Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance




Abstract
:1. Introduction

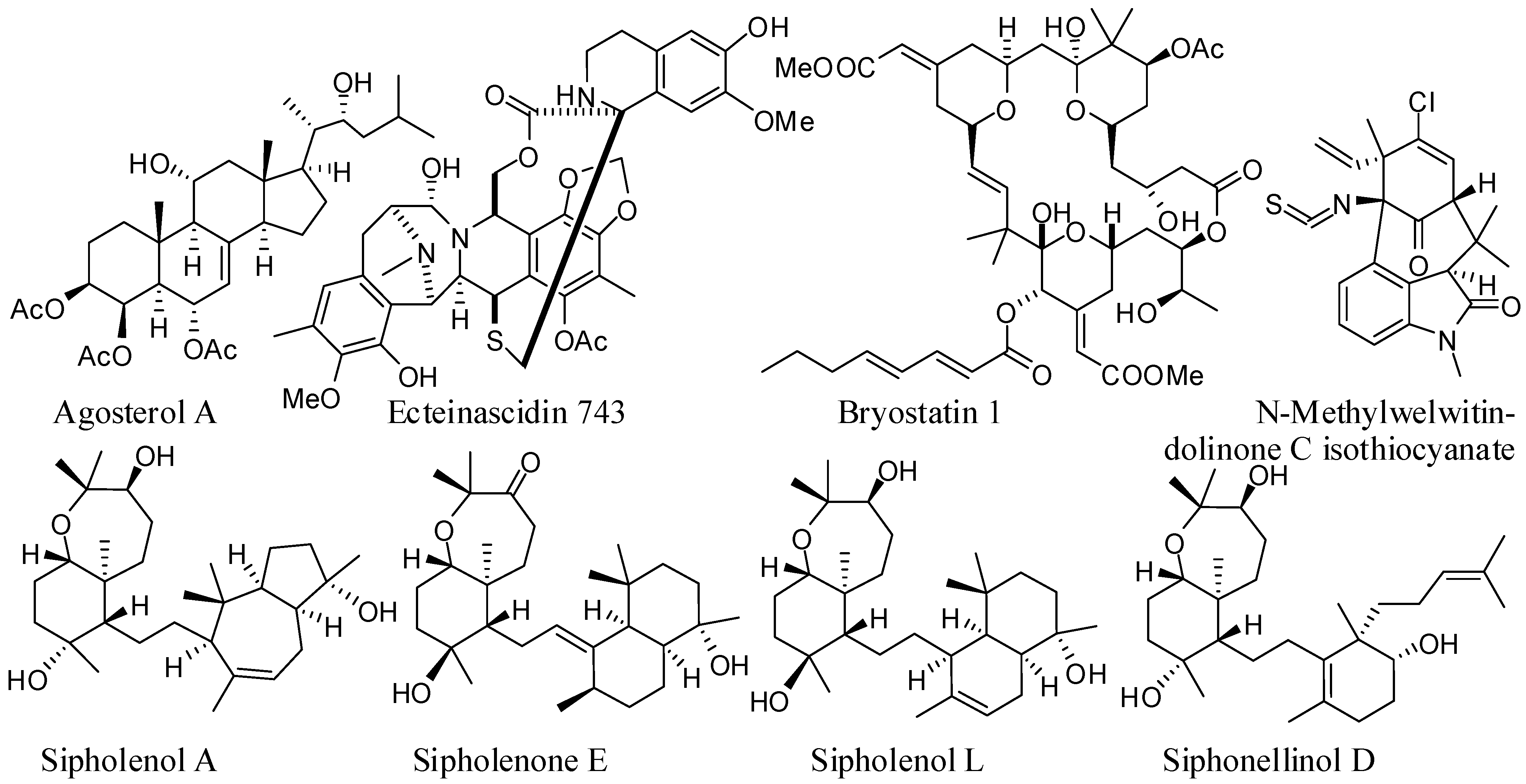{kind=link}
| Compounds | Source organism | Chemical class | Cell lines used | Refences |
|---|---|---|---|---|
| Agosterol A (AG-A) | Spongia sp. | polyhydroxylated sterol acetate | P-gp/MDR1-overexpressing (KB-C2) | [28,29,30] |
| MRP1-overexpressing (KB-CV60) | [28,29,30] | |||
| MRP1-transfected (KB/MRP) | [31] | |||
| Ecteinascidin 743 (ET-743) | Ecteinascidia turbinate | tetrahydroiso-quinolone alkaloid | P-gp/MDR1-overexpressing (KB-8-5, KB-C2) | [32] |
| Sipholenol A | Callyspongia siphonella | sipholane triterpenoids | P-gp/MDR1-overexpressing (KB-C2, KB-V1) | [33,34,35] |
| Sipholenone E Sipholenol L Siphonellinol D | P-gp/MDR1-overexpressing (KB-C2) | [36] | ||
| Bryostatin 1 | Bugula neritina | macrocyclic lactone | P-gp/MDR1-overexpressing (KB-C1, HeLa-MDR1-V185) | [37] |
| N-Methylwelwitin-dolinone C isothiocyanate | Hapalosiphon welwitschii | alkaloid | P-gp/MDR1-overexpressing (SK-VLB-1, MCF-7/ADR) | [38] |
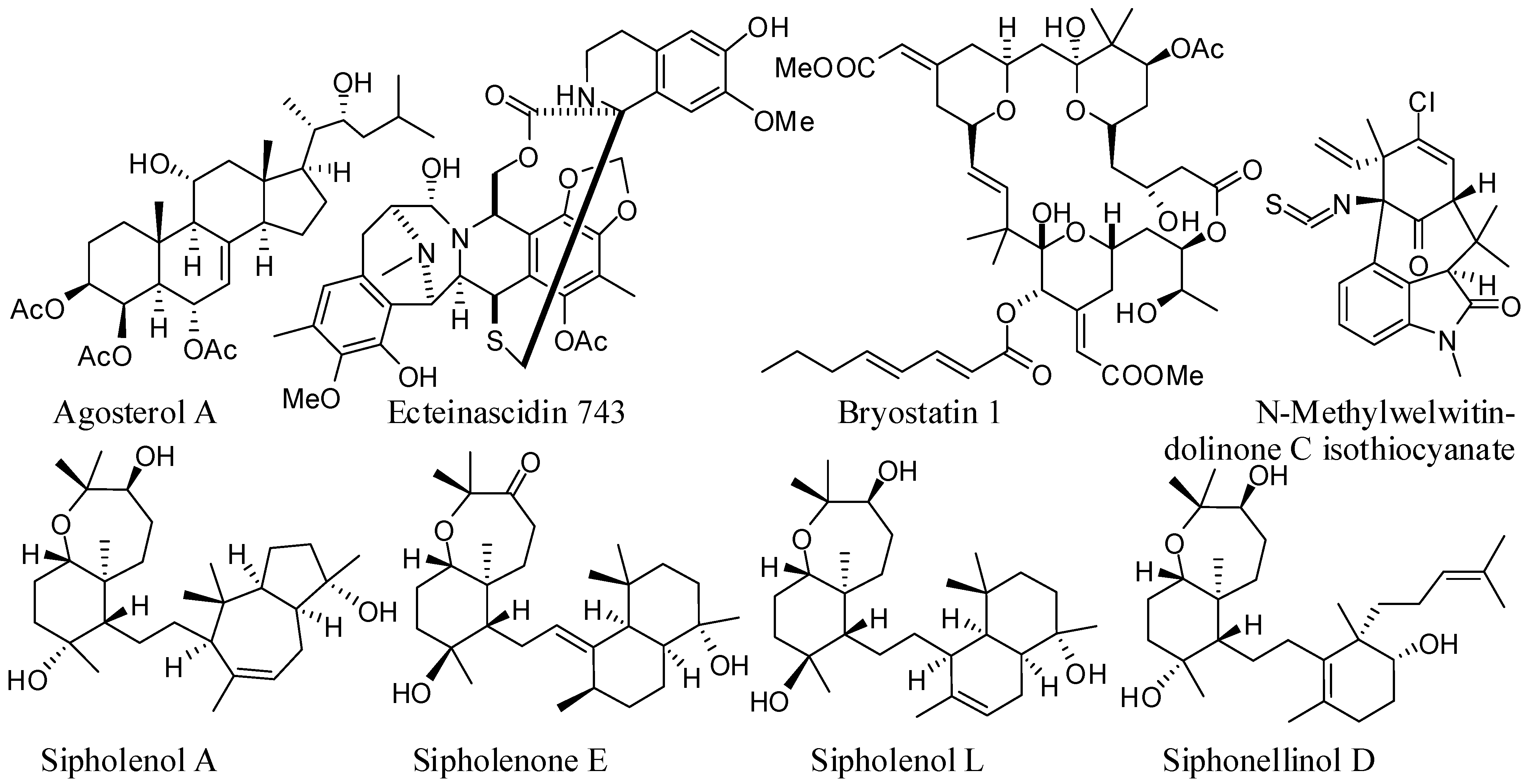
2. Agosterol A (AG-A)
3. Ecteinascidin 743 (ET-743)
4. Sipholane Triterpenoids
5. Bryostatin 1
6. Welwitindolinones
7. Conclusions
Acknowledgements
Conflict of Interest
References
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Paterson, I.; Anderson, E.A. Chemistry. The renaissance of natural products as drug candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef]
- Sithranga, B.N.; Kathiresan, K. Anticancer drugs from marine flora: An overview. J. Oncol. 2010, 2010, 214186. [Google Scholar]
- Hu, G.P.; Yuan, J.; Sun, L.; She, Z.G.; Wu, J.H.; Lan, X.J.; Zhu, X.; Lin, Y.C.; Chen, S.P. Statistical research on marine natural products based on data obtained between 1985 and 2008. Mar. Drugs 2011, 9, 514–525. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Guo, Y.; Kock, K.; Ritter, C.A.; Chen, Z.S.; Grube, M.; Jedlitschky, G.; Illmer, T.; Ayres, M.; Beck, J.F.; Siegmund, W.; et al. Expression of ABCC-type nucleotide exporters in blasts of adult acute myeloid leukemia: Relation to long-term survival. Clin. Cancer Res. 2009, 15, 1762–1769. [Google Scholar] [CrossRef]
- Hopper-Borge, E.; Xu, X.; Shen, T.; Shi, Z.; Chen, Z.S.; Kruh, G.D. Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res. 2009, 69, 178–184. [Google Scholar] [CrossRef]
