1. Introduction
The field of nanomedicine has attracted more and more interest over the last decades, as nanoparticles (NPs) and polymeric structures have been related to biological and pathophysiological questions. Originally, NPs or polymers were designed and used for various purposes, like magnetic resonance imaging (MRI), computed tomography (CT) and optical imaging or simply drug delivery [
1,
2]. Therefore, these materials had to meet different requirements with respect to their applications. On the other hand, NPs and polymers provide an almost ideal platform to combine different modalities such as the combination of drug delivery with functional imaging techniques such as positron emission tomography (PET) or single photon computed tomography (SPECT). In this line, molecular imaging modalities are essential for patient screening/selection and monitoring in personalized therapy approaches. Furthermore, non-invasive molecular imaging techniques are excellent tools to investigate pharmacological profiles and to identify the optimal nanodimensional architecture of the aimed-at drug delivery system. Multimodal hybrid technologies such as PET/CT or PET/MRI were developed giving the chance to examine the pharmacological profiles of NPs or polymers [
3]. Additionally, polymer-drug conjugates, which have already been applied for chemotherapy approaches, were radiolabeled to investigate their biodistribution via PET imaging [
4]. PET, with its possibility to detect and quantify picomolar amounts of a radiotracer, has emerged as one of the most powerful imaging techniques, which underlines its outstanding role for functional imaging of physiological and/or pathophysiological processes.
Thus, novel strategies were explored for the radiolabeling of NPs (inorganic and organic) or polymers to investigate their pharmacodynamics and pharmacokinetics
in vivo in dependence on their architecture, size and structure. To investigate the
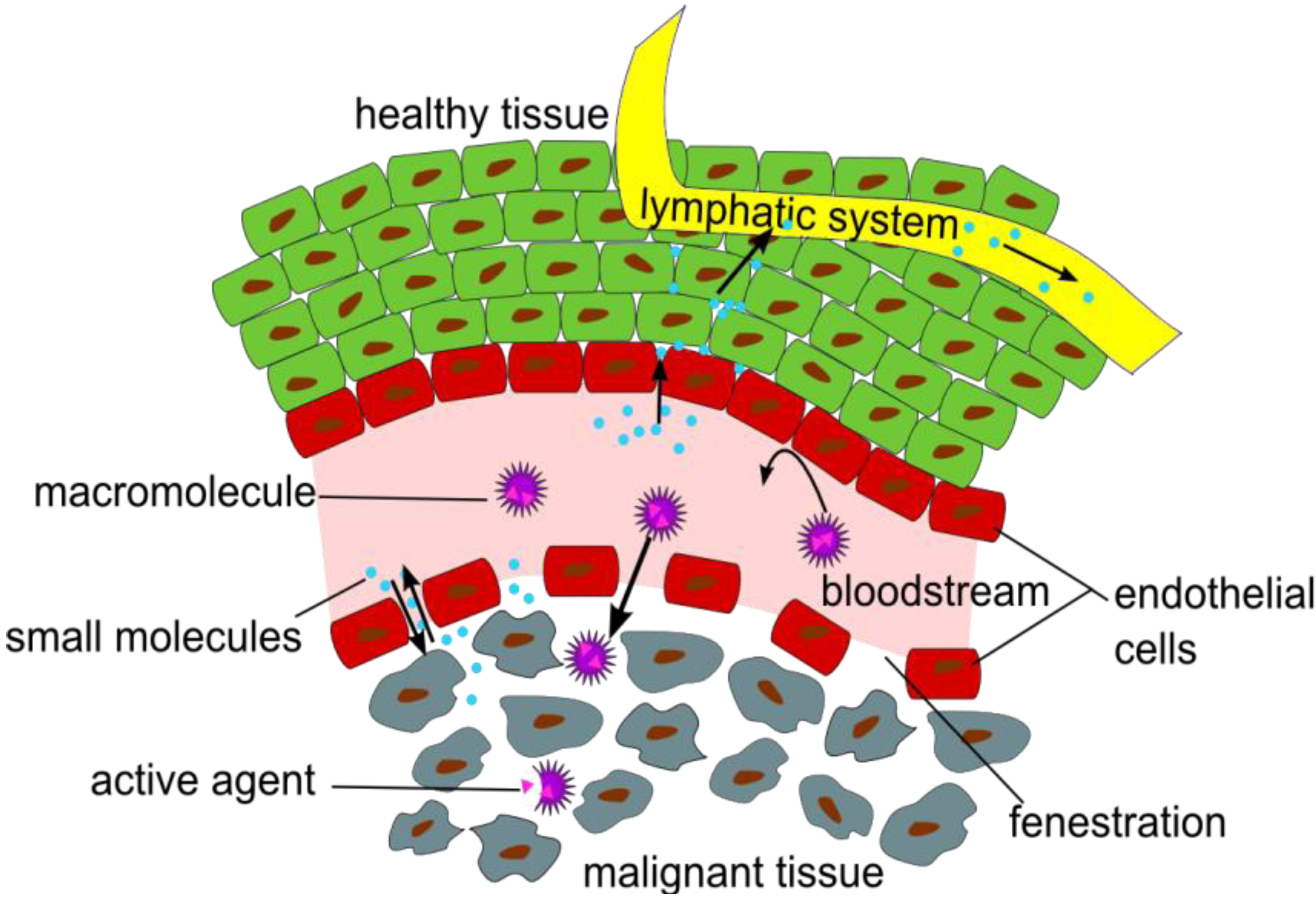
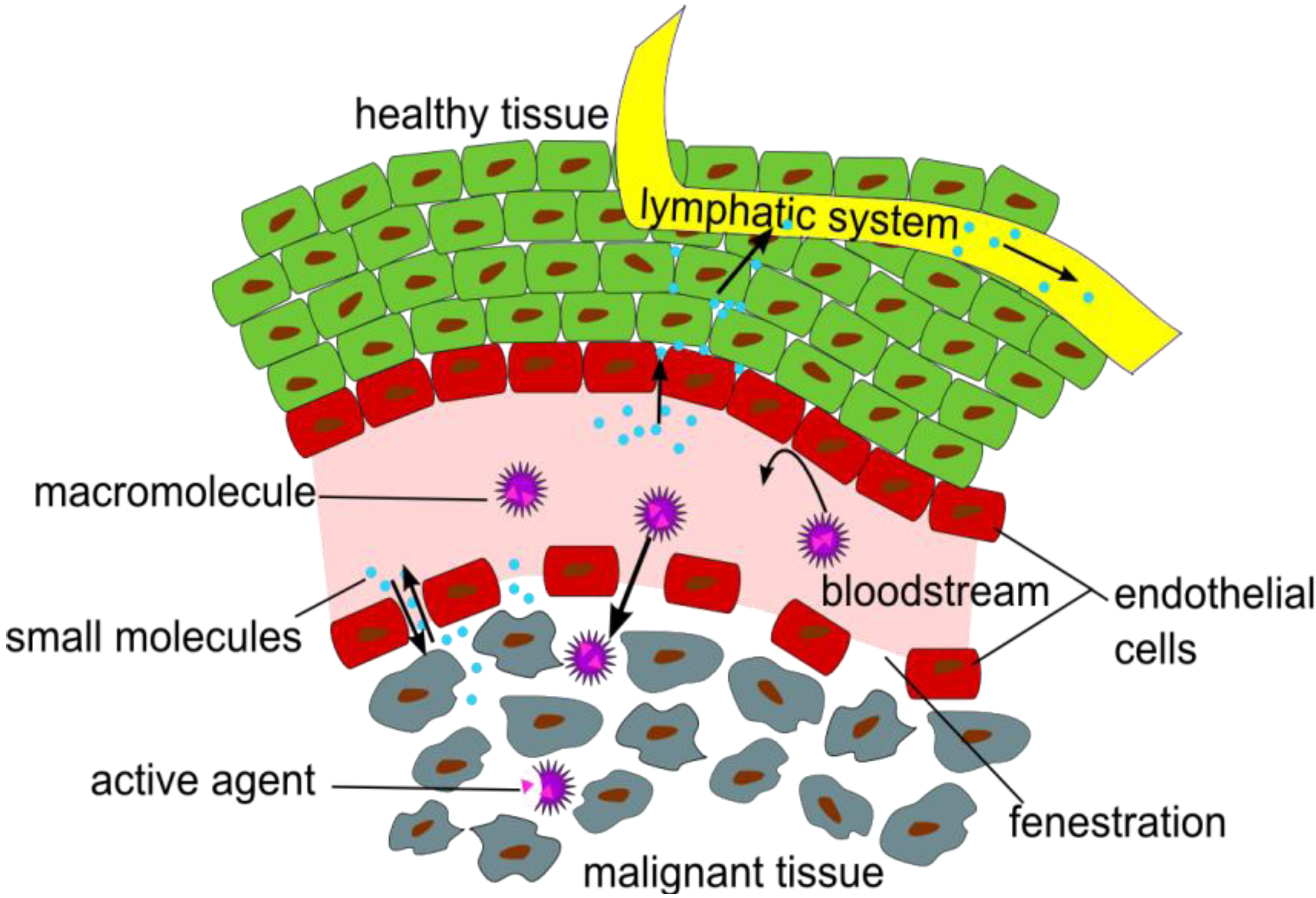
in vivo characteristics of NPs and polymers, it has to be considered how they are interacting with tissues and cells, and especially which time frame allows a suitable visualization of certain effects and functions, like the enhanced permeability and retention (EPR) effect (
Figure 1), which is a passive targeting phenomenon and the mostly used mechanism for the uptake of NP or polymers at oncological target sites in pre-clinical and clinical studies [
5,
6].
Figure 1.
Illustration of the Enhanced Permeation and Retention (EPR) effect of macromolecular structures as drug delivery systems in malignant tissue.
Figure 1.
Illustration of the Enhanced Permeation and Retention (EPR) effect of macromolecular structures as drug delivery systems in malignant tissue.
The EPR effect describes the accumulation of NPs in tumor tissues, due to fenestrations in the blood vessel’s endothelial layer and a significantly reduced lymphatic drainage in the tumor tissue [
7].
Since this review focuses on radiolabeling strategies for polymers and NPs for PET imaging, the radionuclides discussed are positron emitters. Radiolabeling strategies for NPs and polymers using other radionuclides for SPECT or endoradiotherapy are reviewed elsewhere [
8,
9].
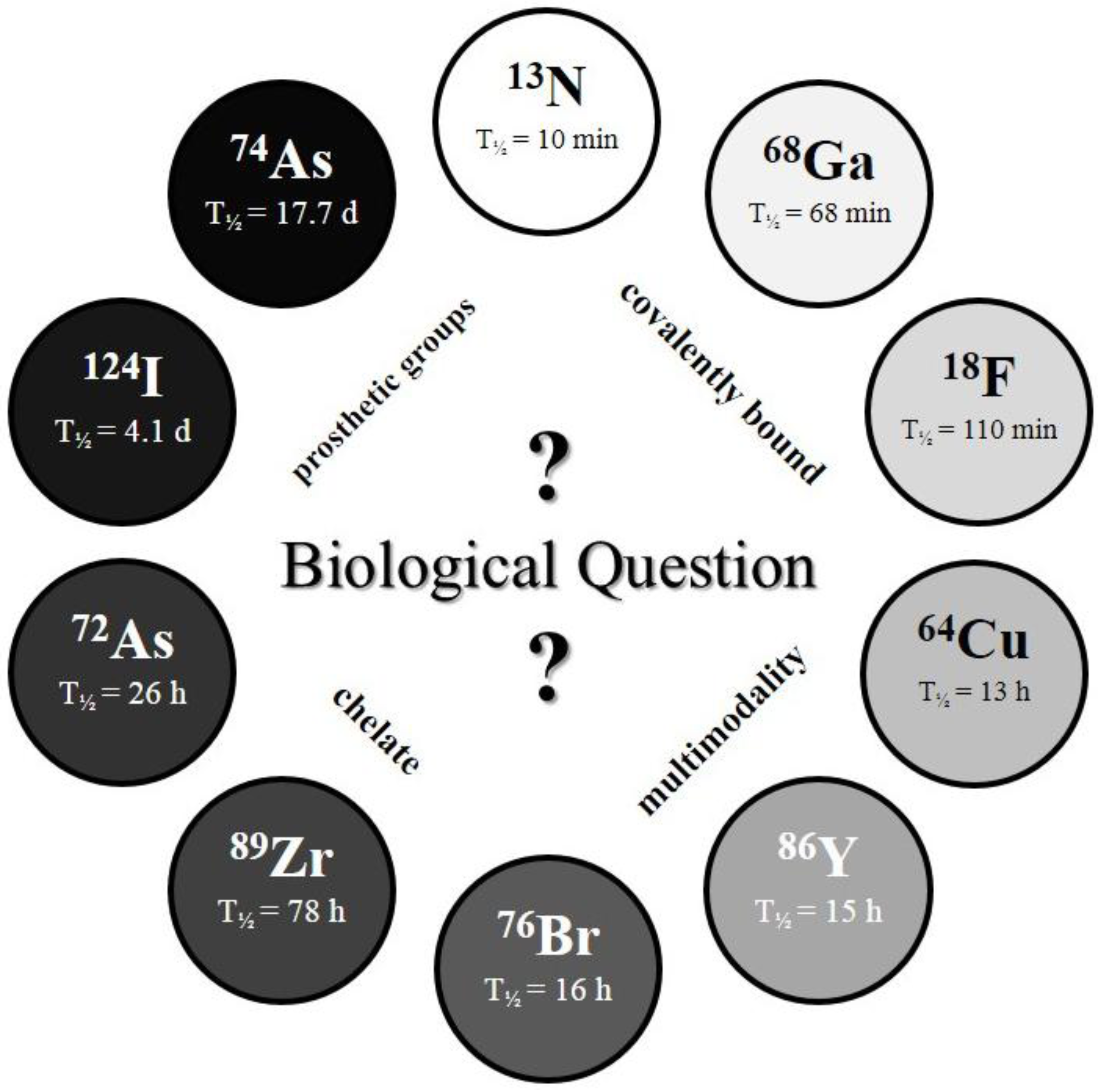
The physical half-life (T
½) of the radionuclide plays a crucial role for measurements in the desired time frame, and it has to be considered which radionuclide or half-life, respectively, is suitable for the investigated question and pharmacokinetic profile (
c.f. Figure 2).
