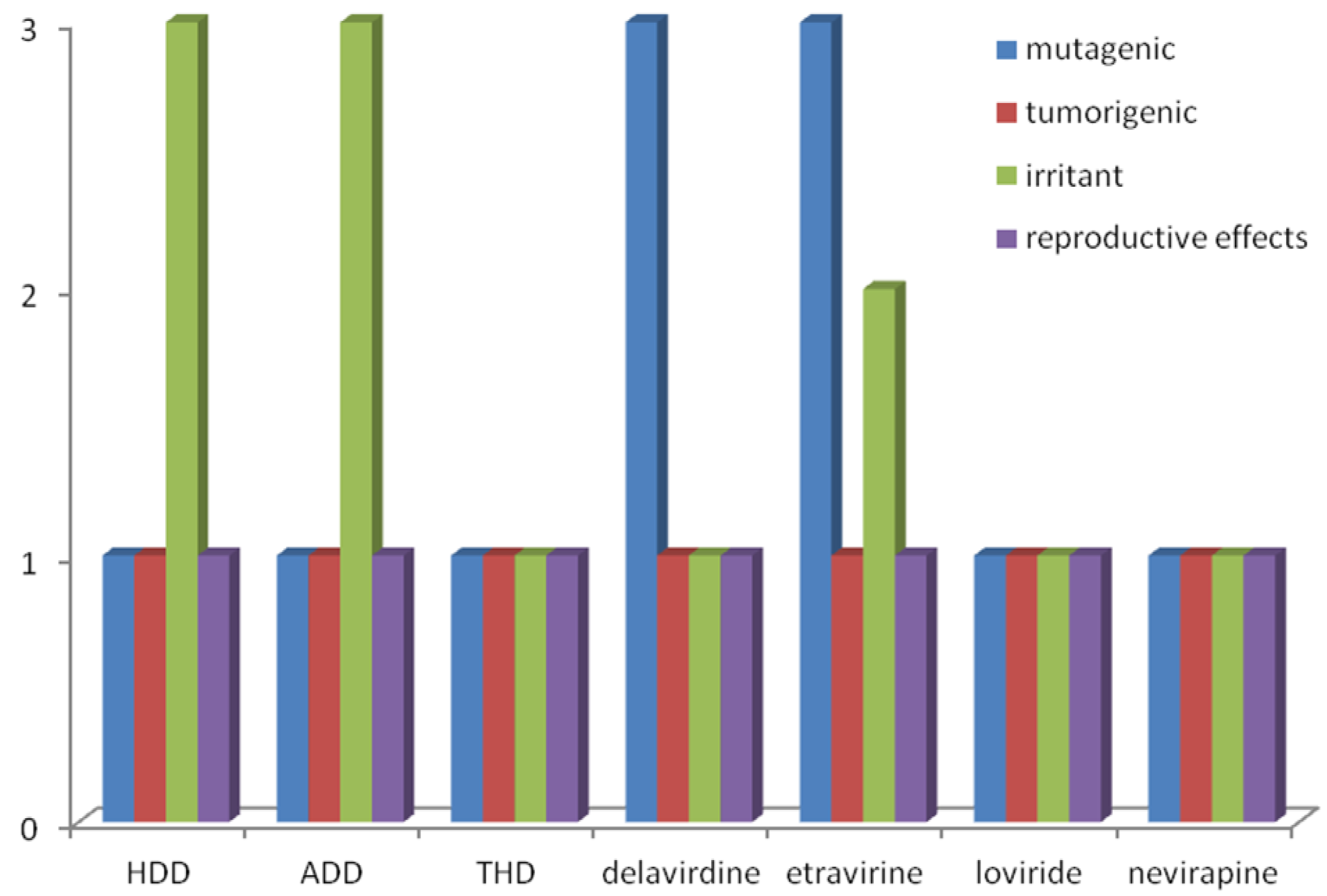
2.1. Structure-Activity Relationship and ADMET Evaluation of Diterpenes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | EC50 (µM) a | IC50 (µM) b | CC50 (µM) a | SI c | µ (D) | EHOMO (eV) | ELUMO (eV) | HOMO-LUMO gap (eV) d | Lipinski’ Rule-of-5 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cLogP | MM (Da) | HBA | HBD | |||||||||
| NVP | 0.4 | 0.5 | >100 | >250 | 2.67 | −8.50 | 2.67 | 11.17 | 2.91 | 260 | 5 | 1 |
| HDD | 40.0 | 10.0 | >200 | >5 | 4.40 | −8.90 | 2.51 | 11.41 | 3.95 | 318 | 3 | 1 |
| ADD | 70.0 | 35.0 | >200 | >2.86 | 5.28 | −8.83 | 2.60 | 11.43 | 4.44 | 360 | 3 | 0 |
| THD | 8.4 | 16.5 | 500 | 59.5 | 1.73 | −8.84 | 4.31 | 13.15 | 3.59 | 322 | 3 | 3 |

2.2. Molecular Docking
2.2.1. Validation of the Docking Performance and Accuracy
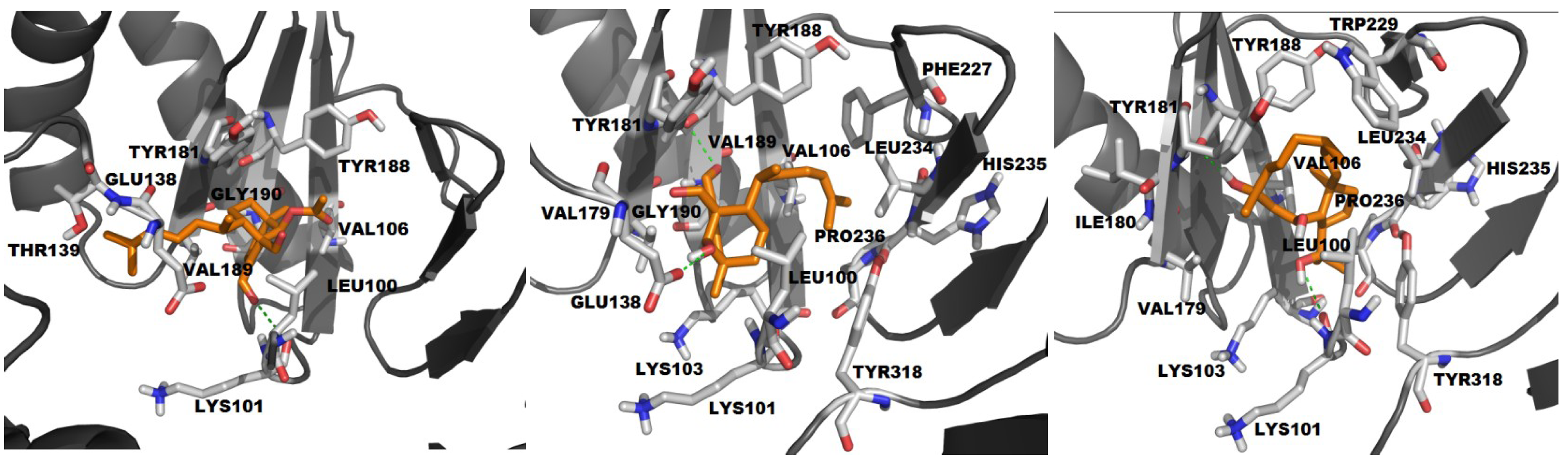
2.2.2. Molecular Docking with HIV-1 RT Wild Type

| # | BE | NI | NC | CE | vdW | H-bond |