- Garcia-Fernandez, L.F.; Reyes, F.; Sanchez-Puelles, J.M. The marine pharmacy: New antitumoral compounds from the sea. Pharm. News 2002, 9, 495–501. [Google Scholar]
- Cuevas, C.; Perez, M.; Martin, M.J.; Chicharro, J.L.; Fernandez-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; et al. Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.Y.; Judson, I.; Van Oosterom, A.; Verweij, J.; Radford, J.; Lorigan, P.; Rodenhuis, S.; Ray-Coquard, I.; Bonvalot, S.; et al. Phase II study of ET-743 in advanced soft tissue sarcomas: A European Organisation for the Research and Treatment of Cancer (EORTC) soft tissue and bone sarcoma group trial. J. Clin. Oncol. 2005, 23, 576–584. [Google Scholar]
- Garcia-Carbonero, R.; Supko, J.G.; Maki, R.G.; Manola, J.; Ryan, D.P.; Harmon, D.; Puchalski, T.A.; Goss, G.; Seiden, M.V.; Waxman, A.; et al. Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: Multicenter phase II and pharmacokinetic study. J. Clin. Oncol. 2005, 23, 5484–5492. [Google Scholar]
- Demetri, G.D.; Chawla, S.P.; Von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: Results of a randomized phase II study of two different schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar]
- McBride, A.; Butler, S.K. Eribulin mesylate: A novel halichondrin B analogue for the treatment of metastatic breast cancer. Am. J. Health Syst. Pharm. 2012, 69, 745–755. [Google Scholar] [CrossRef]
- Gourmelon, C.; Frenel, J.S.; Campone, M. Eribulin mesylate for the treatment of late-stage breast cancer. Expert Opin. Pharmacother. 2011, 12, 2883–2890. [Google Scholar] [CrossRef]
- Mita, A.C.; Hammond, L.A.; Bonate, P.L.; Weiss, G.; McCreery, H.; Syed, S.; Garrison, M.; Chu, Q.S.; DeBono, J.S.; Jones, C.B.; et al. Phase I and pharmacokinetic study of tasidotin hydrochloride (ILX651), a third-generation dolastatin-15 analogue, administered weekly for 3 weeks every 28 days in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 5207–5215. [Google Scholar]
- Bai, R.; Edler, M.C.; Bonate, P.L.; Copeland, T.D.; Pettit, G.R.; Luduena, R.F.; Hamel, E. Intracellular activation and deactivation of tasidotin, an analog of dolastatin 15: Correlation with cytotoxicity. Mol. Pharmacol. 2009, 75, 218–226. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Glode, M.; Du, W.; Kraft, A.; Hudes, G.; Wright, J.; Hussain, M. Phase II study of dolastatin-10 in patients with hormone-refractory metastatic prostate adenocarcinoma. Clin. Cancer Res. 2000, 6, 4205–4208. [Google Scholar]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Invest. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef]