Figure 2.
The Clock-Of-Nuclides showing the positron emitters used for radiolabeling of NPs or polymers, so far. Clockwise starting at 13N (at noon) with the shortest physical half-life and ending at 74As with the longest physical half-life.
Figure 2.
The Clock-Of-Nuclides showing the positron emitters used for radiolabeling of NPs or polymers, so far. Clockwise starting at 13N (at noon) with the shortest physical half-life and ending at 74As with the longest physical half-life.
For measurements within a short (initial) time frame after intravenous administration, short-lived radionuclides have been applied, e.g., fluorine-18 (T
½ = 109.7 min), gallium-68 (T
½ = 67.7 min) or interestingly even nitrogen-13 (T
½ = 9.97 min) [
4,
10,
11]. Exemplarily, fluorine-18, which asks for covalent attachment to the NPs or polymers, is the most widely used PET nuclide and therefore,
18F-labeling strategies are of high interest. Due to its ideal imaging characteristics and good availability,
18F is a very attractive radionuclide for radiolabeling of NPs and polymers. Even if the accumulation, using the EPR effect, is a relatively slow process, first indications about a potential renal clearance or fast metabolism of the NPs or polymers can be obtained by using short-lived radionuclides such as
18F. On the other hand, the multifarious types of NPs and polymers require different coupling strategies, which guarantee fast, stable and high yielding of the respective radiosynthesis/radioconjugation. Numerous possibilities of how a radionuclide can be attached to these systems (either directly or via prosthetic group labeling) are available and essential. In case of
18F, direct radiolabeling is often impossible or provides only low radiochemical yields (RCYs). Consequently, an alternative (indirect) labeling strategy has to be considered, frequently resulting in novel coupling reactions using (novel)
18F-labeled prosthetic groups.
In contrast to the short-lived radionuclides, radiolabeling with longer-lived radionuclides allows a prolonged time frame for scanning. Examples are copper-64 (T
½ = 12.7 h), bromine-76 (T
½ = 16.2 h), iodine-124 (T
½ = 4.1 d) and arsenic-74 (T
½ = 17.8 d) [
12,
13,
14]. Moreover, it has to be considered if the NP or polymer allows radiolabeling
via covalent linkage (e.g., using radioiodine) or
via a bifunctional chelator (BFC) (e.g., using radiometals). Thus, labeling with a radiometal requires a chelator, which forms stable complexes with the radiometal. The most widely used chelators are 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) as examples of macrocyclic chelating agents. Further prominent examples are acyclic chelators like diethylenetriaminepentaacetic acid (DTPA) and deferoxamine (DFO). Additionally, some of the chelating systems enable a
theranostic approach by substituting the diagnostic radionuclide with a therapeutic one, whereas the chelator and the nanodimensional structure remain. Furthermore, it is possible to couple, e.g., NPs, which are MR-active to a chelating system enabling
in vivo tracking by multimodal imaging techniques (e.g., PET/MRI).
Most of the radiolabeled NPs and polymers were subsequently tested in vivo to explore which systems can ultimately serve as for which imaging modality in personalized/individualized therapy approaches. So far, the aim of predominantly preclinical studies is to develop several tools for potential therapy approaches of malignancies and to overcome the current limitations in availability of suitable and individualized (radiolabeled) drug delivery systems.
This review article will summarize information about radiolabeling procedures of NPs or polymers intended for PET imaging and their potential use as drug delivery systems. Additionally, first in vitro/in vivo data are briefly discussed. Furthermore, this article should reveal how the biological question determines the radionuclide selection. In detail, every section has a short introduction followed by a summary table of already used approaches, which includes the type of nanostructure, material, size range of nanostructure, obtained specific activity, reaction time and RCY. Subsequently, the summarized data are discussed and unique characteristics of the radiolabeling and in vivo behavior are highlighted.
2. 18F-Labeling Approaches for Nanoparticles and Polymers
18F is the most commonly used positron emitter and its optimal positron energy (E
β+,max) (635 keV) and high β
+ intensity (97%) are almost ideal for PET imaging. Its half-life of 109.8 min allows for even extensive radiosyntheses and enables shipment of
18F-radiopharmaceuticals or
18F itself. In spite of all these benefits, in the clinics,
18F is mostly used in the form of 2-[
18F]fluoro-2-deoxy-
d-glucose ([
18F]FDG). Recently,
18F has been used as a suitable PET nuclide to track NPs, quantum dots (QDs) or polymers,
in vivo [
4,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24].
Section 2 will give a short overview of the research carried out using
18F-labeling in combination with polymers and NPs and their most important characteristics.
As shown in
Table 1, several different systems have been radiolabeled with
18F over the last years. The short half-life of
18F has, however, undoubtedly limited the number of labeling studies performed. Nevertheless, the early biodistribution data of the first four hours post-injection (p.i.) are crucial with respect to the initial excretion routes and seem to give a good idea of the general pharmacokinetic profile and the potential as a drug delivery system.
Table 1.
An overview of 18F-labeled nanoparticles, polymers and their important characteristics. (n.d. = no data, RCY = radiochemical yield, h.r. = hydrodynamic radii, HPMA = N-(2-hydroxypropyl)methacrylamide, DSPE-PEG2000-NH2 = 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000], Cd = cadmium, Se = selenium, Zn = zinc, S = sulfur, Na = sodium, Y = yttrium, F = fluorine, Yb = ytterbium, Er = erbium, Tm = terbium, Gd = gadolinium, Ce = cerium, O = oxygen, Al = aluminium, Si = silicium).
Table 1.
An overview of 18F-labeled nanoparticles, polymers and their important characteristics. (n.d. = no data, RCY = radiochemical yield, h.r. = hydrodynamic radii, HPMA = N-(2-hydroxypropyl)methacrylamide, DSPE-PEG2000-NH2 = 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000], Cd = cadmium, Se = selenium, Zn = zinc, S = sulfur, Na = sodium, Y = yttrium, F = fluorine, Yb = ytterbium, Er = erbium, Tm = terbium, Gd = gadolinium, Ce = cerium, O = oxygen, Al = aluminium, Si = silicium).
| Nanostructure/system | Material | Size [nm] | Specific activity | Reaction time [min] | RCY [%] | Ref. |
|---|
| phospholipid coated core/shell quantum dot | CdSe/CdZnS DSPE-PEG2000-NH2 | ≥20 | 37–75 MBq/nmol | 145 | n.d | [15] |
| nanoparticles | NaYF4 (co-doped with Yb, Er, Tm, Gd) | 10–20 | n.d. | 10 (only labeling) | 92 | [16] |
| nanoparticle/peptide | gold/CLPFFD (peptide) | 23 (h.r.) | 27 atoms 18F per NP * | 60 (only labeling) | 0.3–0.8 | [17] |
| amino functionalized nanoparticle | CeO2 (ceria) | 5 | n.d | n.d | 17.7 ± 0.3 | [18] |
| hydrophobin functionalized porous silicon | p-type porous silica | 215 ± 54 | 73.4 ± 13.9 MBq/g | 10 (only labeling) | 40.2 ± 0.5 | [19] |
| nanoparticles | Al2O3 (alumina) | n.d | 2.3 ± 0.2 MBq/mg | 6 (only irradiation) | n.d | [20] |
| nanoparticles | gold | n.d | n.d | n.d | n.d | [21] |
| nanoparticles | mesoporous SiO2 (silica) | 100–150 | n.d | n.d | 70 | [22] |
| polymers | HPMA-based block copolymers | n.d | 1.5–2.5 MBq/μmol | n.d | ≥50 | [4] |
| polymers | HPMA-based block copolymers | n.d | n.d | n.d | 10–37 | [23] |
| polymers | HPMA-based block copolymers | 38–113 (h.r.) | n.d | n.d | 5–18 | [24] |
2.1. 18F-Labeled Quantum Dots
QDs are well known for their versatile optical properties and currently, extensive research is focused on new methods to exploit these particles. One unique property for biochemical applications is the fact that QDs often luminesce brightly in the visible area of the spectrum when exposed to UV. Therefore, these particles could be used for multimodality imaging purposes. However, their applicability so far has been very limited due to their composition of toxic metals.
In 2008 Ducongé
et al. [
15] described a novel method for the
18F-labeling of core/shell QDs. The CdSe/CdZnS QDs are well known for their luminescent properties [
25,
26] and the authors described the use of their labeled QDs for PET and optical whole body imaging. After preparation, the QDs were encapsulated with polyethylene glycol (PEG-phospholipid micelles) and this coating was further functionalized with thiol groups. To these functionalities a coupling
via a maleimido-based prosthetic group was performed, which was recently developed for the
18F-labeling of peptides. The maleimido reagent 1-[3(2-
18F]fluoropyridin-3-xyloxy)propyl]pyrrole-2,5-dione ([
18F]FPyME) [
27] has been synthesized within 110 min in 20% yield (not decay corrected). The coupling of the QDs was performed by adding QDs in phosphate buffered saline (PBS) to the dried
18F-labeled maleimido reagent and subsequent vortexing for 15 min. The QDs were purified by gel filtration on a NAP-10 G25 sephadex cartridge, which yielded a 1.5 mL solution of
18F-labeled QDs. The total synthesis took 145 min and a activity concentration of 555–1110 MBq/mL was obtained when the reaction was started with 37 GBq of [
18F]fluoride. The
in vivo experiments showed a long blood circulation time of the QDs with a plasma half-life of 2 h and only small amounts of radioactivity in urine. In comparison to non-PEG-coated QDs, the uptake in liver and spleen could significantly be reduced. In
ex vivo studies, no cadmium was detected in urine, indicating that the small amount of radiation found in the urine correspond to a low-molecular-weight degradation product under the renal threshold, implying that the QDs are not cleared from the body via the renal pathway.
2.2. 18F-Labeled Nanoparticles
NPs exist in a wide variety of sizes and materials, which is advantageous compared to the QDs. They can also be functionalized with different organic groups. These groups can be used to extend circulation time (e.g., PEG) or to conjugate a targeting moiety or a labeling agent. It is further possible to dope NPs with rare earth elements, which are known for their optical and magnetic properties, allowing the particles to luminesce or be used in MRI. Modification of these NPs allows them to serve as multimodality imaging agents.
In 2011 Liu
et al. [
16] developed
18F-labeled rare earth containing NPs. As a basis for their NPs Liu
et al. used NaYF
4, the particles were surface-modified with Gd
3+ by cation exchange for Y
3+ for use in MRI and different rare earth elements (Yb, Er) for use in luminescence studies. [
18F]Fluoride was incorporated into the NPs through interactions with the rare earth ions in an aqueous [
18F]fluoride (170 MBq) solution under sonication for 10 min. The NPs were separated by centrifugation and washed with water (3 times). Radio-TLC indicated an excellent labeling yield of 92%. Surface-modification of the NPs was carried out by coordination of carboxylic acid functions of different (bio)molecules to the surface of the NP. Thus, folic acid for active targeting was bound to the NP as well as oleic acid and aminocaproic acid.