|---|---|---|---|---|---|---|
| ADD | −3.88 | 10 | 5 | 23, 21, 2, 3, 1 | L100, V106, V179, Y181, Y188, V189, G190, E138, T139 | K101 (2.21) |
| HDD | −4.96 | 16 | 5 | 13, 11, 17, 8, 1 | L100, K101, K103, V106, V179, Y181, Y188, V189, G190, F227, L234, H235, P236, Y318 | Y188 (2.73) E138 (1.97) |
| THD | −6.3 | 13 | 1 | 50 | L100, K103, V106, V179, I180, Y181, W229, L234, H235, P236, Y318 | K101 (3.06) Y188 (2.70) |
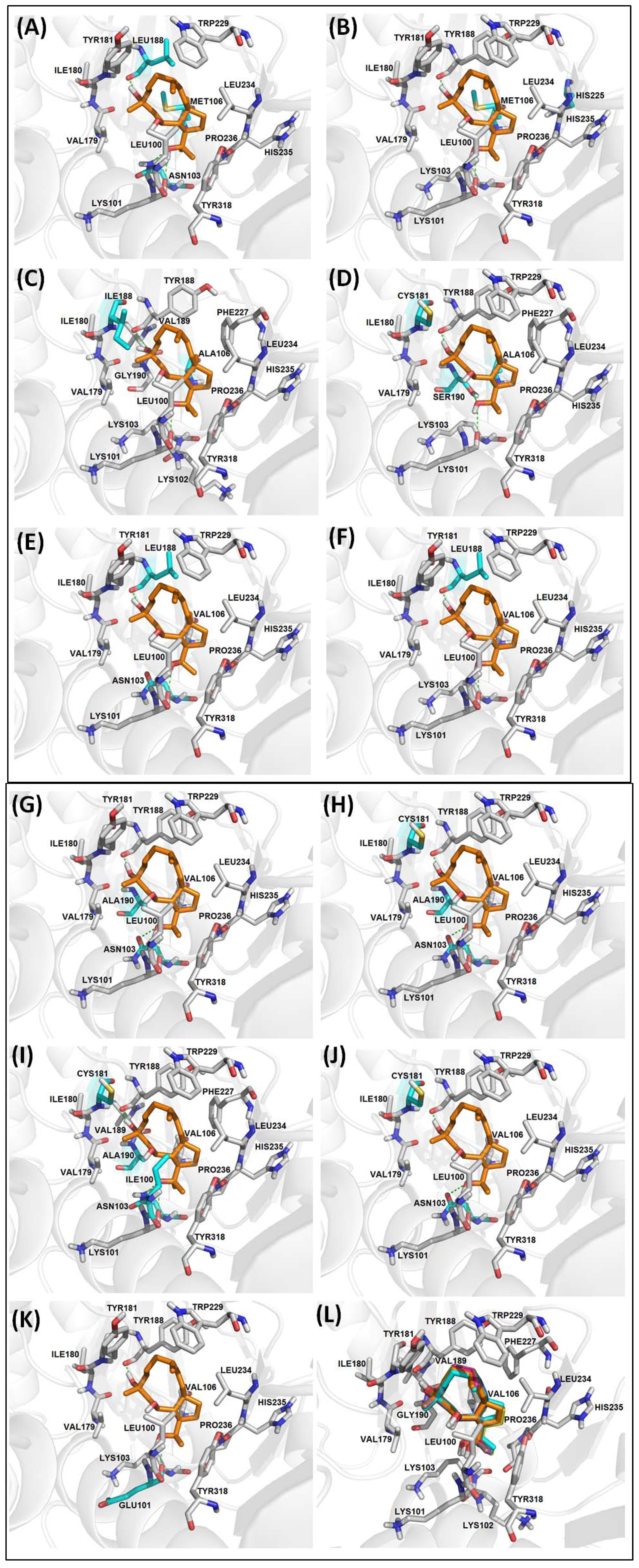
2.2.3. Diterpenes and HIV-1 RT Mutants Complexes Evaluation

| RT | Mutation | BE | NI | NC | CE | vdW | H-bond |
|---|---|---|---|---|---|---|---|
| WT | None | −6.30 | 13 | 1 | 50 | L100, K103, V106, V179, I180, Y181, W229, L234, H235, P236, Y318 | K101 (3.06), Y188 (2.70) |
| RT-1 | K103N, V106M, Y188L | −6.03 | 13 | 2 | 49, 1 | L100, M106, V179, I180, Y181, W229, L234, H235, P236, Y318 | K101 (3.14), N103 (2.77), L188 (2.76) |
