Fucoidan Extract Enhances the Anti-Cancer Activity of Chemotherapeutic Agents in MDA-MB-231 and MCF-7 Breast Cancer Cells

Abstract
:1. Introduction
2. Results and Discussion
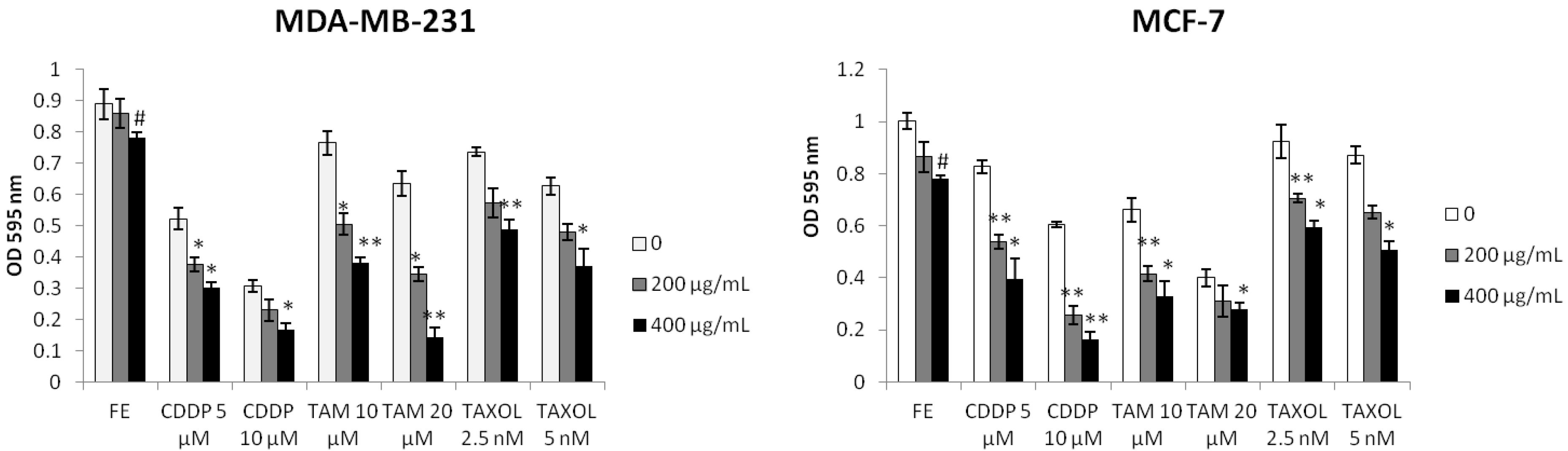
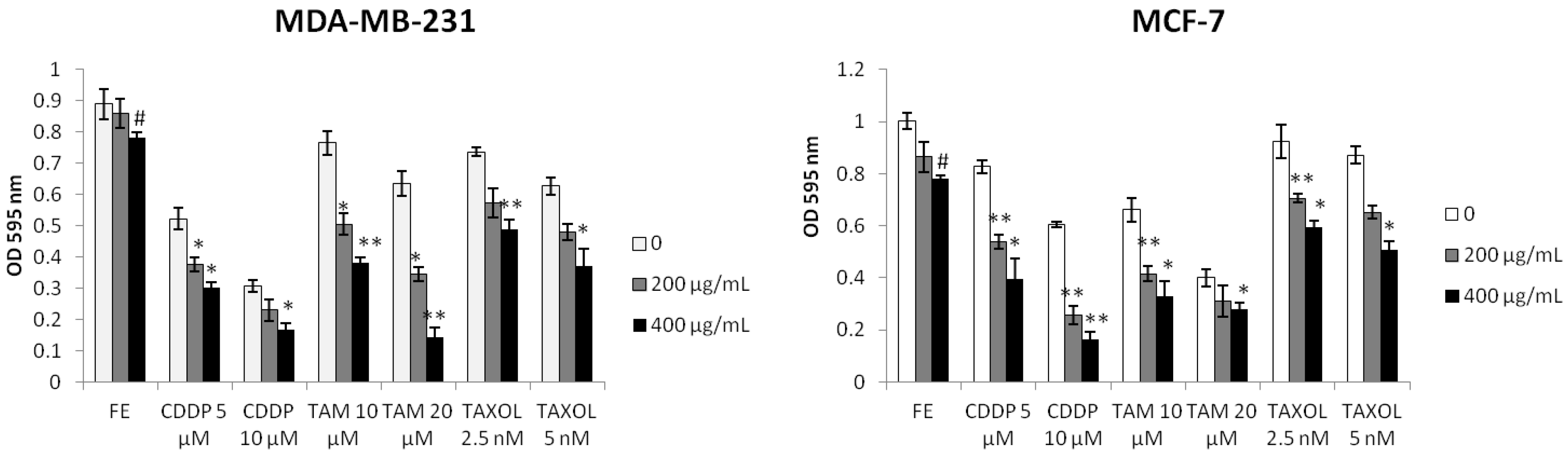
2.1. Enhanced Cytotoxicity by Combination of FE and Chemotherapeutic Agents
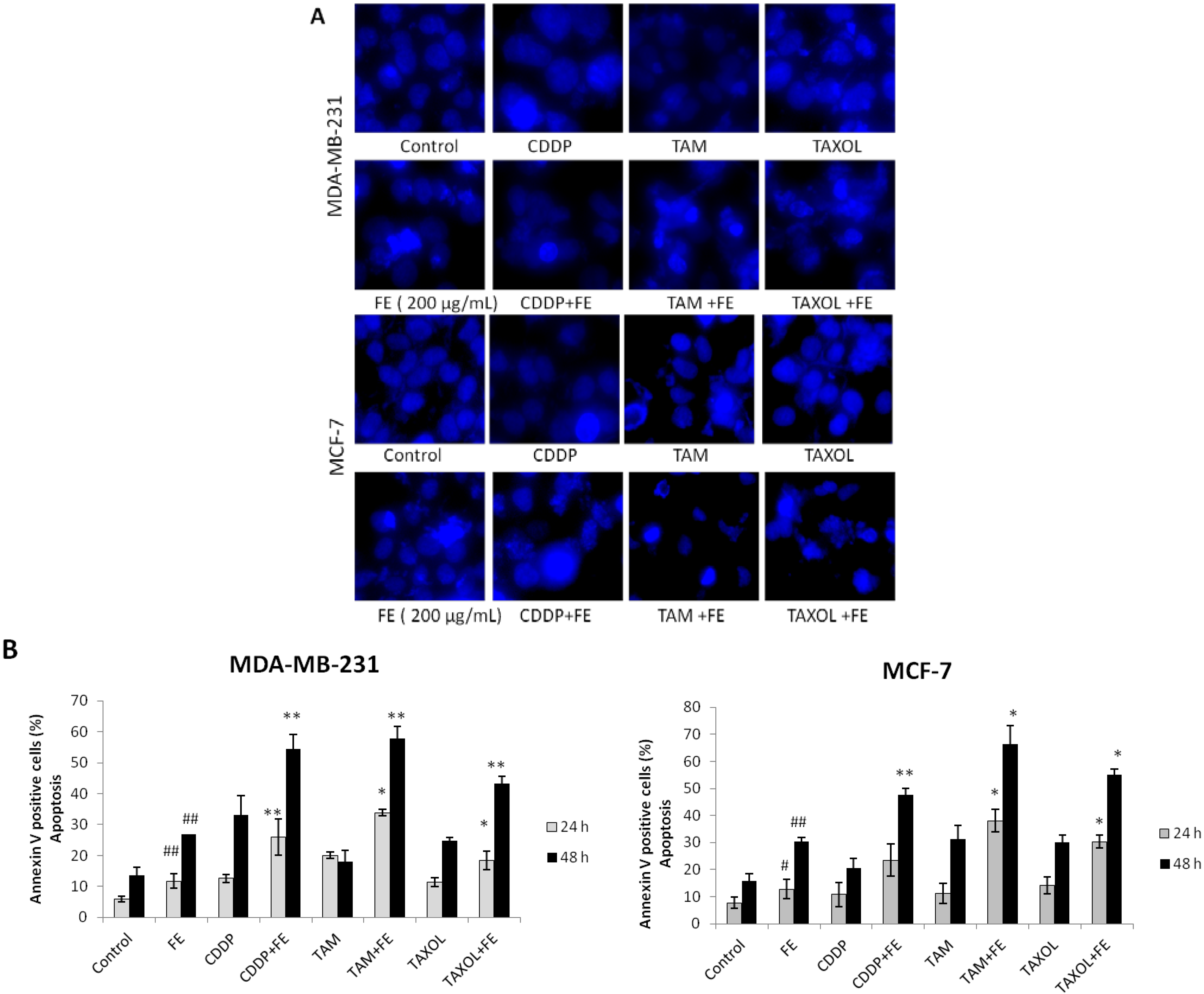
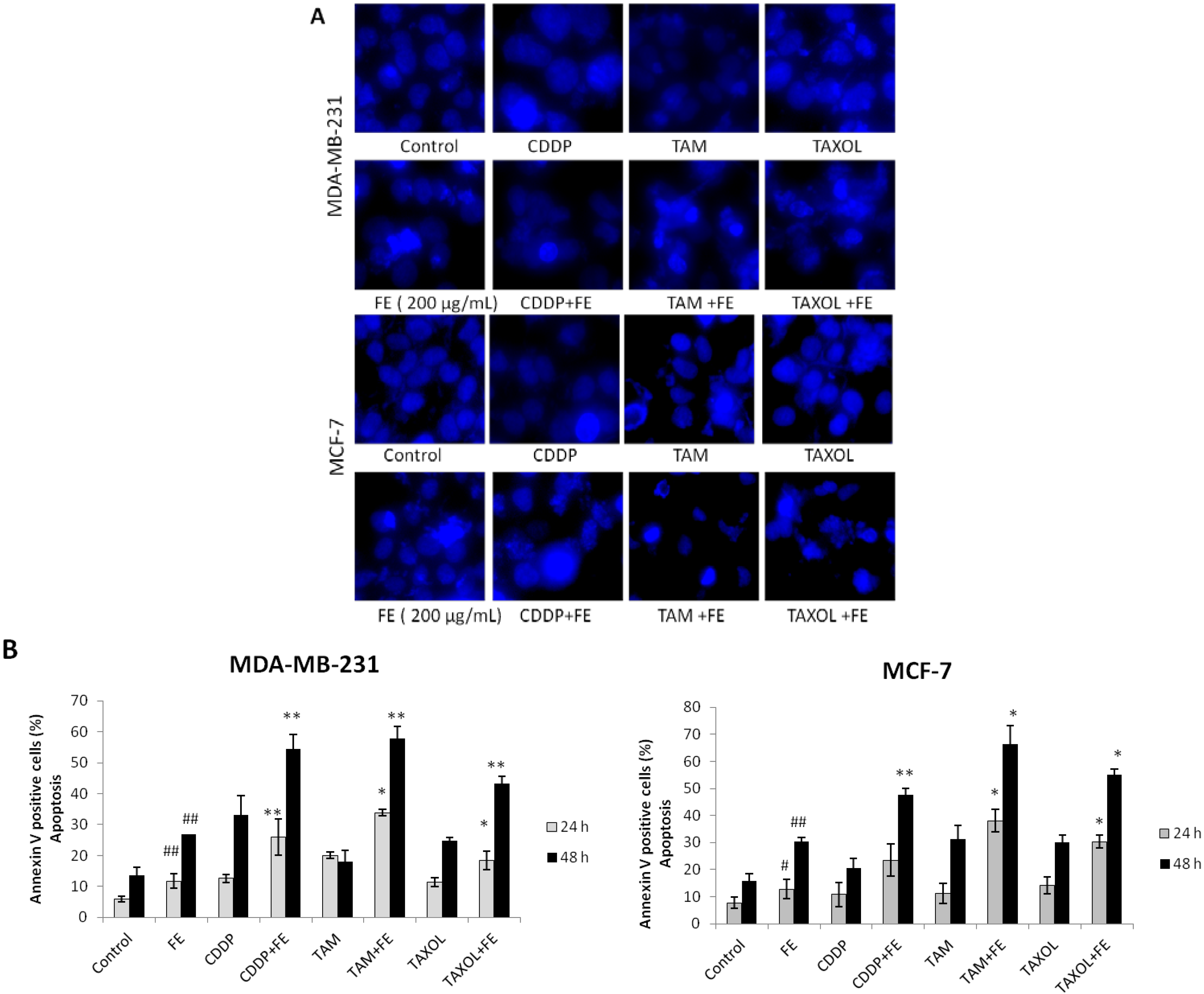
2.2. Synergistic Induction of Apoptosis and Cell Cycle Arrest by FE and Chemotherapeutic Agents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | MDA-MB-231 | MCF-7 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| G1 | S | G2/M | Sub-G1 | G1 | S | G2/M | Sub-G1 | ||
| Control | 65.9 ± 2.8 | 21.9 ± 2.0 | 12.2 ± 1.8 | 1.9 ± 0.2 | 54.6 ± 5.2 | 31.7 ± 2.5 | 13.7 ± 0.5 | 5.0 ± 0.2 | |
| FE (200 μg/mL) | 68.7 ± 1.0 | 18.8 ± 1.6 | 12.5 ± 0.9 | 10.7 ± 0.8 | 57.3 ± 4.8 | 27.5 ± 1.5 | 15.2 ± 1.2 | 13.6 ± 1.0 | |
| CDDP (5 μM) | 13.7 ± 0.3 | 29.4 ± 3.5 | 56.9 ± 3.2 | 17.5 ± 4.3 | 37.1 ± 1.4 | 14.1 ± 1.0 | 48.8 ± 1.5 | 8.9 ± 1.5 | |
| CDDP + FE | 19.9 ± 0.7 | 21.4 ± 0.6 | 58.7 ± 0.4 | 37.8 ± 6.2 | 26.2 ± 2.5 | 29.0 ± 2.6 | 44.8 ± 2.0 | 32.4 ± 3.6 | |
| TAM (10 μM) | 66.6 ± 3.6 | 23.3 ± 2.1 | 10.1 ± 1.6 | 4.9 ± 0.6 | 63.7 ± 2.2 | 18.9 ± 1.5 | 17.4 ± 0.9 | 12.6 ± 0.9 | |
| TAM + FE | 60.8 ± 2.0 | 22.7 ± 1.5 | 16.5 ± 1.5 | 18.3 ± 2.1 | 67.8 ± 2.4 | 17.6 ± 1.6 | 14.6 ± 1.1 | 46.5 ± 4.3 | |
| TAXOL (2.5 nM) | 54.5 ± 2.0 | 20.5 ± 1.5 | 25 ± 0.8 | 6.4 ± 0.4 | 54.2 ± 2.2 | 16.9 ± 1.3 | 29.1 ± 0.8 | 8.4 ± 0.6 | |