- Barr, P.M.; Lazarus, H.M.; Cooper, B.W.; Schluchter, M.D.; Panneerselvam, A.; Jacobberger, J.W.; Hsu, J.W.; Janakiraman, N.; Simic, A.; Dowlati, A.; Remick, S.C. Phase II study of bryostatin 1 and vincristine for aggressive non-Hodgkin lymphoma relapsing after an autologous stem cell transplant. Am. J. Hematol. 2009, 84, 484–487. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Ocio, E.M.; Pandiella, A.; Maiso, P.; Gajate, C.; Garayoa, M.; Vilanova, D.; Montero, J.C.; Mitsiades, N.; McMullan, C.J.; et al. Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res. 2008, 68, 5216–5225. [Google Scholar] [CrossRef]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Kellen, J.A. The reversal of multidrug resistance: An update. J. Exp. Ther. Oncol. 2003, 3, 5–13. [Google Scholar] [CrossRef]
- Ullah, M.F. Cancer multidrug resistance (MDR): A major impediment to effective chemotherapy. Asian Pac. J. Cancer Prev. 2008, 9, 1–6. [Google Scholar]
- Yang, K.; Wu, J.; Li, X. Recent advances in the research of P-glycoprotein inhibitors. Biosci. Trends 2008, 2, 137–146. [Google Scholar]
- He, S.M.; Li, R.; Kanwar, J.R.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar]
- Aoki, S.; Yoshioka, Y.; Miyamoto, Y.; Higuchi, K.; Setiawan, A.; Murakami, N.; Chen, Z.S.; Sumizawa, T.; Akiyama, S.; Kobayashi, M. Agosterol A, a novel polyhydroxylated sterol acetate reversing multidrug resistance from a marine sponge of Spongia sp. Tetrahedron Lett. 1998, 39, 6303–6306. [Google Scholar]
- Aoki, S.; Setiawan, A.; Yoshioka, Y.; Higuchi, K.; Fudetani, R.; Chen, Z.S.; Sumizawa, T.; Akiyama, S.; Kobayashi, M. Reversal of multidrug resistance in human carcinoma cell line by agosterols, marine spongean sterols. Tetrahedron 1999, 55, 13965–13972. [Google Scholar]
- Aoki, S.; Chen, Z.S.; Higasiyama, K.; Setiawan, A.; Akiyama, S.; Kobayashi, M. Reversing effect of agosterol A, a spongean sterol acetate, on multidrug resistance in human carcinoma cells. Jpn. J. Cancer Res. 2001, 92, 886–895. [Google Scholar] [CrossRef]