In vitro cell experiments demonstrated low cytotoxicity,
in vivo studies showed a high uptake almost exclusively in the liver (80.9% ID/g) and spleen (36.6% ID/g) already after 10 min showing a decrease in the liver (53.5% ID/g) and further increase in the spleen (89.9% ID/g) 2 h p.i.. The rapid accumulation in spleen and liver was confirmed by MRI and optical imaging studies, furthermore, MRI studies showed a significant contrast enhancement due to the presence of the NPs. Considering the simple labeling method by just mixing [
18F]fluoride with metallic (rare earth) NPs, the
in vivo-stability is remarkable. An early, but stable bone uptake of ~13% ID/g revealed an initial loss of [
18F]fluoride from the particles.
Guerrero
et al. [
17] synthesized and studied
18F-labeled gold-peptide conjugates. Gold NPs were conjugated to an amphipathic peptide CLPFFD, which showed in a previous study the ability to remove
β-amyloid aggregates, which are involved in Alzheimer’s disease [
28,
29].
N-Succinimidyl-4-[
18F]fluorobenzoate ([
18F]SFB) was prepared as prosthetic group and used to radiolabel the NPs. [
18F]SFB was synthesized starting with drying and activating of
18F in the presence of the amino polyether 1,10-diaza-4,7,13,16,21,24-hexaoxabicyclo[8.8.8]hexacosane (Kryptofix
© K2.2.2) and K
2CO
3. After azeotropic drying, the precursor 4-(
tert-butoxycarbonylmethyl)phenyl trimethylamonium trifluoromethanesulfonate, dissolved in acetonitrile, was added and heated to 90 °C for 10 min. Final deprotection was facilitated by 1M HCl (100 °C, 5 min). [
18F]SFB was purified on a C18-HELA cartridge. The eluate was treated with 25% methanolic tetramethylammonium hydroxide solution. After drying, the activated ester was formed by addition of
N,N,N’,N,’-tetramethyl-
O-(
N-succinimidyl)uronium tetrafluoroborate (TSTU). The product was purified by semipreparative HPLC with a RCY of 37%, a radiochemical purity (RCP) of ≥99% and specific activity of 110 ± 15 GBq/μmol. The labeling of the NPs was performed by first drying the NPs by centrifugation and re-dissolving them in DMSO/sodium citrate, followed by the addition of the NPs to 4.44 ± 1.11 GBq of dry [
18F]SFB. This mixture was stirred for 1 h before sodium citrate was added and centrifugation of the whole mixture was performed. Subsequent washing was repeated until no more radioactivity was released. The residual solid was re-dissolved in a mixture of sodium citrate and Tween80. Guerrero
et al. reported for the final product an average of 27
18F atoms per NP (derived from calculation from the radioactivity-to-mass-ratio) with a labeling yield of 0.8% ± 0.3%. During
in vivo studies it was observed that 120 min p.i. a considerable amount of the NPs was trapped by the reticuloendothelial system (RES) in the spleen. Furthermore, a fast renal clearance was observed leading to a rapid blood concentration drop during the first minutes p.i.
Ceria based NPs carrying an amino functionality were studied by Rojas
et al. [
18] Ceria-NPs were surface modified by silylation with 3-(aminopropyl)triethoxysilane to enable subsequent coupling to [
18F]SFB. The [
18F]SFB was prepared using the standard method already used by Guerrero
et al., with almost identical results, a RCY of 37% ± 5%, a RCP of 98% and a specific activity of 102 ± 7 GBq/μmol. The ceria-NPs (2.0 mg ± 0.3 mg) were labeled by stirring them for 1 h in a DMSO (150 μL) phosphate buffer (100 μL; pH = 7.4) mixture with ~2.6 GBq of [
18F]SFB. After the reaction additional phosphate buffer was added and the mixture was centrifuged. The solid was then washed twice with phosphate buffer by centrifugation and the supernatant was checked for radioactivity, leading to a RCY of 18%. After suspension of the NPs in phosphate buffer, they were injected intravenously in Sprague-Dawley rats. High uptake values in liver, spleen and lungs were immediately observed after administration. At 2 h p.i., almost all particles were cleared from the blood. The remaining particles in the blood pool were further cleared
via renal excretion. Only a very limited uptake in the brain was observed.
Sarparante
et al. [
19] functionalized porous silica NPs with hydrophobin as a self-assembled protein coating to study the
in vitro and
in vivo behavior using PET. Thermally hydrocarbonized porous silicon (THCPSi) NPs were synthesized and labeled with
18F. Labeling was facilitated
via a direct
18F-fluorination using the Si-F-bond formation as driving force. Dry cryptate complex (K.2.2.2./K
2CO
3-system) was dissolved in anhydrous DMF containing 4% (
v/v) acetic acid. The solution was added to 1 mg of the NPs suspended in DMF and the mixture was heated to 120 °C for 10 min. The particles were separated by centrifugation and repeatedly washed in ethanol and water using sonication. Finally, the particles were suspended in ethanol. The radiolabeled particles were covered with HBFII (which is a fungal protein class II hydrophobin). In
in vivo studies (rat model, RAW 264.7 macrophages and HepG2 liver cells), the
18F-HFBII-THCPSi NPs accumulated mainly in liver and spleen. The HFBII coating improved the pharmacokinetics and biodistribution compared to the uncoated variant
18F-THCPSi. However, a slow HFBII-leaching from the coated NPs reduced the benefit of the HFBII coating and restored a fast renal clearance of the NPs.
An elegant direct way of labeling Al
2O
3-NPs with
18F was developed by Pérez-Campaña
et al. [
20]. Al
2O
3-NPs were directly activated in a cyclotron via the nuclear reaction
18O(p,n)
18F. [
18O]Al
2O
3-NPs were obtained by reacting AlCl
3 in ammonia and
18O-enriched (100%) water. A short irradiation time (beam time 6 min, current 5 µA) yielded high amounts of radioactivity (2.3 ± 0.2 MBq/mg, saturation yield = 225.45 MBq/µA) of which 71% ± 4% could be attributed to
18F, the residue being assigned to
13N, which is always formed during
18F-production in a cyclotron
via the
16O(p,α)
13N reaction. Due to the much shorter half-life of
13N (9.7 min) the samples could be left for the
13N to decay without a significant loss of
18F. A second irradiation experiment was carried out with pure Al
2O
3, after irradiation only trace amounts of
18F could be detected as was expected from the natural abundance of
18O (0.201%), showing that there are no side reactions on the alumina. PET scans were carried out in dynamic mode and showed a renal excretion of the NPs, while a long retention of radioactivity in the heart indicated a long blood circulation half-life. PET imaging further revealed a minimal uptake in bones, suggesting that
18F might slowly leak from the NPs. Stability tests in rat serum at 37 °C, however, confirmed a stable
18F-label on the NPs with no degradation over a period of 8 h.
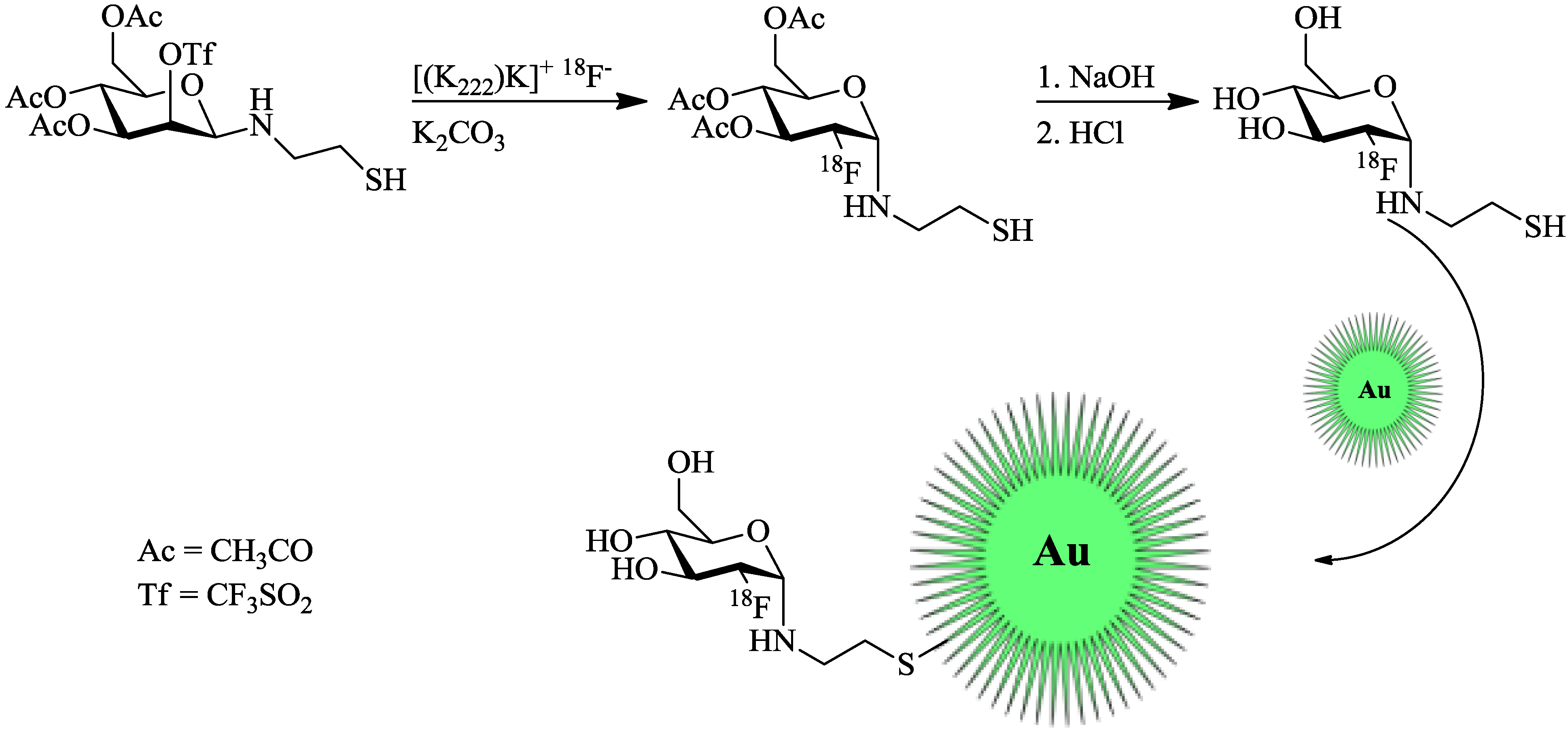
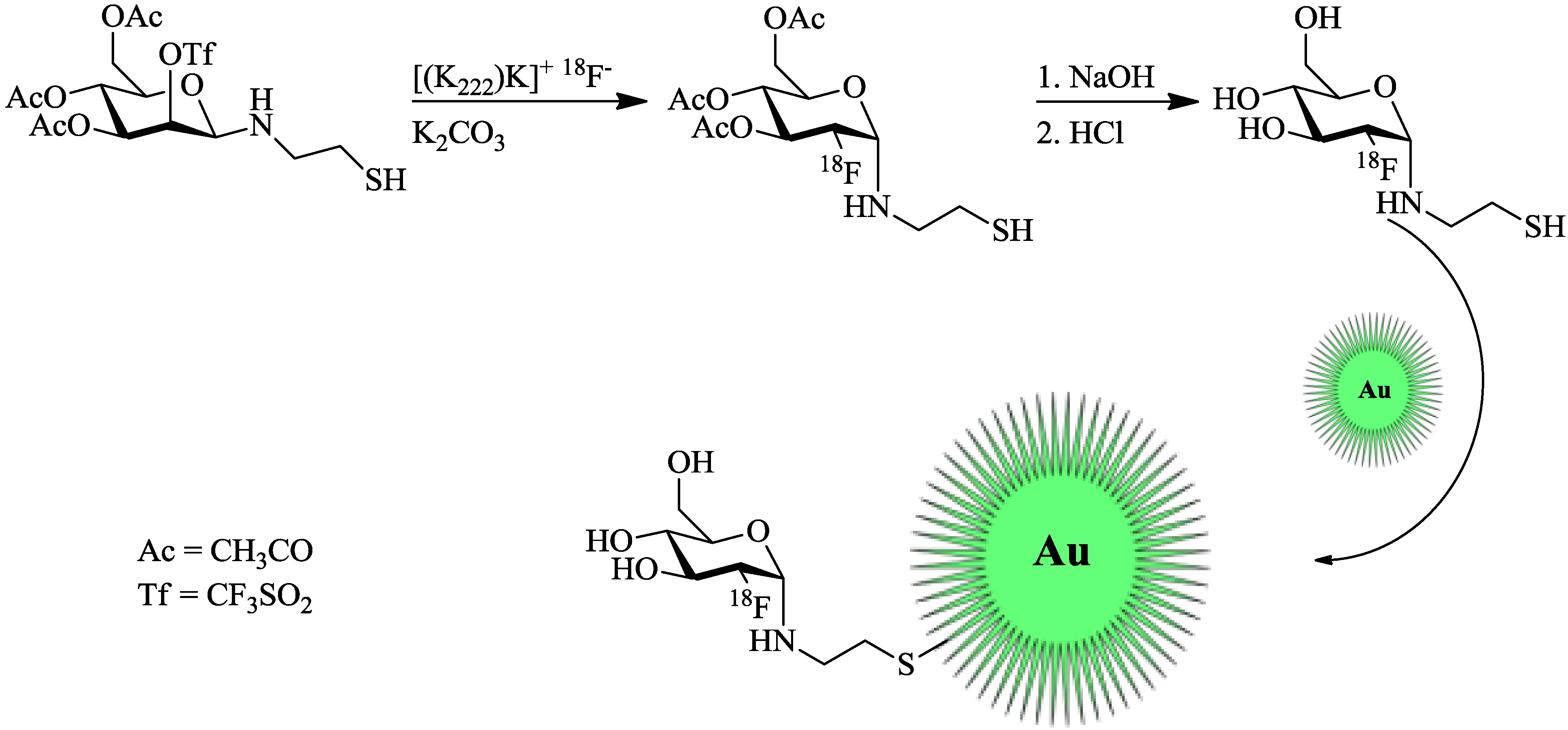
Unak
et al. [
21] worked on radiolabeled gold NPs. However, they only tested them
in vitro in cancer cell cultures (MCF7, human breast adenocarcinoma cell line). Gold NPs were labeled with modified [
18F]FDG. The cysteamine derivative [
18F]FDG-CA was synthesized from a mannose-triflate cysteamine (Man-CA) precursor. Man-CA was produced according to standard methods, which entailed the addition of mannose-triflate to a mixture of cysteamine and NaCNBH
3. Direct
18F-labeling of Man-CA was performed with [
18F]fluoride (7.4 GBq) in the K2.2.2./K
2CO
3 system and DMF at 90 °C for 20 min. Purification was facilitated by elution from different ion exchange columns and a C18 cartridge. Labeling and synthesis of the gold NPs was carried out by mixing the previous prepared [
18F]FDG-CA with 20 mM HAuCl
4 solution and the addition of a NaBH
4 solution (
Figure 3). The mixture was stirred at 60 °C for 2 h, giving the desired
18F-labeled gold NPs. anti-metadherin (anti-MTDH) was conjugated to the particles for an active targeting of metadherin, which is overexpressed in many breast cancer cell-lines. Coupling of anti-MTDH was done by using 1,1'-carbonyldiimidazole as a reagent, involving multiple shaking steps, whereof two were 45 min each. The incorporation of these NPs into MCF7 breast cancer cells was higher than that of free
18F (2%) or [
18F]FDG-CA alone (3%), 30% and 33% respectively for NPs without and with anti-MTDH. Additional apoptosis studies showed the expected decrease
in the apoptotic effect for the NPs (20%), when compared to [
18F]FDG-CA with 30% apoptotic effect, which is known for cysteamine in combination with radiation.