| TAXOL + FE | 53.7 ± 1.9 | 22.1 ± 2.1 | 24.2 ± 1.6 | 25.8 ± 2.4 | 49.8 ± 2.6 | 21.8 ± 1.0 | 28.4 ± 0.7 | 20.6 ± 0.3 | |
2.3. Synergistic Changes in the Expression of Bcl-2 Proteins

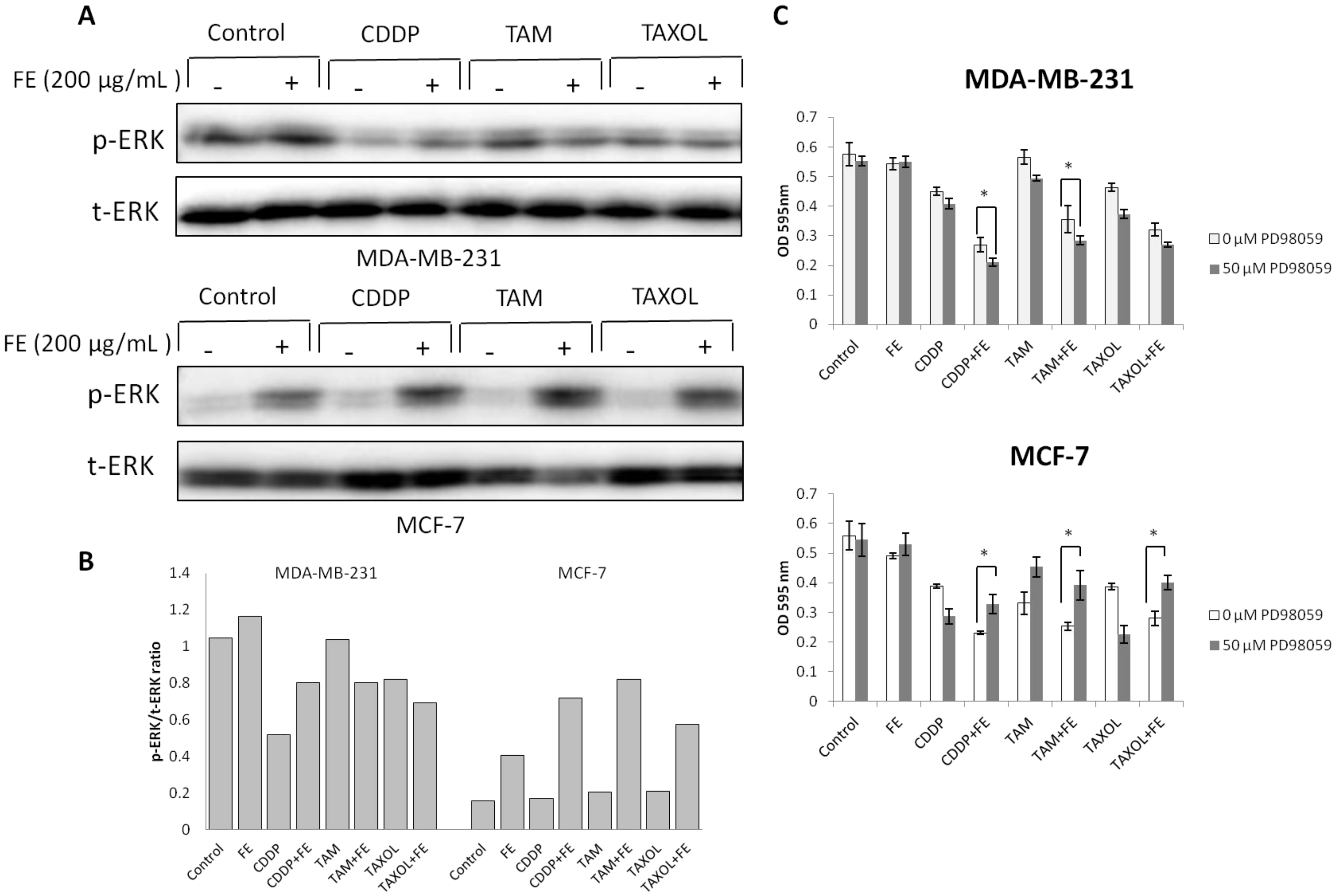
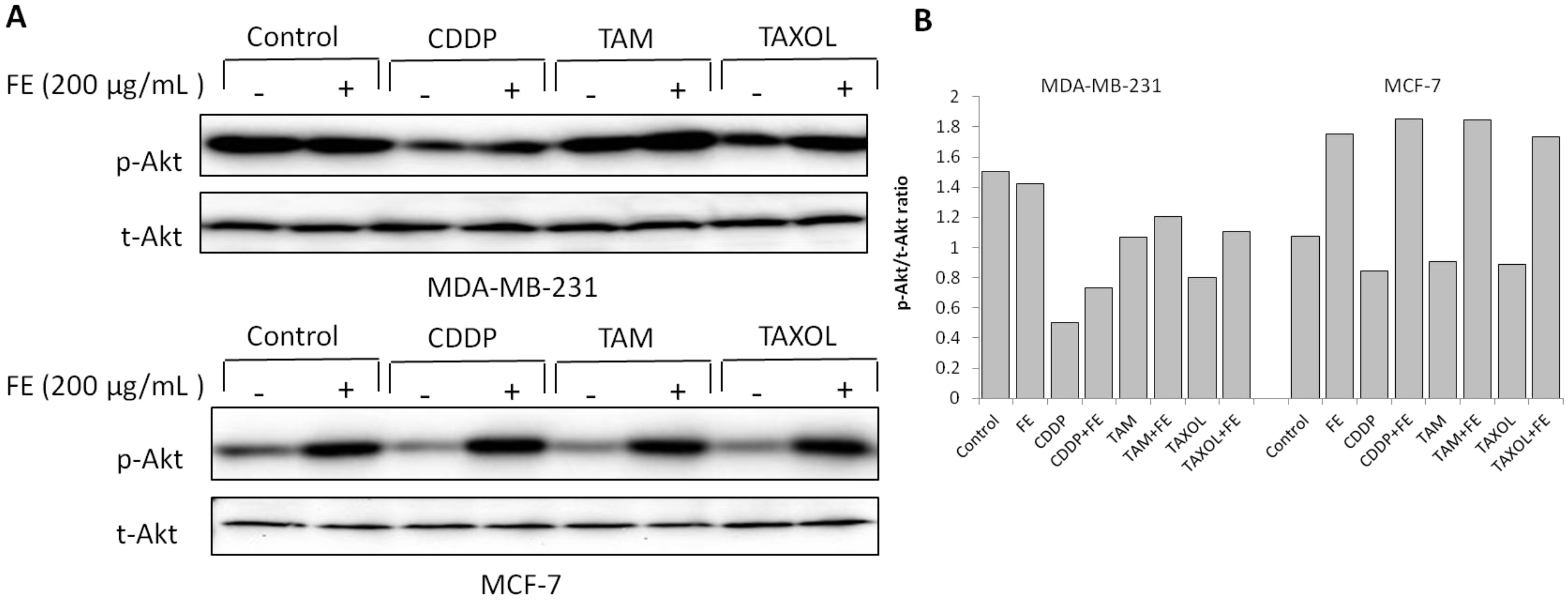
2.4. Modulations in ERK and Akt Phosphorylation
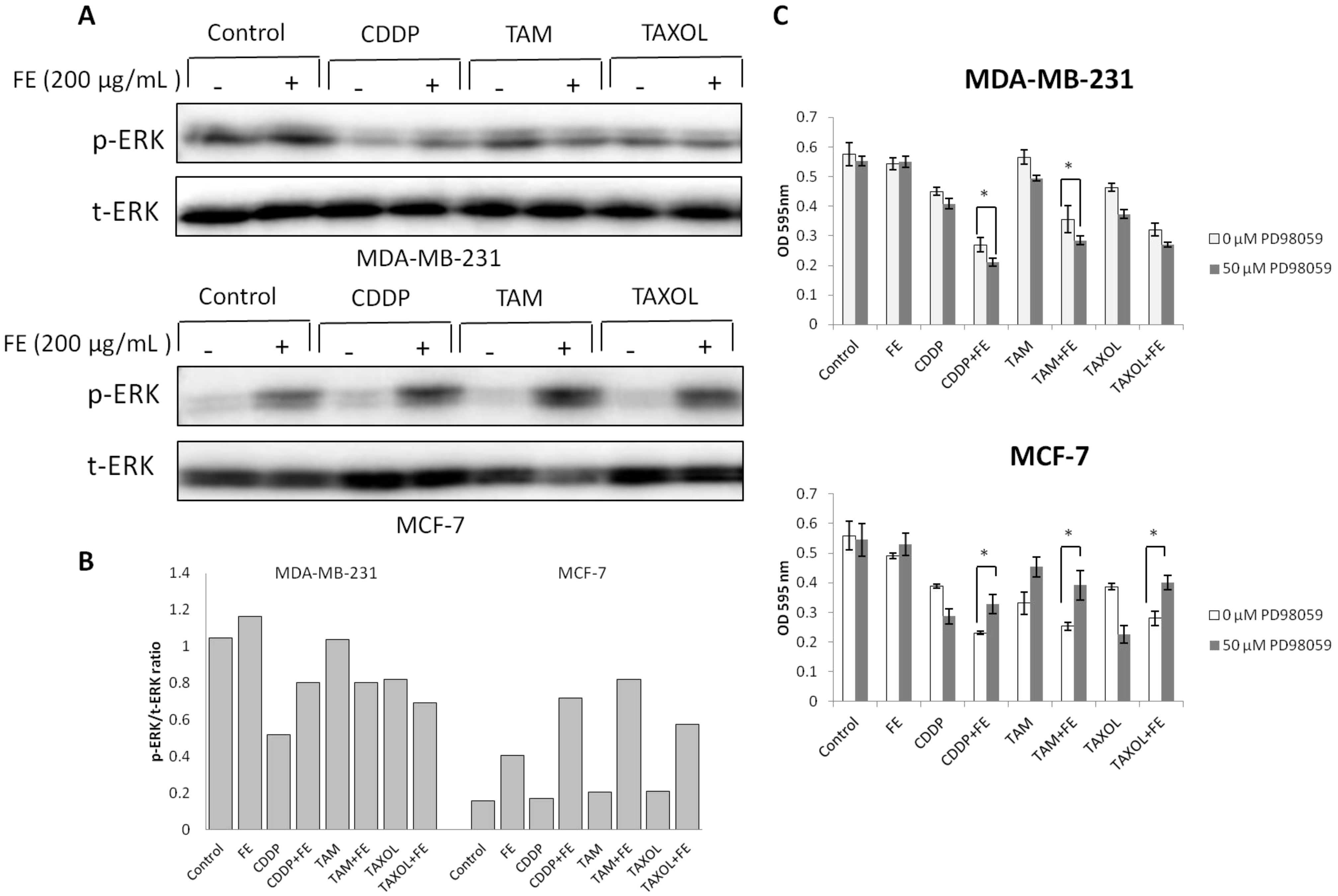
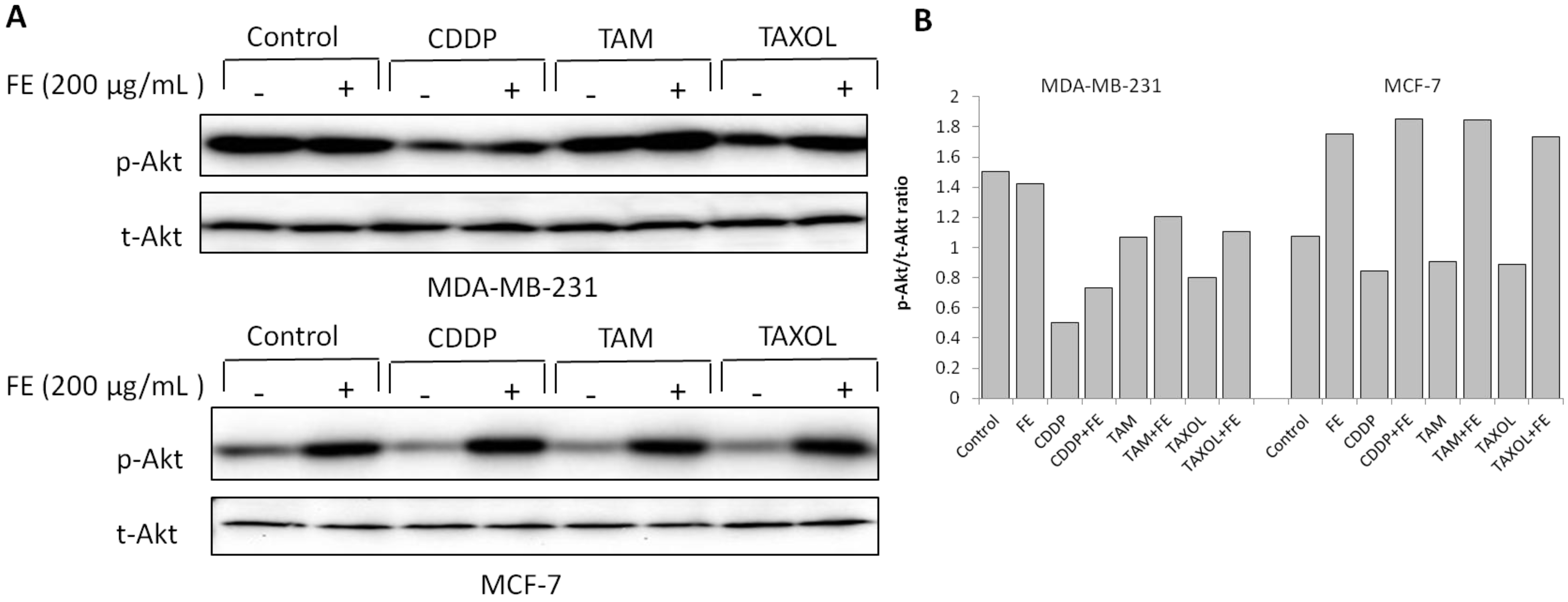
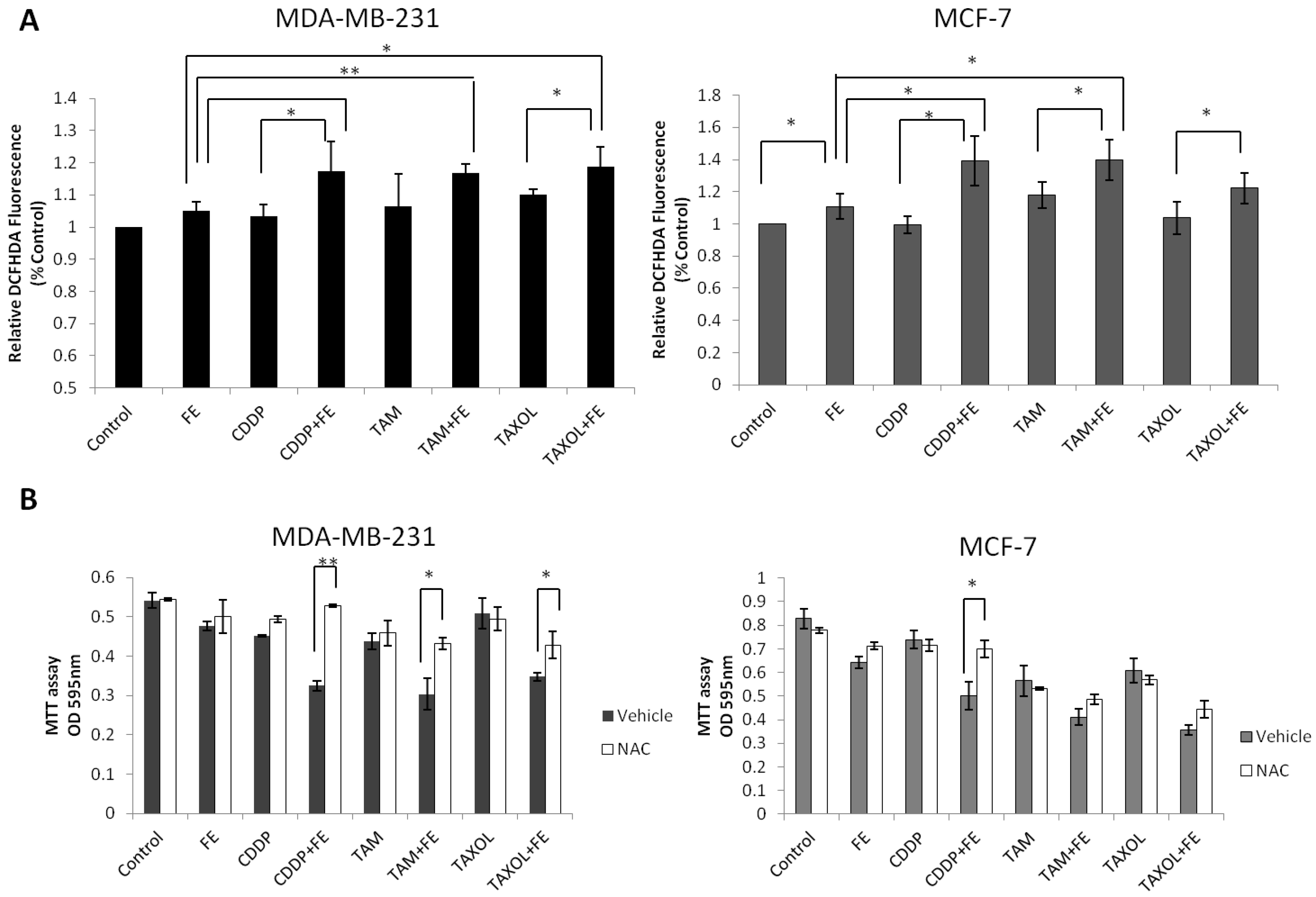
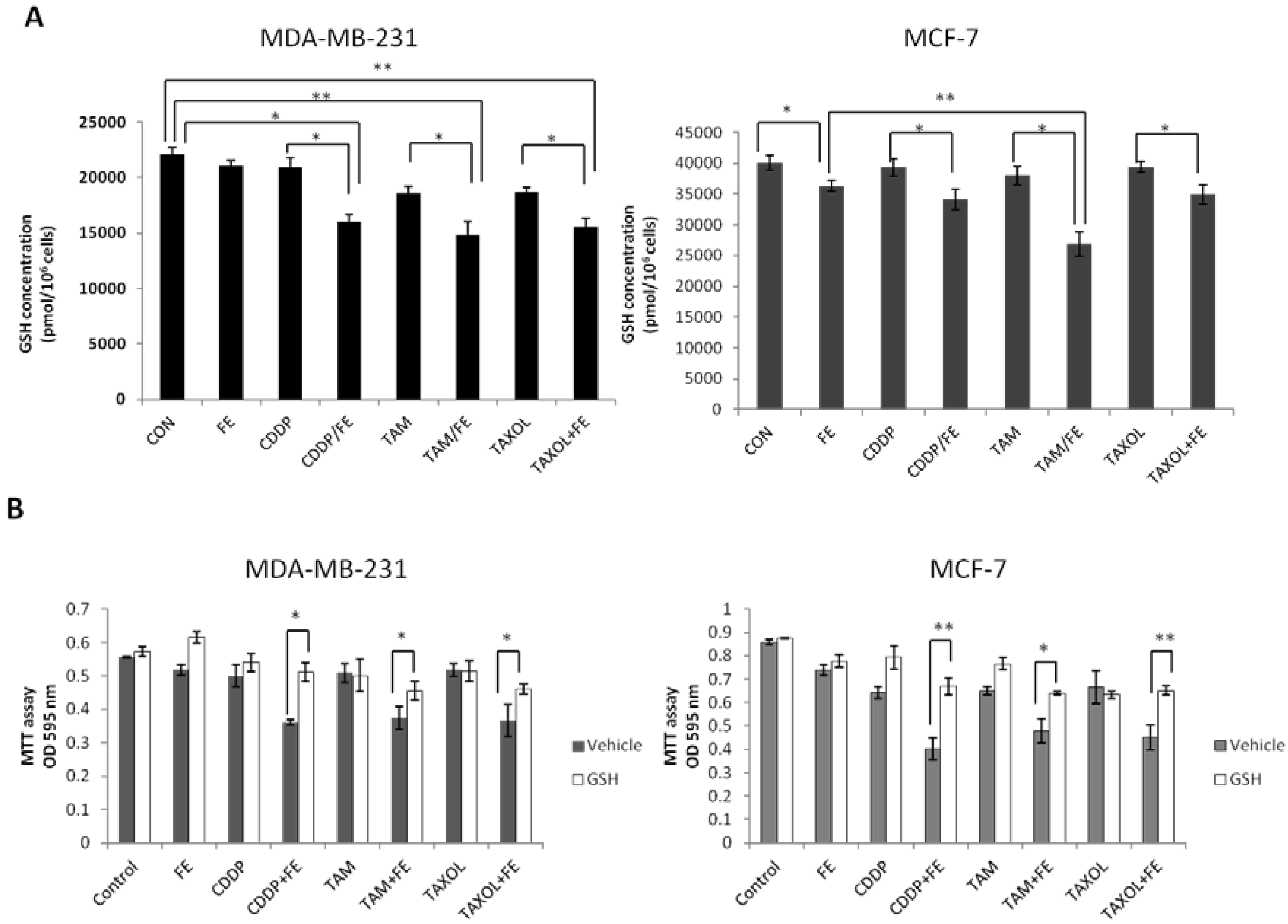
2.5. Synergistic Increases in ROS Levels and Reduction in GSH Levels
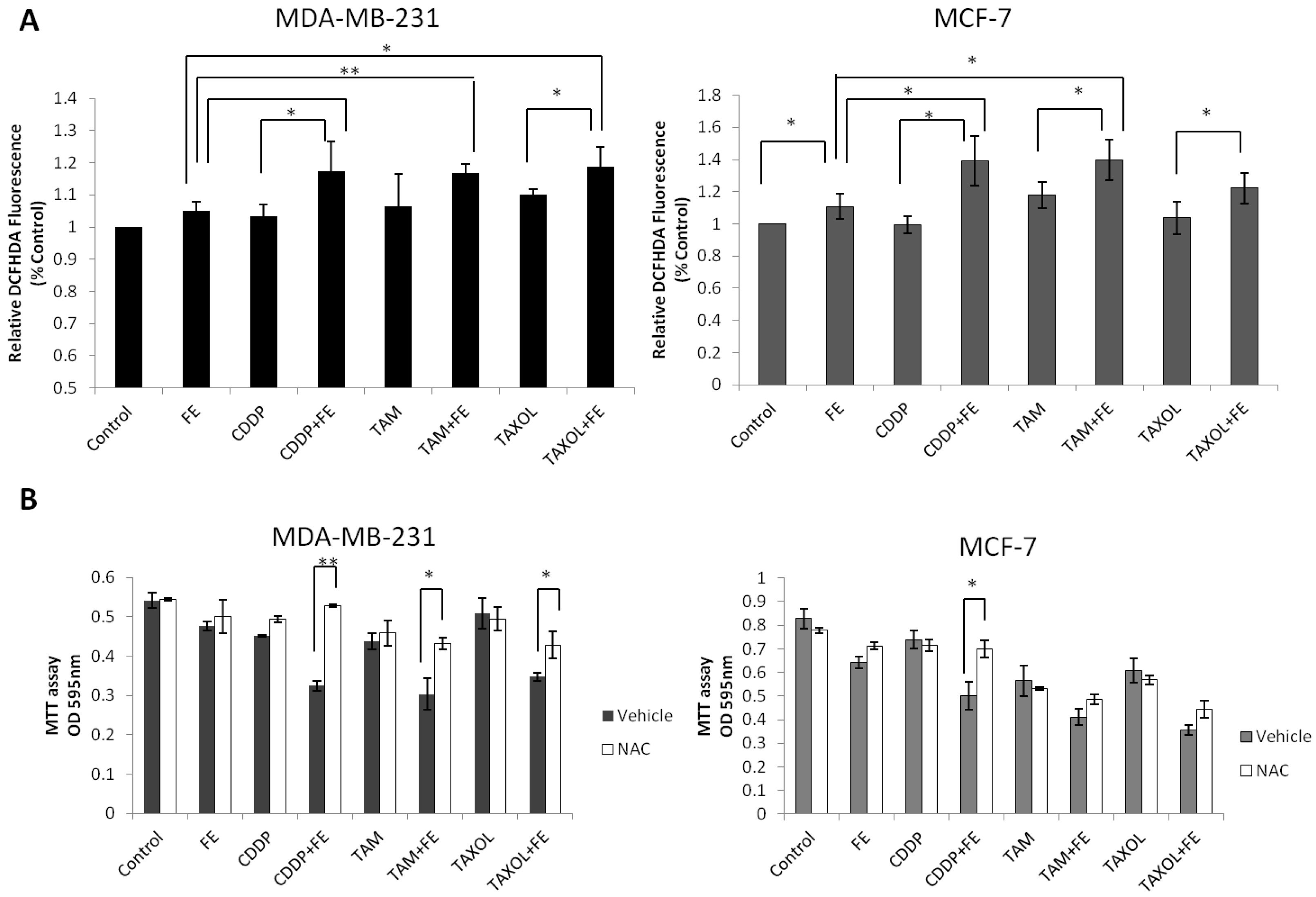
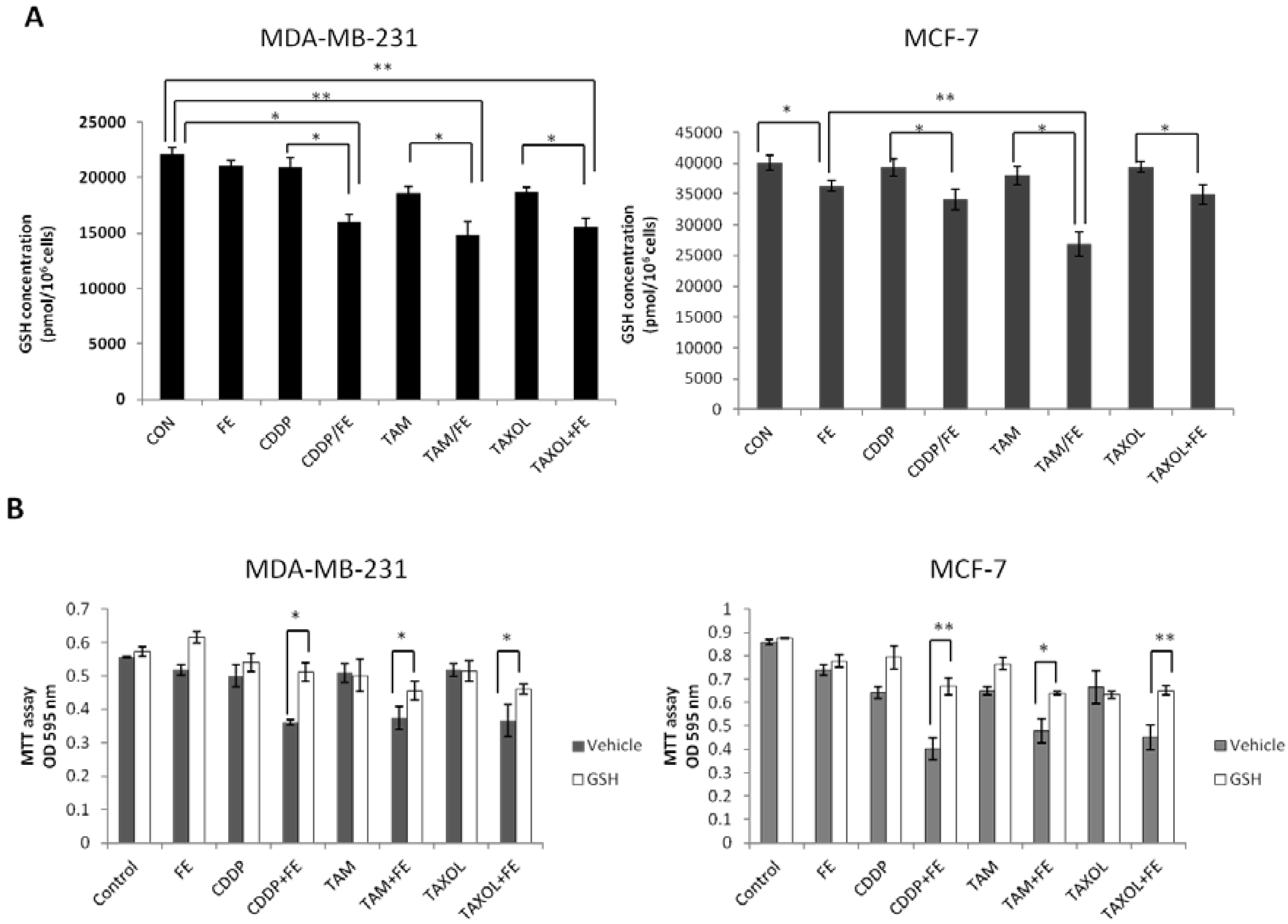
2.6. Global Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Materials
3.3. MTT Assay
3.4. Hoechst Staining
3.5. Annexin V Binding Assay
3.6. Flow Cytometry Analysis
3.7. Western Blot Analysis
3.8. Measurement of Intracellular ROS
3.9. GSH Assay
3.10. Statistical Analysis
4. Conclusions
References
- Cumashi, A.; Ushakova, N.; Preobrazhenskaya, M.; DIncecco, A.; Piccoli, A.; Totani, L.; Tinari, N.; Morozevich, G.E.; Berman, A.E.; Bilan, M.I.; et al. A comparative study of the anti-inflammatory, anticoagulant, antiangiogenic and antiadhesive activities of nine different fucoidans from brown seaweeds. Glycobiology 2007, 17, 541–552. [Google Scholar]
- Ye, J.; Li, Y.P.; Teruya, K.; Katakura, Y.; Ichikawa, A.; Eto, H.; Hosoi, M.; Hosoi, M.; Nishimoto, S.; Shirahata, S. Enzyme-digested fucoidan extracts derived from seaweed Mozuku of Cladosiphonnovae-caledoniae Kylin inhibit invasion and angiogenesis of tumor cells. Cytotechnology 2005, 47, 117–126. [Google Scholar]