- Chen, Z.S.; Aoki, S.; Komatsu, M.; Ueda, K.; Sumizawa, T.; Furukawa, T.; Okumura, H.; Ren, X.Q.; Belinsky, M.G.; Lee, K.; Kruh, G.D.; Kobayashi, M.; Akiyama, S. Reversal of drug resistance mediated by multidrug resistance protein (MRP) 1 by dual effects of agosterol A on MRP1 function. Int. J. Cancer 2001, 93, 107–113. [Google Scholar] [CrossRef]
- Zewail-Foote, M.; Hurley, L.H. Ecteinascidin 743: A minor groove alkylator that bends DNA toward the major groove. J. Med. Chem. 1999, 42, 2493–2497. [Google Scholar] [CrossRef]
- Jain, S.; Laphookhieo, S.; Shi, Z.; Fu, L.W.; Akiyama, S.; Chen, Z.S.; Youssef, D.T.; van Soest, R.W.; El Sayed, K.A. Reversal of P-glycoprotein-mediated multidrug resistance by sipholane triterpenoids. J. Nat. Prod. 2007, 70, 928–931. [Google Scholar] [CrossRef]
- Jain, S.; Abraham, I.; Carvalho, P.; Kuang, Y.H.; Shaala, L.A.; Youssef, D.T.; Avery, M.A.; Chen, Z.S.; El Sayed, K.A. Sipholane triterpenoids: Chemistry, reversal of ABCB1/P-glycoprotein-mediated multidrug resistance, and pharmacophore modeling. J. Nat. Prod. 2009, 72, 1291–1298. [Google Scholar] [CrossRef]
- Shi, Z.; Jain, S.; Kim, I.W.; Peng, X.X.; Abraham, I.; Youssef, D.T.; Fu, L.W.; El Sayed, K.A.; Ambudkar, S.; Chen, Z.S. Sipholenol A, a marine-derived sipholane triterpene, potently reverses P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Cancer Sci. 2007, 98, 1373–1380. [Google Scholar] [CrossRef]
- Abraham, I.; Jain, S.; Wu, C.P.; Khanfar, M.A.; Kuang, Y.; Dai, C.L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; El Sayed, K.; Chen, Z.S. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef]
- Spitaler, M.; Utz, I.; Hilbe, W.; Hofmann, J.; Grunicke, H.H. PKC-Independent modulation of multidrug resistance in cells with mutant (V185) but not wild-type (G185) P-glycoprotein by bryostatin 1. Biochem. Pharmacol. 1998, 56, 861–869. [Google Scholar] [CrossRef]
- Smith, C.D.; Zilfou, J.T.; Stratmann, K.; Patterson, G.M.; Moore, R.E. Welwitindolinone analogues that reverse P-glycoprotein-mediated multiple drug resistance. Mol. Pharmacol. 1995, 47, 241–247. [Google Scholar]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Kanzaki, A.; Takebayashi, Y.; Ren, X.Q.; Miyashita, H.; Mori, S.; Akiyama, S.; Pommier, Y. Overcoming multidrug drug resistance in P-glycoprotein/MDR1-overexpressing cell lines by ecteinascidin 743. Mol. Cancer Ther. 2002, 1, 1327–1334. [Google Scholar]