Figure 3.
Radiolabeling of thiol-functionalized Au-NPs using a maleimido-[
18F]FDG. [
18F]FDG was produced in accordance with the standard protocol [
21].
Figure 3.
Radiolabeling of thiol-functionalized Au-NPs using a maleimido-[
18F]FDG. [
18F]FDG was produced in accordance with the standard protocol [
21].
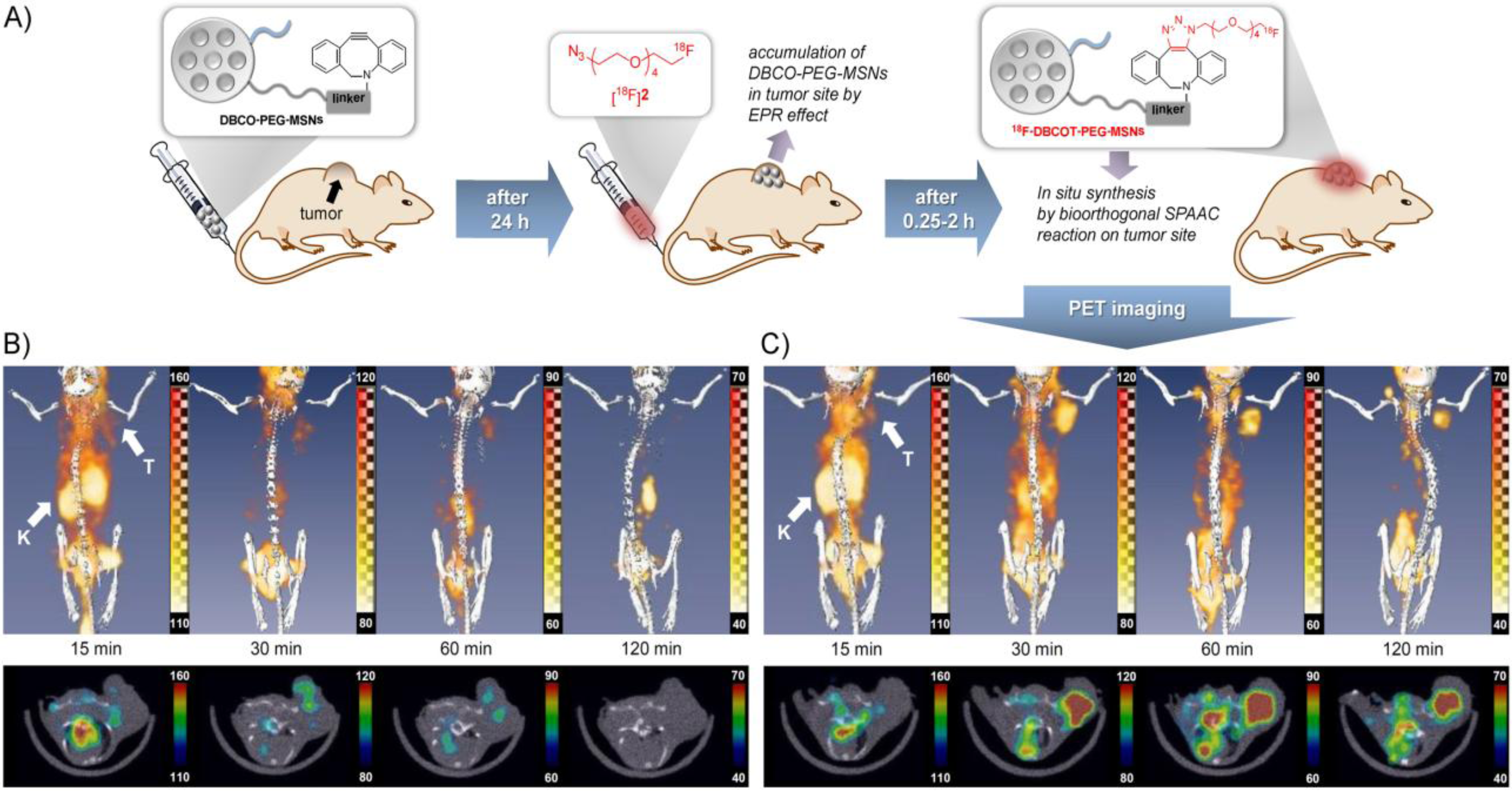
Recently, Lee
et al. [
22] described a bioorthogonal labeling strategy where they applied a copper-free click reaction
in vivo for
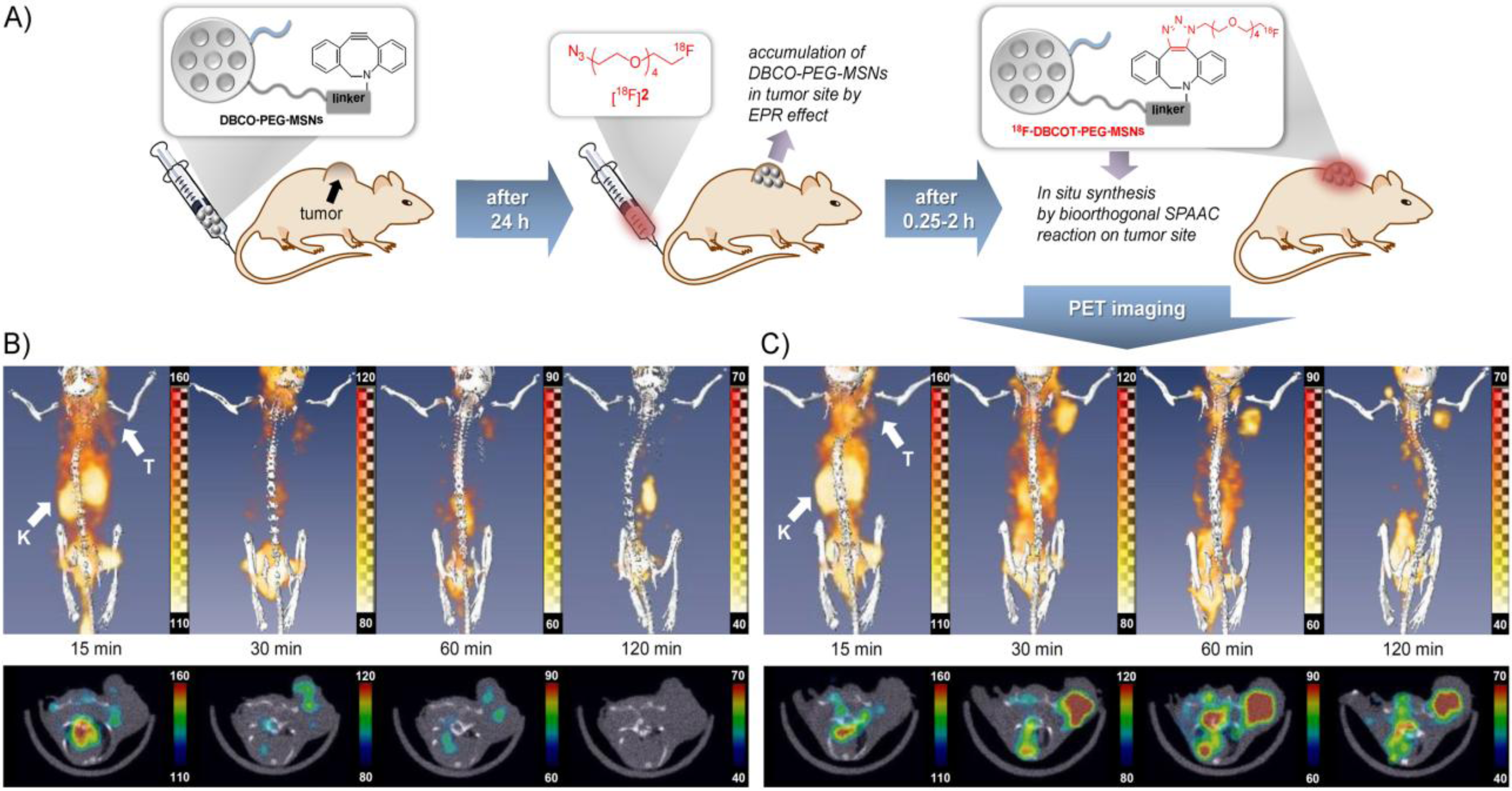
18F-(pre)labeling of NPs (
Figure 4). The radiotracer used in this experiment consisted of ω-[
18F]fluoropentaethylene glycolic azide with a specific activity of 42 GBq/μmol. NPs where modified with azadibenzocyclooctyne (DBCO) and PEG. The reaction was first tested in PBS at 36.7 °C to test if the copper-free click reaction has a chance to work
in vivo, this gave almost quantitative yields within 15–20 min. The modified NPs were injected and allowed 24 h to accumulate in tumors via the EPR effect. After 24 h, the radiotracer was injected and PET images were acquired. A control group was measured, which was given only the radiolabeled product and not the NPs. The comparison between the two groups showed similar uptake in all tissues except for the tumor, whereas the pre-targeted animals showed a much higher tumor uptake than the non-targeted animals which confirms that the copper-free click reaction proceeds efficiently
in vivo. Furthermore, it was found that increasing the amount of radiolabeled compound also increased the tumor uptake and thereby the signal-to-noise ratio.
2.3. 18F-Labeled Polymers
Polymers can appear in many different compositions and architectures. The fact that polymers are derived from organic materials means that they can easily be modified using organic chemistry techniques and that
18F is covalently bound to the molecule, limiting the possibility of leaching. Polymers can be modified to carry targeting agents and/or their architecture can be varied. Some of the known polymers are biodegradable
in vivo with no toxicity at all. Furthermore, several different polymer/drug conjugates are known, e.g., pHPMA-doxorubicin [
30], and about twelve have been approved for the market and about fifteen additional conjugates have entered clinical trials to treat different diseases [
31].