| RT-2 | V106M, P225H | −6.14 | 13 | 1 | 50 | L100, K103, M106, V179, I180, Y181, W229, L234, H235, P236, Y318 | K101 (3.08), Y188 (2.75) |
| RT-3 | V106A, Y181I | −5.07 | 15 | 5 | 47 | L100, K102, K103, V179, I180, I181, V189, G190, F227, L234, H235, P236, Y318 | K101 (2.97), Y188 (2.57) |
| RT-4 | V106A, Y181C, G190S | −6.01 | 13 | 1 | 50 | L100, K103, V179, I180, C181, W229, F227, L234, H235, P236, Y318 | K101 (3.28), Y188 (2.73), S190 (3.21) |
| RT-5 | K103N, Y188L | −5.41 | 13 | 1 | 50 | L100, K103, V106, V179, I180, Y181, W229, L234, H235, P236, Y318 | K101 (2.67), L188 (2.70) |
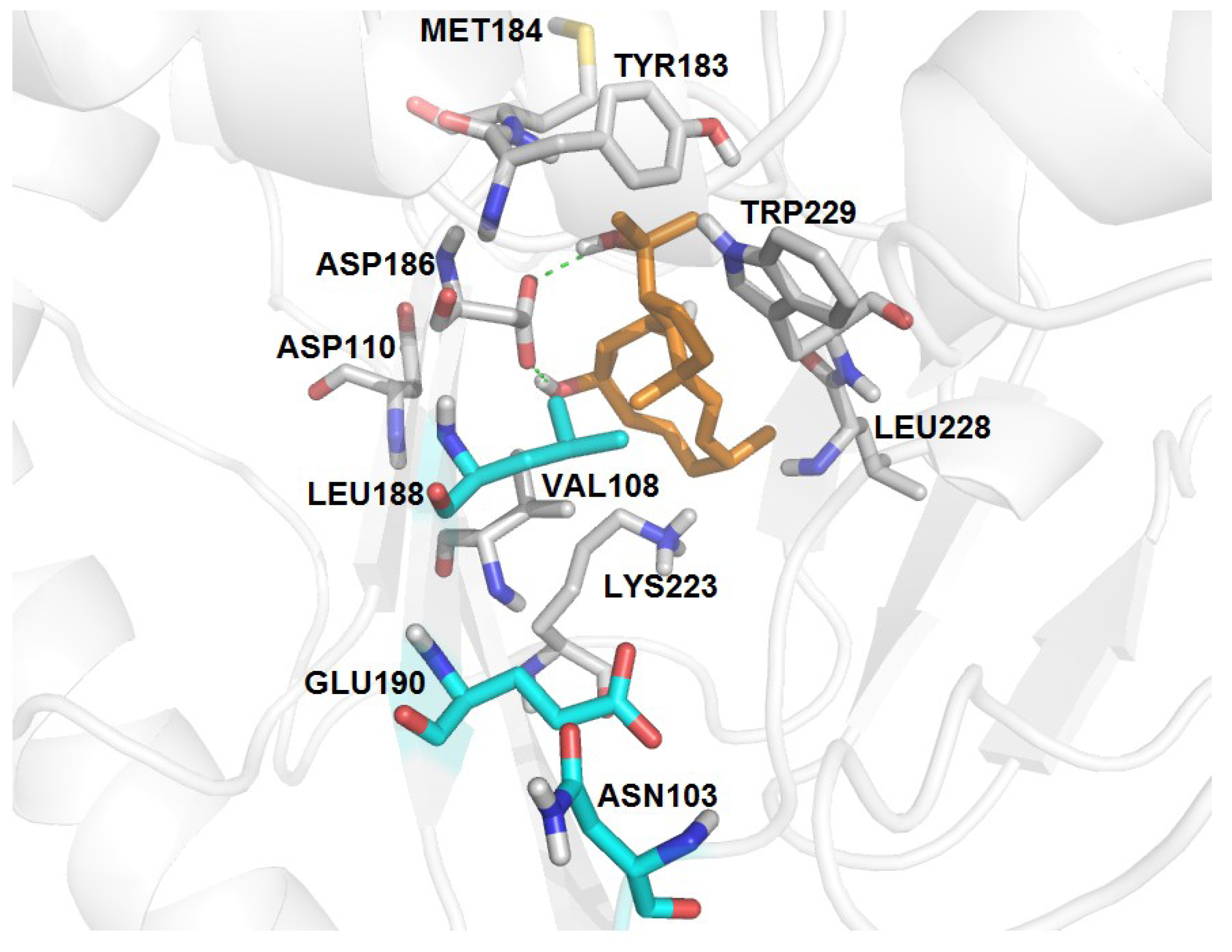
| RT-6 | K103N, Y188L, G190E | −3.18 | 07 | 13 | 16 | V108, Y183, M184, K223, L228, W29 | D186 (2.80, 3.14) |
| RT-7 | Y188L | −5.65 | 13 | 2 | 49, 1 | L100, K103, V106, V179, I180, Y181, W229, L234, H235, P236, Y318 | K101 (2.94), L188 (2.74) |
| RT-8 | K103N, G190A | −6.13 | 14 | 1 | 50 | L100, V106, V179, I180, Y181, A190, W229, L234, H235, P236, Y318 | K101 (3.09), N103(2.95), Y188(2.78) |
| RT-9 | K103N, Y181C, G190A | −5.92 | 14 | 1 | 50 | L100, V106, V179, I180, C181, A190, W229, L234, H235, P236, Y318 | K101 (3.10), N103 (2.97), Y188 (2.71) |
| RT-10 | L100I, K103N, Y181C, G190A | −5.99 | 16 | 2 | 35, 15 | I100, V106, V179, I180, Y181, V189, A190, F227, W229, L234, H235, P236, Y318 | K101 (2.98), N103 (2.59), Y188 (2.68) |
| RT-11 | K103N, Y181C | −5.72 | 13 | 1 | 50 | L100, V106, V179, I180, C181, W229, L234, H235, P236, Y318 | K101 (3.09), N103 (2.98), Y188 (2.71) |
| RT-12 | K101E | −6.03 | 13 | 1 | 50 | K103, V106, V179, I180, Y181, W229, L234, H235, P236, Y318 | E101(3.06), Y188(2.76) |

3. Experimental Section
3.1. Diterpene Structures
3.2. Molecular Docking
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- WHO Progress Report 2011: Global HIV/AIDS Response. Available online: http://www.who.int/hiv/pub/progress_report2011/en/index.html (accessed on 14 August 2012).