- Fitton, J.H. Therapies from fucoidan; multifunctional marine polymers. Mar. Drugs 2011, 9, 1731–1760. [Google Scholar] [CrossRef]
- Aisa, Y.; Miyakawa, Y.; Nakazato, T.; Shibata, H.; Saito, K.; Ikeda, Y.; Kizaki, M. Fucoidan induces apoptosis of human HS-sultan cells accompanied by activation of caspase-3 and downregulation of ERK pathways. Am. J. Hematol. 2005, 78, 7–14. [Google Scholar] [CrossRef]
- Haneji, K.; Matsuda, T.; Tomita, M.; Kawakami, H.; Ohshiro, K.; Uchihara, J.N.; Masuda, M.; Takasu, N.; Tanaka, Y.; Ohta, T.; et al. Fucoidan extracted from Cladosiphon okamuranus Tokida induces apoptosis of human T cell leukemia virus type 1 infected T cell lines and primary adult T-cell leukemia cells. Nutr. Cancer 2005, 52, 189–201. [Google Scholar] [CrossRef]
- Hyun, J.H.; Kim, S.C.; Kang, J.I.; Kim, M.K.; Boo, H.J.; Kwon, J.M.; Koh, Y.S.; Hyun, J.W.; Park, D.B.; Yoo, E.S.; et al. Apoptosis inducing activity of fucoidan in HCT-15 colon carcinoma cells. Biol. Pharm. Bull. 2009, 32, 1760–1764. [Google Scholar] [CrossRef]
- Nagamine, T.; Hayakawa, K.; Kusakabe, T.; Takada, H.; Nakazato, K.; Hisanaga, E.; Iha, M. Inhibitory effect of fucoidan on Huh7 hepatoma cells through downregulation of CXCL12. Nutr. Cancer 2009, 61, 340–347. [Google Scholar] [CrossRef]
- Zhang, Z.; Teruya, K.; Eto, H.; Shirahata, S. Fucoidan extract induces apoptosis in MCF-7 cells via a mechanism involving the ROS-dependent JNK activation and mitochondria-mediated pathways. PLoS One 2011, 6, e27441. [Google Scholar]