Figure 4.
(
A) Pre-targeting/labeling protocol for
in vivo click reaction. (
B) 3D PET images (upper row) and transversal slides (lower row) of a U87 MG tumor-bearing mouse injected with ω-[
18F]fluoro-pentaethylene glycolic azide without pretargeting. (
C) 3D PET images (
upper row) and transversal slides (
lower row) of a U87 MG tumor-bearing mouse injected with ω-[
18F]fluoro-pentaethylene glycolic azide with pretargeting using DBCO-PEG-NPs. Reprinted with permission from S.B. Lee
et al. [
22]; Copyright 2013 John Wiley and Sons.
Figure 4.
(
A) Pre-targeting/labeling protocol for
in vivo click reaction. (
B) 3D PET images (upper row) and transversal slides (lower row) of a U87 MG tumor-bearing mouse injected with ω-[
18F]fluoro-pentaethylene glycolic azide without pretargeting. (
C) 3D PET images (
upper row) and transversal slides (
lower row) of a U87 MG tumor-bearing mouse injected with ω-[
18F]fluoro-pentaethylene glycolic azide with pretargeting using DBCO-PEG-NPs. Reprinted with permission from S.B. Lee
et al. [
22]; Copyright 2013 John Wiley and Sons.
The polymers discussed in the following section were all labeled using the prosthetic group radiolabeling method for HPMA-based polymers described by Herth
et al. in 2009 [
4]. Generally, for the radiolabeling
18F was first dried and activatedas cryptate complex (K2.2.2/K
2CO
3-system). The precursor ethylene-1,2-ditosylate was added in acetonitrile and heated for 3 min in a sealed vial to complete the
18F-labeling. Purification from crude mixture was facilitated by HPLC (Lichrosphere RP18-EC5, acetonitrile/water). The HPLC-fraction containing the 2-[
18F]fluoroethyl-1-tosylate ([
18F]FETos) was diluted with water (1:4 HPLC fraction/water) and loaded on a C18-Sep-Pak cartridge (Waters, Milford, MA, USA), dried by a nitrogen stream and eluted with DMSO, ready for further coupling reactions. The whole preparation takes around 40 min with a RCY from 60% to 80%. Labeling of the polymer was typically done by dissolving about 3 mg of polymer in DMSO followed by the addition of 1 μL of 5N NaOH solution and addition to the [
18F]FETos solution. The coupling reaction was performed at temperatures between 80 and 150 °C for 20 min. Radio-size exclusion chromatography (SEC) was used to separate the smaller compounds from the labeled polymer. The SEC is performed with physiological saline solution and hence, the collected polymer fraction is directly available for
in vivo studies.
Herth and co-workers described the labeling of an HPMA-polymer by using [
18F]FETos, which was coupled to the hydroxyl group of a tyramine moiety which was previously incorporated in the polymer during the polymer analogous reaction [
4]. Several polymers of different molecular weights were labeled with
18F and a systematic study was done to find the optimal
18F-labeling conditions. Best results were obtained using 120 °C for 10 min in DMSO. Moreover, at 60 °C 20% RCY could be obtained within 20 min leading the authors to conclude that labeling at room temperature would be possible. The authors further reported that the compound was cleared from the body predominantly via the urine and that initially after injection some metabolism was visible.
In 2011 a follow-up was made to this study by Allmeroth and Moderegger
et al. [
23] where different polymers were synthesized and
18F-labeled. The polymers were either HPMA homopolymers or HPMA-
ran-LMA (LMA = lauryl methacrylate) copolymers. Different molecular weight (M
W) polymers were synthesized with sizes ranging from 12,000 to 130,000 g/mol. The obtained RCY in this study varied and a trend was noticeable that the RCY decreased with increasing M
W, the RCY also decreased when HPMA-
ran-LMA copolymer was used instead of the HPMA-homopolymer. During
in vivo studies, a clear difference was noticeable between the high and low M
W polymers. Expectedly, the low M
W polymers tended to renal excretion with 12% ID/g in kidneys while for the high M
W polymers this was reduced to 6.4% ID/g. The authors found a difference in circulation time between the homo- and
ran-LMA-co-polymer with a similar M
W. The HPMA-
ran-LMA copolymer had a much longer blood circulation time with around 30% ID/g for the low M
W and 60% ID/g for the high M
W polymer still present in the blood at 2 h p.i.
HPMA-
b-LMA block-copolymers were employed in 2013 [
24] to study the effect of different grades of PEGylation. This work was specifically focused on trying to avoid aggregate formation and reducing protein interactions by coating the polymer with amino-functionalized PEG
2000 fragments.
18F-Labeling of the polymers was again carried out with [
18F]FETos, however, this time Cs
2CO
3 was used as base instead of K2.2.2/K
2CO
3. The authors came to the conclusion that the introduction of the PEG
2000 as well as the degree of PEGylation has a great influence on the pharmacokinetics of the polymer. Thus, the polymer with only a small percentage of PEGylation still showed a fast clearance via the liver and a short blood circulation time. An increase in the PEGylation grade correlated with an increase in the blood circulation time and an increased tumor uptake in the Walker-256 carcinoma (rat model).
18F-Labeling of NPs and polymers is just at the very beginning, but the first examples have already shown their potential in both the suitability of the radiochemistry and the benefit of the pharmacological information from PET imaging. There is still a broad range of methods for the 18F-introduction to be explored for their applicability to macromolecular drug delivery systems and further studies and research will quickly follow.
In the next section, the radiolabeling for NPs and polymers and their preliminary preclinical evaluation using the metallic PET nuclides 68Ga and 64Cu is summarized and discussed.
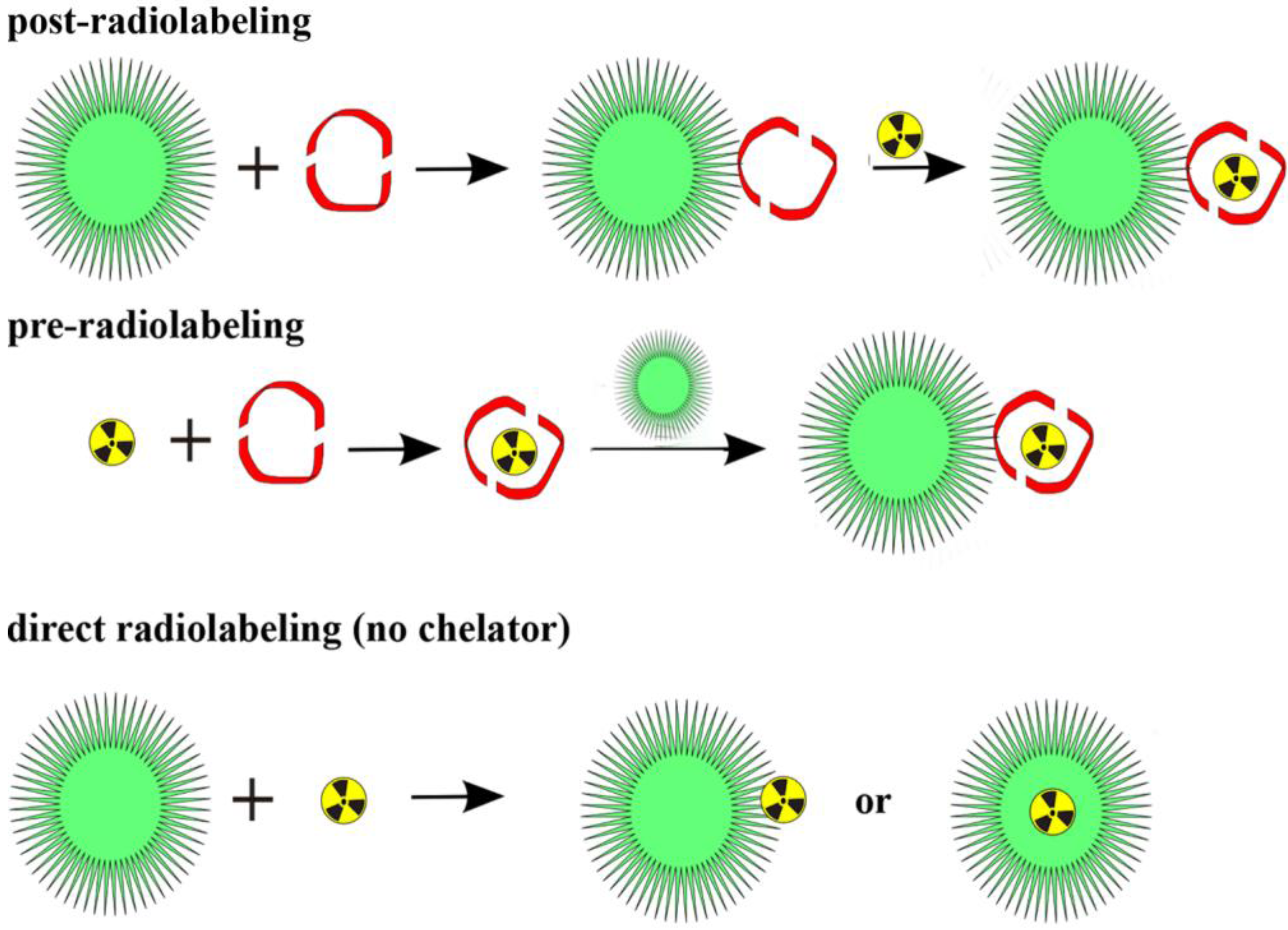
3. Labeling of Polymers and Nanoparticles with 64Cu and 68Ga
There are only a few radiometals suitable for PET, including
64Cu,
68Ga and
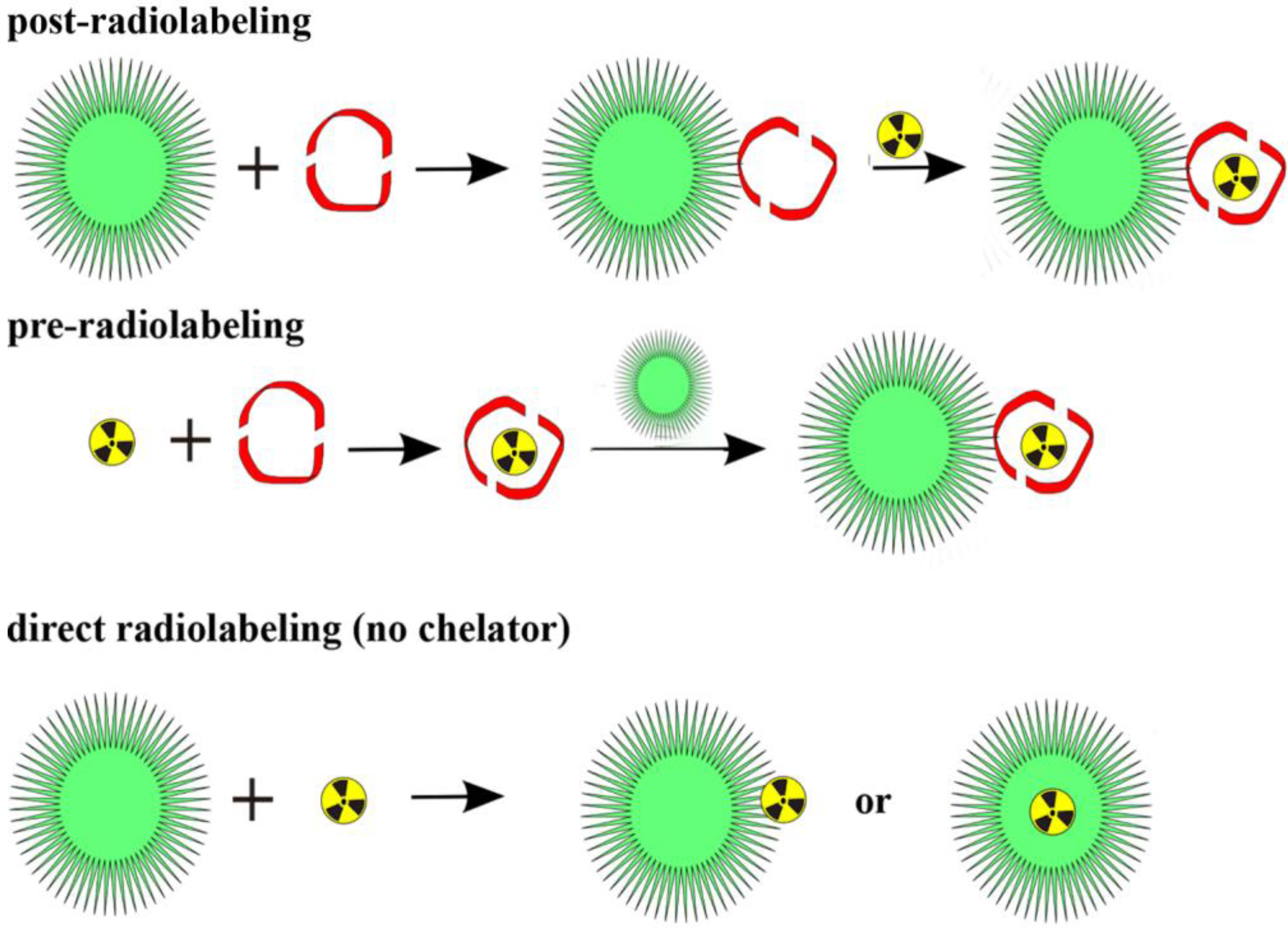
89Zr, which are attached to (polymeric and nanodimensional) molecules via BFCs. Thus, the chemistry is quite different from that of covalently bound radionuclides. There are two main roots to label a polymeric or nanodimensional system (
Figure 5). Either the label is attached to the whole polymer/particle, which has been synthesized in advance, or one compound (in the case of metals this is a BFC) is labeled first and subsequently the polymer/particle is formed. Whether to use the so called pre- or postradiolabeling [
32,
33] is dependent on several factors, above all the half-life of the chosen radionuclide. In this section, the approaches in
64Cu and
68Ga-(nano)chemistry are discussed.