- Ammaranond, P.; Sanguansittianan, S. Mechanism of HIV antiretroviral drugs progress toward drug resistance. Fundam. Clin. Pharm. 2012, 26, 146–161. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat. Rev. Micro. 2012, 10, 279–290. [Google Scholar] [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar]
- Castro, H.C.; Loureiro, N.I.V.; Pujol-Luz, M.; Souza, A.M.T.; Albuquerque, M.G.; Santos, D.O.; Cabral, L.M.; Frugulhetti, I.C.; Rodrigues, C.R. HIV-1 reverse transcriptase: A therapeutical target in the spotlight. Curr. Med. Chem. 2006, 13, 313–324. [Google Scholar] [CrossRef]
- Tan, J.J.; Cong, X.J.; Hu, L.M.; Wang, C.X.; Jia, L.; Liang, X.-J. Therapeutic strategies underpinning the development of novel techniques for the treatment of HIV infection. Drug Discov. Today 2010, 15, 186–197. [Google Scholar] [CrossRef]
- La Regina, G.; Coluccia, A.; Silvestri, R. Looking for an active conformation of the future HIV type-1 non-nucleoside reverse transcriptase inhibitors. Antivir. Chem. Chemother. 2010, 20, 213–237. [Google Scholar] [CrossRef]
- Corbett, J.W.; Ko, S.S.; Rodgers, J.D.; Jeffrey, S.; Bacheler, L.T.; Klabe, R.M.; Diamond, S.; Lai, C.M.; Rabel, S.R.; Saye, J.A.; et al. Expanded-spectrum nonnucleoside reverse transcriptase inhibitors inhibit clinically relevant mutant variants of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 1999, 43, 2893–2897. [Google Scholar]
- Hopkins, A.L.; Ren, J.; Tanaka, H.; Baba, M.; Okamato, M.; Stuart, D.I.; Stammers, D.K. Design of MKC-442 (Emivirine) analogues with improved activity against drug-resistant HIV mutants. J. Med. Chem. 1999, 42, 4500–4505. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Wang, D.P.; Tirado-Rives, J.; Jorgensen, W.L. Validation of a model for the complex of HIV-1 reverse transcriptase with sustiva through computation of resistance profiles. J. Am. Chem. Soc. 2000, 122, 12898–12900. [Google Scholar]
- El-Brollosy, N.R.; Jørgensen, P.T.; Dahan, B.; Boel, A.M.; Pedersen, E.B.; Nielsen, C. Synthesis of novel N-1 (Allyloxymethyl) analogues of 6-benzyl-1-(ethoxymethyl)-5-isopropyluracil (MKC-442, Emivirine) with improved activity against HIV-1 and its mutants. J. Med. Chem. 2002, 45, 5721–5726. [Google Scholar] [CrossRef]
- Das, K.; Clark, A.D.; Lewi, P.J.; Heeres, J.; de Jonge, M.R.; Koymans, L.M.H.; Vinkers, H.M.; Daeyaert, F.; Ludovici, D.W.; Kukla, M.J.; et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [Google Scholar] [CrossRef]
- Himmel, D.M.; Das, K.; Clark, A.D.; Hughes, S.H.; Benjahad, A.; Oumouch, S.; Guillemont, J.; Coupa, S.; Poncelet, A.; Csoka, I.; et al. Crystal structures for HIV-1 reverse transcriptase in complexes with three Pyridinone derivatives: A new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J. Med. Chem. 2005, 48, 7582–7591. [Google Scholar] [CrossRef]
- Rodríguez-Barrios, F.; Balzarini, J.; Gago, F. The molecular basis of resilience to the effect of the Lys103Asn mutation in non-nucleoside HIV-1 reverse transcriptase inhibitors studied by targeted molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 7570–7578. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Ruiz-Caro, J.; Tirado-Rives, J.; Basavapathruni, A.; Anderson, K.S.; Hamilton, A.D. Computer-Aided design of non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2006, 16, 663–667. [Google Scholar] [CrossRef]