- Ale, M.T.; Maruyama, H.; Tamauchi, H.; Mikkelsen, J.D.; Meyer, A.S. Fucose containing sulfated polysaccharides from brown seaweeds inhibit proliferation of melanoma cells and induce apoptosis by activation of caspase-3 in vitro. Mar. Drugs 2011, 9, 2605–2621. [Google Scholar] [CrossRef]
- Koyanagi, S.; Tanigawa, N.; Nakagawa, H.; Soeda, S.; Shimeno, H. Oversulfation of fucoidan enhances its anti-angiogenic and antitumor activities. Biochem. Pharmacol. 2003, 65, 173–179. [Google Scholar]
- Alekseyenko, T.V.; Zhanayeva, S.Y.; Venediktova, A.A.; Zvyagintseva, T.N.; Kuznetsova, T.A.; Besednova, N.N.; Korolenko, T.A. Antitumor and antimetastatic activity of fucoidan, a sulfated polysaccharide isolated from the Okhotsk Sea Fucus evanescens brown alga. Bull. Exp. Bio. Med. 2007, 143, 730–732. [Google Scholar] [CrossRef]
- Coombe, D.R.; Parish, C.R.; Ramshaw, I.A.; Snowden, J.M. Analysis of the inhibition of tumour metastasis by sulphated polysaccharides. Int. J. Cancer 1987, 39, 82–88. [Google Scholar] [CrossRef]
- Nishimoto, S.J. Clinical improvement in cancer patients through alternative medicine, mainly fucoidan. J. Int. Soc. Life Inf. Sci. 2004, 22, 497–505. [Google Scholar]
- Lee, N.Y.; Ermakova, S.P.; Zvyagintseva, T.N.; Kang, K.W.; Dong, Z.G.; Choi, H.S. Inhibitory effects of fucoidan on activation of epidermal growth factor receptor and cell transformation in JB6 Cl41 cells. Food Chem. Toxicol. 2008, 46, 1793–1800. [Google Scholar] [CrossRef]