Figure 5.
Three general radiolabeling approaches using metallic radionuclides and nanoparticles.
Figure 5.
Three general radiolabeling approaches using metallic radionuclides and nanoparticles.
3.1. Radiolabeling with 64Cu
Copper-64 with a half-life of 12.7 h, its a positron energy E
β+,max of 655 keV and an β
+ intensity of 17% [
34] is a frequently used PET nuclide which is available via different production routes. The most common one is the
64Ni(p,n)
64Cu nuclear reaction providing good yields of up to 241MBq/µAh [
33,
34,
35].
As a radiometal, copper-64 requires a BFC for attaching it to biomolecules and forming stable complexes. The most popular one is DOTA or derivatives of this macrocycle. Originally, DOTA was designed for lanthanides (e.g., Gd
3+), but it can be used for a wide range of (radio)metals as well. Since DOTA has four carboxylic functions on the side-chains of the macrocycle bearing four nitrogens, leading to a deformed octahedral complexation of the Cu
2+-ion, which is preferred due to the Jahn-Teller effect [
36] thereby leaving two of the acidic functions “free”. Thus, one is available for the coupling to a molecule (in our case NPs or polymers) and the other one allows further derivatization or acts as additional hydrophilic group. Additional macrocyclic chelators are TETA (1,4,8,11-tetraazacyclotetradecane-
N,N',N'',N'''-tetraacetic acid) and NOTA or derivatives of those.
The general
64Cu-labeling conditions in literature are very similar: Since the product of the bombardment at the accelerator (after target dissolving and work-up) in every case is CuCl
2 in hydrochloric acid, this species is transferred into the copper(II)acetate by adding ammonium acetate buffer. Subsequently, the solution of the polymer/particle is added and the mixture is heated for 30–240 min [
32,
37] to 40–43 °C [
32,
37,
38,
39,
40,
41,
42] or to 80–95 °C [
43,
44,
45,
46,
47]. The pH-value varies from nearly neutral (7.4 [
40]) to slightly acidic 5.5 [
47,
48,
49,
50,
51].
In the same manner, purification methods of crude labeling mixtures are quite similar. The excess of copper is removed by either adding another chelating agent (in most cases DTPA [
37,
40,
41,
42,
43,
46,
50], but also EDTA is used [
44,
48,
50]), bySEC (with PD10 [
38,
40] or by a filter centrifugation [
47,
49,
50,
51]).
Table 2 gives an overview on the different approaches in radiolabeling NPs and polymers with
64Cu and
68Ga.
Table 2.
An overview of different approaches of 64Cu-labeled nanoparticles and polymers and important parameters. (n.d. = no data, RCY = radiochemical yield, RCP = radiochemical purity, PAA-PMA = polyacrylic acid – polymethacrylic acid, PS-PAA = polystyrene – polyacrylic acid, PMMA = polymethylmethacrylic acid, PMASI = polymethacryloxy-succinimide).
Table 2.
An overview of different approaches of 64Cu-labeled nanoparticles and polymers and important parameters. (n.d. = no data, RCY = radiochemical yield, RCP = radiochemical purity, PAA-PMA = polyacrylic acid – polymethacrylic acid, PS-PAA = polystyrene – polyacrylic acid, PMMA = polymethylmethacrylic acid, PMASI = polymethacryloxy-succinimide).
| Nanostructure/ system | Material | Size [nm] | Chelator | Labeling time | T [°C] | pH | RCY (RCP) [%] | Specific activity | Ref. |
|---|
| organic polymer (star or arm) | mPEG (metoxy-terminated PEG) | 25–70 | DOTA | n.d. | n.d. | n.d. | n.d. (≥95%) | ≥3700 kBq/µg | [52] |
| core-shell arm or star copolymers | PEG,
N,N-dimethylacrylamide, N-acryloxysuccinimide | 5–70 | DOTA | 1 h | 80 | n.d. | n.d. (≥95%) | 185-370 GBq/mg | [43] |
| organic polymers | PAA-PMA, PEG (folic acid) | 20 | TETA | 2.5 h | 43 | 7.4 | 15%–20% (95%) | n.d. | [40] |
| inorganic QDs | silicone | 15 | DOTA | n.d. | n.d. | 5.5 | 78% | n.d. | [48] |
| inorganic NPs | iron oxide | 30 | DOTA | 1 h | 40 | 6.5 | n.d. | n.d. | [39] |
| organic polymers | PS-PAA | 13–47 | DOTA | 2 h | 43 | n.d. | n.d. | n.d. | [42] |
| organic polymers | poly(
t-butyl acrylate), PEG, methyl acrylate, styrene | 18–37 | TETA | 2–4 h | 43 | n.d. | n.d. | n.d. | [37] |
| organic polymers | PMMA, PMASI, PEG | 10–20 | DOTA | 1 h | 80 | n.d. | n.d. (≥95%) | 0.4–0.8 MBq/µg | [46] |
| organic polymer | CANF (C-atrial natriuretic factor) comb | 20, 22 | DOTA | 1 h | 80 | n.d. | 60:5% ± 7.3% | n.d | [45] |
| organic polymer | CANF (C-atrial natriuretic factor) comb | n.d. | DOTA | 1 h | 80 | n.d. | n.d. | n.d. | [44] |
| inorganic NPs | iron oxide | 68 | NOTA | 40 min | 40 | 6.5 | n.d. | n.d. | [40] |
| inorganic NPs | silicone | 77 | DOTA | 1 h | | 5.5 | n.d. | n.d. | [49] |
| organic polymers | glycol chitosan | 300 | DOTA | 30 min | 40 | n.d. | ≥98% | 11 MBq/mg | [32] |
| QDs | CdSe | 12; 21 | DOTA | 1 h | 37 | 5.5 | ≥95% | ≥37 GBq/µmol | [51] |
| inorganic NP | dextranated iron oxide | 20 | DTPA | 25 min | 95 | 5.5 | n.d. | 370 MBq/mg Fe | [47] |
| QDs | CdSe and InAs | 2; 12 | DOTA | 1 h | 37 | 0.1 5.5 | n.d. | n.d. | [53] |
| inorganic NPs | iron oxide | 20 | DOTA | 1 h | 37 | 5.5 | 94% (≥95%) | 2–4 GBq/mmol | [40,50] |
As mentioned before, two general radiolabeling approaches are available, pre- and postradiolabeling. Several examples are described and discussed below.
3.1.1. Post-Radiolabeling
Most of the labeling procedures described here, are “post-radiolabeling” processes [
32,
33], meaning that the polymeric or nanoparticular system is formed first, a chelator is coupled to this system and in the last step the radiolabel is introduced. This procedure has been applied to various nanodimensional structures, for organic polymers as well as for inorganic NPs.
64Cu-Labeled Organic Polymers
Pressly and coworkers attached DOTA to a C-type atrial natriuretic factor (CANF) functionalized Comb-copolymer for targeting the natriuretic peptide clearance receptor in prostate cancer [
44,
45]. After the self-assembly of the DOTA-CANF-Comb-copolymer to a particle,
64Cu-radiolabeling was done by adding approximately 5 pmol of the readily prepared particles to a solution of 185 MBq
64Cu at pH 5.5 at 80 °C. Subsequently, both purification methods were applied, EDTA-challenge as well as a desalting column. The
64Cu-labeled CANF-Comb-copolymers were tested in CWR22 tumor mice and compared with a non-targeted version,
64Cu-Comb-copolymer. The targeted version showed significantly higher tumor uptake of
~9% ID/g (24 h) than
64Cu-Comb-copolymer (~3% ID/g, 24 h). Moreover, the uptake of the
64Cu-CANF-Comb-copolymers decreased in all non-targeted tissues and organs over time (1-24 h), while the tumor accumulation increased over time from ~4% to ~9% ID/g. Under blockade conditions (100-fold excess of inactiveCANF-Comb-copolymer) the tumor uptake was significantly reduced, and thus CANF-specificity was confirmed [
44].
Welch
et al. have chosen a similar root, by coupling DOTA to a polymer via amide formation, and performed radiolabeling with
64Cu after self-assembling [
52]. It was demonstrated that the shape of the polymer (
star- or
arm-shaped) has a stronger influence on its biodistribution in BLAB/C mice than the size or molecular weight, respectively. Thus, in contrast to the arm polymer, the star polymer was found to have an extended blood circulation time. Additionally, the arm polymer showed a greater uptake in liver and spleen than the star polymer [
52].
Rossin
et al. combined the passive targeting that NPs automatically undergo with active targeting by attaching folic acid to their shell-cross-linked micelles. Thus, they could ratify the EPR-effect, but at the same time they could not see a clear difference between the folate-conjugated tracer and the polymer without targeting-vector [
40]. TETA was used as chelating agent and conjugated to shell-cross-linked nanoparticles (SCKs) composed from an amphiphilic block-copolymer. The SCKs were mixed with 185 MBq
64Cu and reacted at slightly increased temperatures for 2.5 h, before free copper was removed by a DTPA-challenge, giving a RCP of ≥95%. Although there was no obvious difference between the targeted and the non-targeted micelles in tumor uptake (human KB cells, female nu/nu mice), the liver uptake increased due to the functionalization with folic acid [
40].
64Cu-Labeled Inorganic Nanoparticles
In most cases, the inorganic substances that are described are silicon-based or iron-based NPs. In addition, some groups exploit the physical properties of the metals, such as their magnetic behavior or their suitability as MRI or CT contrast agents. Such NPs show potential as dual modality imaging agents. A combination of a morphological imaging technique (MRI/CT) and PET as a functional imaging technique is one of the most potent imaging systems.
Tu
et al. coated manganese-doped silicon QDs with dextrane and coupled a DO3A-derivative as chelator.
64Cu-Radiolabeling was facilitated in acetate buffer (pH 5.5), followed by an EDTA-challenge. After centrifuge filtration, a RCY of 78% was achieved.
In vivo PET images show that the QDs were retained in the bladder and liver for over 1 h p.i. and can still be found in the liver at 48 h p.i. In general, a rapid blood clearance was observed (<2.5% ID/g 10 min p.i. in the blood) [
48]. Originally, DOTA-NHS-ester was applied as chelator, but the activated ester did not couple to the particles’ surface. The authors assumed that the side chain was slightly too short and designed a DO3A-derivative with a propylamine chain. As a result, the coupling worked in satisfying yields and enabled radiolabeling [
48].
Huang
et al. loaded mesoporous silica nanoprobes with a near infrared (NIR)-dye, and labeled the nanostructures with two different metal ions, which were Gd
3+ (T
1-contrast agent in MRI) and
64Cu
2+ for PET imaging, respectively. As a chelating agent DOTA was employed on the one hand, and on the other hand the authors exploited the fact that both the copper and the gadolinium embed into the surface pores. The labeling conditions were 1 h of constant shaking at a pH of 5.5 and 40 °C. Stability tests showed that the procedure delivers a highly stable radiotracer, which exhibits excellent uptakes in the sentinel lymph node (SLN), which could be demonstrated in PET imaging using BALB/C mice carrying 4T1 tumors [
49,
54].
Non-iron and non-silica-based inorganic particles have been radiolabeled by Schipper
et al. to investigate the biodistribution in living mice of CdSe-QDs, which carry DOTA at their surfaces. They labeled the particles at a pH of 5.5, 37 °C and for 1 h and gained excellent RCYs of ≥95% and quite high specific activities of ≥37 GBq/µmol.