- Ren, J.; Nichols, C.E.; Stamp, A.; Chamberlain, P.P.; Ferris, R.; Weaver, K.L.; Short, S.A.; Stammers, D.K. Structural insights into mechanisms of non-nucleoside drug resistance for HIV-1 reverse transcriptases mutated at codons 101 or 138. FEBS J. 2006, 273, 3850–3860. [Google Scholar] [CrossRef]
- Gagnon, A.; Amad, M.H.; Bonneau, P.R.; Coulombe, R.; DeRoy, P.L.; Doyon, L.; Duan, J.; Garneau, M.; Guse, I.; Jakalian, A.; et al. Thiotetrazole alkynylacetanilides as potent and bioavailable non-nucleoside inhibitors of the HIV-1 wild type and K103N/Y181C double mutant reverse transcriptases. Bioorg. Med. Chem. Lett. 2007, 17, 4437–4441. [Google Scholar] [CrossRef]
- Gerber, P.; Dutcher, J.D.; Adams, E.V.; Sherman, J.H. Protective effect of seaweed extracts for chicken embryos infected with influenza B or mumps virus. Proc. Soc. Exp. Biol. Med. 1958, 99, 590–593. [Google Scholar] [CrossRef]
- Bongiorni, L.; Pietra, F. Marine natural products for industrial applications. Chem. Ind. 1996, 2, 54–58. [Google Scholar]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar]
- Balunas, M.J.; Kinghorn, A.D. Drug discovery from medicinal plants. Life Sci. 2005, 78, 431–441. [Google Scholar] [CrossRef]
- Paula, J.C.D.; Vallim, M.A.; Teixeira, V.L. What are and where are the bioactive terpenoids metabolites from Dictyotaceae (Phaeophyceae). Rev. Bras. Farmacogn. 2011, 21, 216–228. [Google Scholar] [CrossRef]
- Pereira, H.S.; Leão-Ferreira, L.R.; Moussatché, N.; Teixeira, V.L.; Cavalcanti, D.N.; Costa, L.J.; Diaz, R.; Frugulhetti, I.C.P.P. Antiviral activity of diterpenes isolated from the Brazilian marine alga Dictyota menstrualis against human immunodeficiency virus type 1 (HIV-1). Antivir. Res. 2004, 64, 69–76. [Google Scholar]
- Pata, J.D.; Stirtan, W.G.; Goldstein, S.W.; Steitz, T.A. Structure of HIV-1 reverse transcriptase bound to an inhibitor active against mutant reverse transcriptases resistant to other nonnucleoside inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 10548–10553. [Google Scholar] [CrossRef]
- Barbosa, J.P.; Teixeira, V.L.; Villaça, R.; Pereira, R.C.; Abrantes, J.L.; Frugulhetti, I.C.P.P. A dolabellane diterpene from the Brazilian brown alga Dictyota pfaffii. Biochem. Syst. Ecol. 2003, 31, 1451–1453. [Google Scholar] [CrossRef]
- Cirne-Santos, C.C.; Teixeira, V.L.; Castello-Branco, L.R.; Frugulhetti, I.C.P.P.; Bou-Habib, D.C. Inhibition of HIV-1 replication in human primary cells by a dolabellane diterpene isolated from the marine algae Dictyota pfaffii. Planta Medica 2006, 72, 295–299. [Google Scholar] [CrossRef]
- Zhan, P.; Chen, X.; Li, D.; Fang, Z.; de Clercq, E.; Liu, X. HIV-1 NNRTIs: Structural diversity, pharmacophore similarity, and implications for drug design. Med. Res. Rev. 2011, 33. [Google Scholar] [CrossRef]
- Cirne-Santos, C.C.; Souza, T.M.L.; Teixeira, V.L.; Fontes, C.F.L.; Rebello, M.A.; Castello-Branco, L.R.R.; Abreu, C.M.; Tanuri, A.; Frugulhetti, I.C.P.P.; Bou-Habib, D.C. The dolabellane diterpene Dolabelladienetriol is a typical noncompetitive inhibitor of HIV-1 reverse transcriptase enzyme. Antivir. Res. 2008, 77, 64–71. [Google Scholar] [CrossRef]
- Organic Chemistry Portal—OSIRIS Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 14 February 2013).