- Jin, J.O.; Song, M.G.; Kim, Y.N.; Park, J.I.; Kwak, J.Y. The mechanism of fucoidan-induced apoptosis in leukemic cells: Involvement of ERK1/2, JNK, glutathione and nitric oxide. Mol. Carcinog. 2010, 49, 771–782. [Google Scholar]
- Ikeguchi, M.; Yamamoto, M.; Arai, Y.; Maeta, Y.; Ashida, K.; Katano, K.; Miki, Y.; Kimura, T. Fucoidan reduces the toxicities of chemotherapy for patients with unresectable advanced or recurrent colorectal cancer. Oncol. Lett. 2011, 2, 319–322. [Google Scholar]
- Zemani, F.; Benisvy, D.; Galy, F.I.; Lokajczyk, A.; Colliec, J.S.; Uzan, G.; Fischer, A.M.; Boisson, V.C. Low-molecular-weight fucoidan enhances the proangiogenic phenotype of endothelial progenitor cells. Biochem. Pharmacol. 2005, 70, 1167–1175. [Google Scholar] [CrossRef]
- Alkhatib, B.; Freguin, B.C.; Lallemand, F.; Henry, J.P.; Litzler, P.Y.; Marie, J.P.; Richard, V.; Thuillez, C.; Plissonnier, D. Low molecular weight fucan prevents transplant coronaropathy in rat cardiac allograft model. Transpl. Immunol. 2006, 16, 14–19. [Google Scholar] [CrossRef]
- Lake, A.C.; Vassy, R.; Di, B.M.; Lavigne, D.; Le, V.C.; Perret, G.Y.; Letourneur, D. Low molecular weight fucoidan increases VEGF165-induced endothelial cell migration by enhancing VEGF165 binding to VEGFR-2 and NRP1. J. Biol. Chem. 2006, 281, 37844–37852. [Google Scholar]
- Freguin, B.C.; Alkhatib, B.; David, N.; Lallemand, F.; Henry, J.P.; Godin, M.; Thuillez, C.; Plissonnier, D. Low molecular weight fucoidan prevents neointimal hyperplasia after aortic allografting. Transplantation 2007, 83, 1234–1241. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef]