In vivo microPET images and biodistribution data showed a delayed uptake in the organs of the RES (liver and spleen) if the QDs are coated with PEG or peptide(s). Additionally, the group could show that hydrodynamic diameters of their QDs (12 nm and 21 nm) had no influence on the biodistribution [
51,
53].
3.1.2. Pre-Radiolabeling
Recently, Dong-Eun Lee
et al. followed a very interesting approach to radiolabel glycol chitosan nanoparticles (CNPs) [
32]. Copper-free click chemistry was applied to attach a
64Cu-radiolabeled alkyne complex to azide-functionalized CNPs
in vivo. The general labeling procedure was (due to DOTA as chelator) 30 min at 40 °C, where a DOTA-DBCO-conjugate was labeled with excellent RCYs of more than 98% within 30 min at 40 °C. PET imaging in SCC/-tumor-bearing mice showed promising results with an excellent tumor-to-background contrast. Furthermore, a significant uptake in liver and kidneys was observed.
3.2. Radiolabeling with 68Ga
The amount of reports about
68Ga-labeled polymers and NPs is much smaller than for
64Cu.
Table 3 gives an overview. Gallium-68 decays with a half-life of 67.71 min under β
+-decay into stable zinc-68. It has 89% positron branching accompanied by 3.22% γ-emission [
55]. Its positron energy E
β+,mean of 740 keV [
55] is ideal for PET imaging and provides a high spatial resolution.
Table 3.
An overview of the different approaches of 68Ga-labeled nanoparticles and polymers and crucial parameters. (n.d. = no data, RCY = radiochemical yield, RCP = radiochemical purity, CAN = cerium-ammonium-nitrate).
Table 3.
An overview of the different approaches of 68Ga-labeled nanoparticles and polymers and crucial parameters. (n.d. = no data, RCY = radiochemical yield, RCP = radiochemical purity, CAN = cerium-ammonium-nitrate).
| Nanostructure/ system | Material | Size [nm] | Chelator | Labeling time [min] | T [°C] | pH | RCY (RCP) [%] | Specific activity | Ref. |
|---|
| organic nanogels | PEG | 250–270 | NODAGA | 15 | RT | 4.5 | ≥99% | ≥1500 GBq/g | [56] |
| inorganic NP | iron oxide, oleanic acid | 60 | NOTA | 20 | RT | 5.0–5.5 | n.d. | n.d. | [57] |
| inorganic NP | γ-Fe2O3, CAN; PEG-coat | 44–55 | NODAGA | 30 | 60 | 3.5 | 84% ± 6% | n.d. | [11] |
| superparamagnetic NPs | iron oxide amino-silane coated | 100 | none | 20 | 70 | n.d. | (≥95%) | 358 MBq/nmol | [58] |
| organic polymer | poly-glycidyl-methacrylate(poly-2,3-epoxy-propylmethacrylate) | 144 | none | 15 | 82–60 | n.d. | n.d. | 0.2 MBq/mg | [59] |
In contrast to copper-64, gallium-68 is a generator-produced nuclide. It is the daughter of germanium-68 which has a half-life of 270.8 d and decays under electron capture into gallium-68 [
55]. Thus, as a major advantage no on-site cyclotron is required and the costs for nuclide production are much lower.
Gallium, as well as copper, requires a chelating agent, which can be DOTA (frequently used for copper), but also NOTA which is a highly potent chelator for the smaller gallium(III)-cation. So far, only NOTA and its derivatives have been used to radiolabel organic and inorganic nanodimensional systems with gallium-68.
Due to the fact that the chelator is the same (or at least a derivative of NOTA) the quite different labeling methods are surprising. Sing
et al. nearly had quantitative RCYs by labeling nanogels after 15 min at room temperature [
56], whereas Locatelli and coworkers used much higher temperatures and twice as much time and did not reach more than 90% RCY [
11]. Both used NODAGA as chelating system. Noteworthy, the labeled structures are of fundamental difference. Locatelli
et al. described inorganic NPs composed of γ-Fe
3O
4 and cerium ammonium nitrate, whereas Singh
et al. investigated (organic) polymeric nanogels.
Locatelli
et al. also performed PET imaging studies with male Sprague Dawley rats to find out that the uptake in liver, spleen and lungs is quite high. A high uptake and long retention in the heart indicates a long blood circulation time [
11].
In contrast to the radiocopper chemistry, in the radiogallium labeling chemistry methods like DTPA-challenge are rather exotic. Purification methods and procedures for
68Ga-radiotracers are commonly based on SPE (solid phase extraction) cartridges as well as SEC. Due to the time-consuming procedure, the latter is unfavorable. However, Singh
et al. employed PD10-columns to separate the
68Ga-labeled product from the crude reaction mixture, and Locatelli
et al. used ultracentrifugation for purification [
11,
56].
Kim
et al. applied very mild labeling conditions. By adjusting the pH to 5.0–5.5 [
57] they are at the upper limit of the suitable pH-range for
68Ga-labeling reactions. Oleanic acid conjugated iron oxide NPs (IONPs) (carrying again NOTA) were successfully radiolabeled within 20 min at room temperature. As a result, a potent dual imaging agent for MRI and PET was prepared. They show very nicely a significant uptake in the tumor for both imaging systems, MRI and PET [
57].
A quite interesting work was reported by Stelter
et al., where they did not use any chelating agent at all. They simply utilized the fact that primary amines can form stable complexes with gallium(III) and coated their particles with aminosilanes. For radiolabeling, 130 MBq
68Ga were added for 20 min at 70 °C. Afterwards a DTPA-challenge which is common in copper-radiochemistry was employed. Hence, they could on the one hand eliminate the surplus
68Ga and on the other hand ensure the stability of their compound against transchelation. They gained excellent RCPs (≥95%) and demonstrated feasibility in
in vivo small animal PET studies in Wistar rats. The compound only accumulated in liver and spleen [
58].
A similar approach,
i.e., without chelator, was published by Cartier
et al. in 2007. They used EPMA-particles (poly-2,3-epoxypropylmethacrylate) and 300 MBq
68Ga for radiolabeling and started at temperatures of about 82 °C and let it drop to 60 °C during a period of 15 min. After purification via PD10, the specific activities were 0.2 MBq/mg lattice. Injection into Wistar rats and PET imaging showed that this compound accumulates in the liver 1 h p.i [
59].
In gallium-chemistry, no pre-radiolabeling approach like in copper-chemistry has been published, yet. Of course, the much shorter half-life of the gallium-68 plays the predominant role, here. However, only the development of faster labeling methods and rapid coupling/conjugation chemistry would enable radiolabeling via the pre-radiolabeling approach for 68Ga. On the other hand, the existing labeling strategies enable the development of pre-targeting approaches based on 68Ga, which is particularly suitable for the application with NPs and polymers.
4. Labeling of Polymers and Nanoparticles with Other Positron Emitters
As already mentioned before, a broad variety of different radionuclides with a wide range of half-lives available for NPs and polymers is of paramount interest. Such a pool of nuclides is crucial to meet the imaging requirements for following a slow pharmacological process like the EPR effect with PET measurements over a longer period (
i.e., days). On the other hand, data about the initial biodistribution and pharmacokinetics have already proven a high significance for the applicability of a potential drug delivery system. Besides, the major radionuclides
18F,
64Cu and
68Ga, several other positron emitters have been applied for radiolabeling of NPs and polymers (
Table 4). Remarkably, the corresponding half-lives are spread over a very wide range from minutes to weeks, and thus offer very interesting possibilities with a great flexibility.
Table 4.
Further positron emitters used for radiolabeling of polymers and nanoparticles and their decay properties [
13,
60,
61,
62].
Table 4.
Further positron emitters used for radiolabeling of polymers and nanoparticles and their decay properties [13,60,61,62].
| Positron Emitter | Half-life | Decay Properties (%) | β+,max-energy [MeV] | Production Route | Daughter (T½) |
|---|
| 13N | 9.97 min | β+ (100) | 1.19 | 16O(p,α)13N | 13C (stable) |
| 11C | 20.4 min | β+ (99.8)/EC (0.2)/ | 0.96 | 14N(p,α)11C | 11B (stable) |
| 86Y | 14.7 h | β+ (33)/EC (66)/γ | 3.14 | 86Sr(p,n)86Y | 86Sr (stable) |
| 76Br | 16.2 h | β+ (55)/EC (45)/γ | 3.94 | 76Se(p,n)76Br | 76Se (stable) |
| 76Se(d,2n)76Br |
| 72As | 26.0 h | β+ (88)/EC (22) | 3.33 | 72Se/72As (generator) | 72Ge (stable) |
| 89Zr | 3.3 d | β+ (23)/EC (77)/γ | 1.81 | 89Y(p,n)89Zr | 89Y (stable) |
| 124I | 4.18 d | β+ (23)/EC (77)/γ | 0.901 | 124Te(p,n)124I | 124Te (stable) |
| 74As | 17.8 d | β+ (29)/β- (61) | 1.54 | 74Ge(p,n)74As | 74Ge (stable) |
The PET radionuclides discussed in this section are quite different in their half-lives, and they are similarly diverse in their (radio)chemistry. Consequently, the following examples show nicely how the choice of the radionuclide/-chemistry and nanostructures interact. Furthermore, the broad range of half-lives provides
in vivo data from the first initial minutes up to several days.
Table 5 gives an overview on the different combinations of radionuclides and NPs or polymers discussed in this section.
Table 5.
An overview of radiolabeled nanoparticles and polymers using various positron emitters. (n.d. = no data, RCY = radiochemical yield (decay corrected), HPMA = N-(2-hydroxypropyl)-methacrylamide, PEO = polyethyleneoxide, h.d. = hydrodynamic radii).
Table 5.
An overview of radiolabeled nanoparticles and polymers using various positron emitters. (n.d. = no data, RCY = radiochemical yield (decay corrected), HPMA = N-(2-hydroxypropyl)-methacrylamide, PEO = polyethyleneoxide, h.d. = hydrodynamic radii).