- Amador, M.L.; Jimeno, J.; Paz-Ares, L.; Cortes-Funes, H.; Hidalgo, M. Progress in the development and acquisition of anticancer agents from marine sources. Ann. Oncol. 2003, 14, 1607–1615. [Google Scholar] [CrossRef]
- Carter, N.J.; Keam, S.J. Trabectedin: A review of its use in the management of soft tissue sarcoma and ovarian cancer. Drugs 2007, 67, 2257–2276. [Google Scholar] [CrossRef]
- Pettit, G.R. The bryostatins. Prog. Chem. Org. Nat. Prod. 1991, 57, 153–195. [Google Scholar]
- Kraft, A.S.; Smith, J.B.; Berkow, R.L. Bryostatin, an activator of the calcium phospholipid-dependent protein kinase, blocks phorbol ester-induced differentiation of human promyelocytic leukemia cells HL-60. Proc. Natl. Acad. Sci. USA 1986, 83, 1334–1338. [Google Scholar] [CrossRef]
- Jetten, A.M.; George, M.A.; Pettit, G.R.; Rearick, J.I. Effects of bryostatins and retinoic acid on phorbol ester- and diacylglycerol-induced squamous differentiation in human tracheobronchial epithelial cells. Cancer Res. 1989, 49, 3990–3995. [Google Scholar]
- Isakov, N.; Galron, D.; Mustelin, T.; Pettit, G.R.; Altman, A. Inhibition of phorbol ester-induced T cell proliferation by bryostatin is associated with rapid degradation of protein kinase C. J. Immunol. 1993, 150, 1195–1204. [Google Scholar]
- Chambers, T.C.; McAvoy, E.M.; Jacobs, J.W.; Eilon, G. Protein kinase C phosphorylates P-glycoprotein in multidrug resistant human KB carcinoma cells. J. Biol. Chem. 1990, 265, 7679–7686. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abraham, I.; El Sayed, K.; Chen, Z.-S.; Guo, H. Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance. Mar. Drugs 2012, 10, 2312-2321. https://doi.org/10.3390/md10102312
Abraham I, El Sayed K, Chen Z-S, Guo H. Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance. Marine Drugs. 2012; 10(10):2312-2321. https://doi.org/10.3390/md10102312
Chicago/Turabian StyleAbraham, Ioana, Khalid El Sayed, Zhe-Sheng Chen, and Huiqin Guo. 2012. "Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance" Marine Drugs 10, no. 10: 2312-2321. https://doi.org/10.3390/md10102312
APA StyleAbraham, I., El Sayed, K., Chen, Z.-S., & Guo, H. (2012). Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance. Marine Drugs, 10(10), 2312-2321. https://doi.org/10.3390/md10102312