- Mandlekar, S.; Kong, A.N.T. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001, 6, 469–477. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death: Where the cell cycle and apoptosis come together. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarder, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef]
- Tudor, G.; Aguilera, A.; Halverson, D.O.; Laing, N.D.; Sausville, E.A. Susceptibility to drug-induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels. Cell Death Differ. 2000, 7, 574–586. [Google Scholar] [CrossRef]
- Wendel, H.G.; Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cardo, C.C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef]
- Tokita, Y.; Nakajima, K.; Mochida, H.; Iha, M.; Nagamine, T. Development of a fucoidan-specific antibody and measurement of fucoidan in serum and urine by sandwich ELISA. Biosci. Biotechnol. Biochem. 2010, 74, 350–357. [Google Scholar] [CrossRef]
- Varin, E.; Denoyelle, C.H.; Brotin, E.; Meryet-Figuière, M.; Giffard, F.; Abeilard, E.; Goux, D.; Gauduchon, P.; Icard, P.; Poulain, L. Downregulation of Bcl-xL and Mcl-1 is sufficient to induce cell death in mesothelioma cells highly refractory to conventional chemotherapy. Carcinogenesis 2010, 31, 984–993. [Google Scholar] [CrossRef]
- Littlejohn, J.E.; Cao, X.B.; Miller, S.D.; Ozvaran, M.K.; Jupiter, D.; Zhang, L.; Rodarte, C.; Smythe, W.R. Bcl-xL antisense oligonucleotide and cisplatin combination therapy extends survival in SCID mice with established mesothelioma xenografts. Int. J. Cancer 2008, 123, 202–208. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Zhuang, S.; Schnellmann, R.G. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef]
- Mccubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Biochim. Biophys. Acta 2006, 1773, 1263–1284. [Google Scholar]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef]
- Zheng, A.P.; Kallio, A.; Harkonen, P. Tamoxifen-induced rapid death of MCF-7 breast cancer cells is mediated via extracellularly signal-regulated kinase signaling and can be abrogated by estrogen. Endocrinology 2007, 148, 2764–2777. [Google Scholar] [CrossRef]
- Taherian, A.; Mazoochi, T. Different expression of extracellular signal-regulated kinases (ERK) 1/2 and phospho-ERK proteins in MDA-MB-231 and MCF-7 cells after chemotherapy with doxorubicin or docetaxel. Iran. J. Basic Med. Sci. 2011, 15, 669–677. [Google Scholar]
- Moelling, K.; Schad, K.; Bosse, M.; Zimmermann, S.; Schweneker, M. Regulation of Raf-Akt cross-talk. J. Biol. Chem. 2002, 277, 31099–31106. [Google Scholar]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef]
- Simoncini, T.; Hafezi, M.A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Kahlert, S.; Nuedling, S.; Eickels, M.V.; Vetter, H.; Meyer, R.; Grohé, C. Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J. Biol. Chem. 2000, 275, 18447–18453. [Google Scholar]