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Das, K.; Lewi, P.J.; Hughes, S.H.; Arnold, E. Crystallography and the design of anti-AIDS drugs: Conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophy. Mol. Biol. 2005, 88, 209–231. [Google Scholar] [CrossRef]
- Almerico, A.M.; Tutone, M.; Lauria, A. Docking and multivariate methods to explore HIV-1 drug-resistance: A comparative analysis. J. Comput. Aided Mol. Des. 2008, 22, 287–297. [Google Scholar] [CrossRef]
- De Béthune, M.P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antivir. Res. 2010, 85, 75–90. [Google Scholar] [CrossRef]
- Messiaen, P.; Verhofstede, C.; Vandenbroucke, I.; Dinakis, S.; Van Eygen, V.; Thys, K.; Winters, B.; Aerssens, J.; Vogelaers, D.; Stuyver, L.J.; et al. Ultra-deep sequencing of HIV-1 reverse transcriptase before start of an NNRTI-based regimen in treatment-naive patients. Virology 2012, 426, 7–11. [Google Scholar] [CrossRef]
- Wang, D.P.; Rizzo, R.C.; Tirado-Rives, J.; Jorgensen, W.L. Antiviral drug design: Computational analyses of the effects of the L100I mutation for HIV-RT on the binding of NNRTIs. Bioorg. Med. Chem. Lett. 2001, 11, 2799–2802. [Google Scholar] [CrossRef]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef]
- Wang, J.; Liang, H.; Bacheler, L.; Wu, H.; Deriziotis, K.; Demeter, L.M.; Dykes, C. The non-nucleoside reverse transcriptase inhibitor efavirenz stimulates replication of human immunodeficiency virus type 1 harboring certain non-nucleoside resistance mutations. Virology 2010, 402, 228–237. [Google Scholar] [CrossRef]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef]
- RCSB PDB Protein Data Bank. Available online: http://www.rcsb.org/pdb (accessed on 25 February 2013).
- Ding, J.; Das, K.; Moereels, H.; Koymans, L.; Andries, K.; Janssen, P.A.J.; Hughes, S.H.; Arnold, E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Mol. Biol. 1995, 2, 407–415. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miceli, L.A.; Teixeira, V.L.; Castro, H.C.; Rodrigues, C.R.; Mello, J.F.R.; Albuquerque, M.G.; Cabral, L.M.; De Brito, M.A.; De Souza, A.M.T. Molecular Docking Studies of Marine Diterpenes as Inhibitors of Wild-Type and Mutants HIV-1 Reverse Transcriptase. Mar. Drugs 2013, 11, 4127-4143. https://doi.org/10.3390/md11114127
Miceli LA, Teixeira VL, Castro HC, Rodrigues CR, Mello JFR, Albuquerque MG, Cabral LM, De Brito MA, De Souza AMT. Molecular Docking Studies of Marine Diterpenes as Inhibitors of Wild-Type and Mutants HIV-1 Reverse Transcriptase. Marine Drugs. 2013; 11(11):4127-4143. https://doi.org/10.3390/md11114127
Chicago/Turabian StyleMiceli, Leonardo A., Valéria L. Teixeira, Helena C. Castro, Carlos R. Rodrigues, Juliana F. R. Mello, Magaly G. Albuquerque, Lucio M. Cabral, Monique A. De Brito, and Alessandra M. T. De Souza. 2013. "Molecular Docking Studies of Marine Diterpenes as Inhibitors of Wild-Type and Mutants HIV-1 Reverse Transcriptase" Marine Drugs 11, no. 11: 4127-4143. https://doi.org/10.3390/md11114127