- Tsai, E.M.; Wang, S.C.; Lee, J.N. Akt activation by estrogen in estrogen receptor-negative breast cancer cells. Cancer Res. 2001, 61, 8390–8392. [Google Scholar]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis and carcinogenesis. Arch. Toxicol. 2008, 82, 273–299. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar]
- Swamy, S.M.; Huat, B.T. Intracellular glutathione depletion and reactive oxygen species generation are important in alpha-hederin-induced apoptosis of P388 cells. Mol. Cell. Biochem. 2003, 245, 127–139. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Z.; Teruya, K.; Yoshida, T.; Eto, H.; Shirahata, S. Fucoidan Extract Enhances the Anti-Cancer Activity of Chemotherapeutic Agents in MDA-MB-231 and MCF-7 Breast Cancer Cells. Mar. Drugs 2013, 11, 81-98. https://doi.org/10.3390/md11010081
Zhang Z, Teruya K, Yoshida T, Eto H, Shirahata S. Fucoidan Extract Enhances the Anti-Cancer Activity of Chemotherapeutic Agents in MDA-MB-231 and MCF-7 Breast Cancer Cells. Marine Drugs. 2013; 11(1):81-98. https://doi.org/10.3390/md11010081
Chicago/Turabian StyleZhang, Zhongyuan, Kiichiro Teruya, Toshihiro Yoshida, Hiroshi Eto, and Sanetaka Shirahata. 2013. "Fucoidan Extract Enhances the Anti-Cancer Activity of Chemotherapeutic Agents in MDA-MB-231 and MCF-7 Breast Cancer Cells" Marine Drugs 11, no. 1: 81-98. https://doi.org/10.3390/md11010081
APA StyleZhang, Z., Teruya, K., Yoshida, T., Eto, H., & Shirahata, S. (2013). Fucoidan Extract Enhances the Anti-Cancer Activity of Chemotherapeutic Agents in MDA-MB-231 and MCF-7 Breast Cancer Cells. Marine Drugs, 11(1), 81-98. https://doi.org/10.3390/md11010081