| Positron Emitter (T½) | Nanostructure/ System | Material | Size [nm] | Labeling Time [min] | T [°C] | RCY (RCP) [%] | Specific Activity | Ref. |
|---|
| 13N (9.97 min) | nanoparticle | Al2O3 (alumina) | 10–10,000 | 6 (beam time) | n.d. | 1.9 MBq/mg | 1.9 MBq/mg | [10] |
| 11C (20.4 min) | nanoparticle | iron oxide-COOH | 16 | 5 min (methylation) | 125 | 0.3 | n.d. | [63] |
| iron oxide-NH2 | 16 | 2.3 |
| silica-NH2 | 32 | 3.2 |
| platinum-COOH | 2.5 | 7.6 |
| 86Y (14.7 h) | nanotube | carbon | 47 ± 17 | 30 | 60 | n.d. (90) | 555 GBq/g | [64] |
| 76Br (16.2 h) | polymer/dendri-mer | PEO | 12 (h.r) | 20 | RT | n.d. (95) | 190 kBq/µg | [65] |
| 72As (26.0 h) | polymer | HPMA | n.d. | 60–120 | 30–70 | 20–90 | 100 kBq/µmol | [12] |
| 74As (17.8 d) |
| 89Zr (3.3 d) | nanoparticle | | | | | | 592 GBq/g | [66] |
| 124I (4.18 d) | nanoparticle | iron oxide | | | | | | [67] |
Pérez-Campaña
et al. irradiated commercial Al
2O
3-NPs with a proton beam (16 MeV) and utilized the
16O(p,α)
13N nuclear reaction to produce
13N-labeled NPs [
10]. In the same manner, they already produced
18F-labeled NPs via the
18O(p,n)
18F nuclear reaction on
18O-enriched Al
2O
3-NPs and demonstrated the feasibility of this approach [
20]. Furthermore, the
in vivo data (Sprague-Dawley rats) revealed an uptake plateau of the
18F-labeled NPs in organs after 1 h. Consequently, the cost-intensive
18O-enrichment of Al
2O
3-NPs can be avoided by facilitating the
16O(p,α)
13N nuclear reaction and the shorter-lived radionuclide
13N (9.97 min). NPs of 10, 40, 150 nm and 10 µm were applied. The short half-life of 9.97 min enabled an
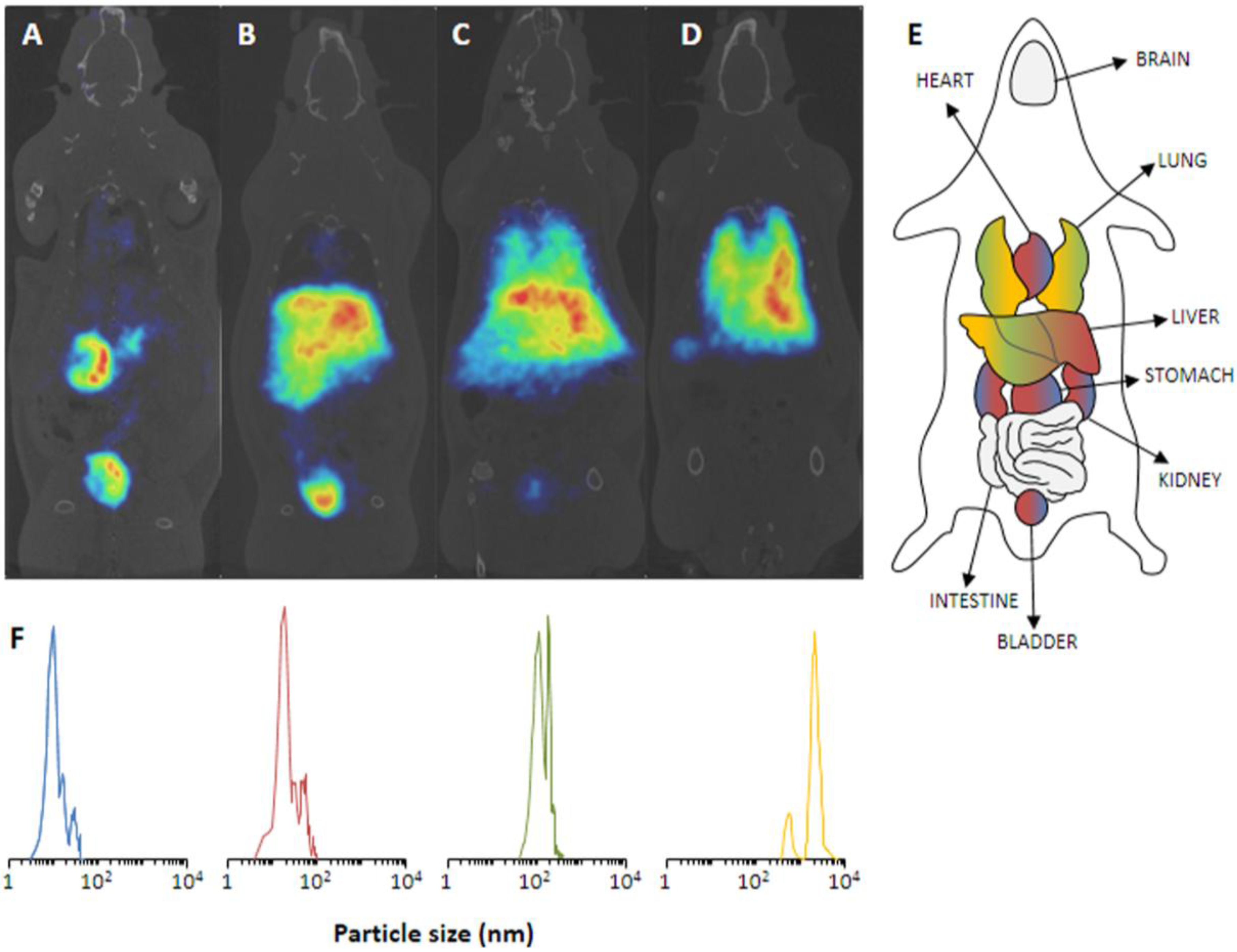
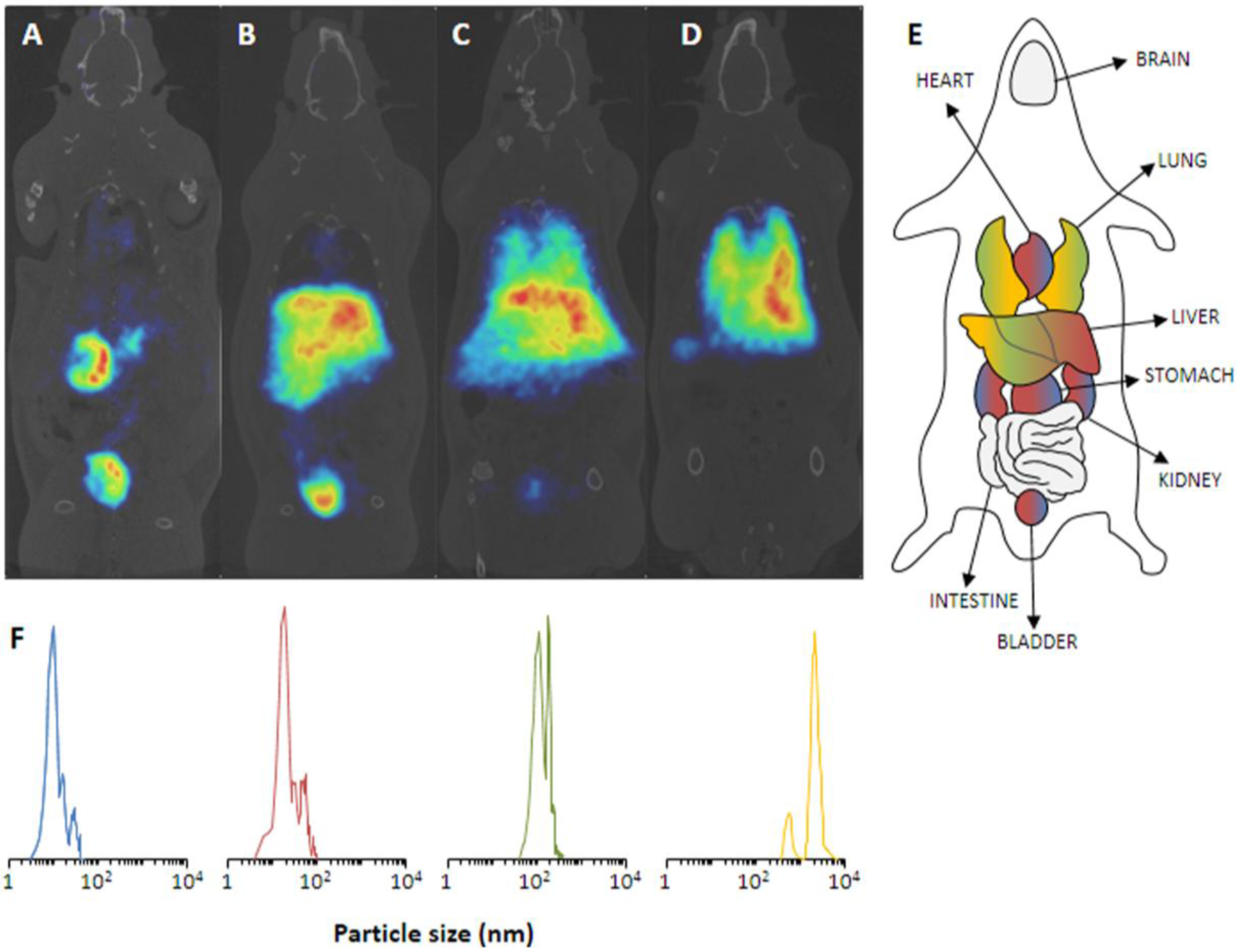
in vivo PET imaging over a period of 68 min, thus, sufficient to track the pharmacokinetics and biodistribution of those NPs. The NPs were irradiated as solid in an aluminum capsule for 6 min with a 16 MeV proton beam of 5 µA. Subsequent suspension of the NPs into physiological saline and centrifugation already gave the injectable solution.
In vivo PET studies in Sprague-Dawley rats show an expected biodistribution in strong dependence on the particles’ size (
Figure 6). Accordingly, the smallest NPs of 10 nm exhibited an exclusively renal excretion with all radioactivity in kidneys and bladder/urine. With increasing size, the renal glomerular filtration cut-off was quickly reached and
13N-NPs of 40 nm still showed a renal fraction, but already a high uptake in liver. The tendency to lung accumulation increased with the larger particles, hence, 10 µm NPs were only found in lungs.
Figure 6.
(
A–
D) PET images of
13N-nanoparticles of different size in Sprague-Dawley rats (60 min p.i.). (
A) 10 nm, (
B) 40 nm, (
C) 150 nm, (
D) 10 µm. (
E) Schematic anatomical overview with localization of important organs. (
F) The corresponding particles size distribution of the employed NPs. Reprinted with permission from Pérez-Campaña C.
et al. [
10]; Copyright 2013 American Chemical Society.
Figure 6.
(
A–
D) PET images of
13N-nanoparticles of different size in Sprague-Dawley rats (60 min p.i.). (
A) 10 nm, (
B) 40 nm, (
C) 150 nm, (
D) 10 µm. (
E) Schematic anatomical overview with localization of important organs. (
F) The corresponding particles size distribution of the employed NPs. Reprinted with permission from Pérez-Campaña C.
et al. [
10]; Copyright 2013 American Chemical Society.
For the development of NPs for dual imaging (PET and MRI), Sharma
et al. radiolabeled iron oxide NPs with the short-lived PET-radionuclide carbon-11 (T
½ = 20.4) [
63]. Additionally, they used silica and platinum NPs to establish the
11C-labeling procedure. The radiolabeling was based on the commonly used
11C-methylation via [
11C]methyl iodide, which was produced on a commercial radiosynthesis module (Microlab, GE Medical Systems, Wauwatosa, WI, USA).
11C-methylation of the different NPs was enabled by surface modification with either amino functionalities or carboxylic acids, leading to the NPs (size) iron oxide-COOH (16 nm), iron oxide-NH2 (16 nm), silica-NH
2 (32 nm) and platinum-COOH (2.5 nm). The
11C-methylation was facilitated by heating to 125 °C in DMF or DMSO for 5 min. RCY were varying from 0.3% to 7.6% in dependence of the NPs (see
Table 5). The
11C-labeled NPs were purified only by washing and centrifugation. The low RCY for iron oxide NPs was assumed to be a result of particle agglomeration and low ligand density on the NP’s surface. However, the
11C-labeled NPs were stable (>95%) in plasma at 37 °C for at least 120 min. In proof of concept
in vivo studies using the
11C-labeled magnetic iron oxide NPs in mice, dual imaging of the liver showed perfect matches for PET and MRI in fused images.
A PET radionuclide with a convenient half-life of 14.7 h is
86Y. McDevitt and co-workers used
86Y for the radiolabeling of carbon nanotubes (CNT) and studied the
in vivo behavior in nude mice [
64]. The surface of single walled carbon nanotubes was amino-functionalized and derivatized using
p-NCS-DOTA for a stable thiourea formation with primary amino functions. The DOTA-CNT were mixed with a [
86Y]YCl
3-solution (296 MBq) for 30 min (60 °C, pH 5.5). The
86Y-DOTA-CNT was isolated via SEC (P6 resin (BioRad)).
86Y-DOTA-CNT was obtained in good radiochemical purities of ≥90% and with a specific activity of 555 GBq/g. The
86Y-labeled nanotubes were applied to healthy nude mice to study their
in vivo behavior.
In vivo PET imaging at 3 h p.i. showed major uptake in liver, kidneys and spleen, which was confirmed by
ex vivo biodistribution studies 24 h p.i. giving 15.2% ± 1.5%, 5.96% ± 1.20% and 0.82% ± 0.04% ID/g, respectively. No further background activity was observed, only a minor accumulation in bones was detectable. Interestingly, the authors compared the
in vivo data of two different administration protocols, intravenous and intraperitoneal injection. Both methods gave almost similar results in kidneys and spleen, only the liver uptake being higher after i.v. administration.
With an almost similar half-life (16.2 h), the radiohalogen
76Br was employed by Almutairi
et al. for radiolabeling of a dendritic polymer [
65]. Based on a core-shell architecture, an eight-branched polyethylene oxide (PEO) dendrimer was synthesized via NHS-esters. The dendrimer was functionalized with tyrosine moieties for a direct electrophilic radiobromination. Furthermore, the PEO-branches were coupled to cRDG peptides (~5 RDG/dendrimer) for active targeting the α
vβ
3 intergin for angiogenesis imaging.
76Br-Radiobromination was facilitated in phosphate buffer (pH 7) in the presence of chloramine-T (CAT). After 20 min radiolabeling, purification via SEC (HiTrap cartridge, GE healthcare) gave the product in high radiochemical purities of ≥95%. In
in vitro cell assays, the multivalent cRDG-dendrimer provided a 50fold increase in affinity (avidity) due to the multivalency with an IC
50 value of 0.18 nM instead of 10.4 nM (mono-cRDG peptide). Similarly, a 6-fold increase in endocytosis was observed using the multivalent dendrimers. The
76Br-labeled dendrimers were further applied to mice with a hind limb ischemia.
In vivo PET imaging 24 h p.i. showed high uptake in the diseased regions and
ex vivo biodistribution studies 4 h, 24 h and 48 h p.i. indicated a relatively fast renal clearance, whereby a prolonged retention in kidneys was assigned to a specific binding/uptake of the RDG-groups.
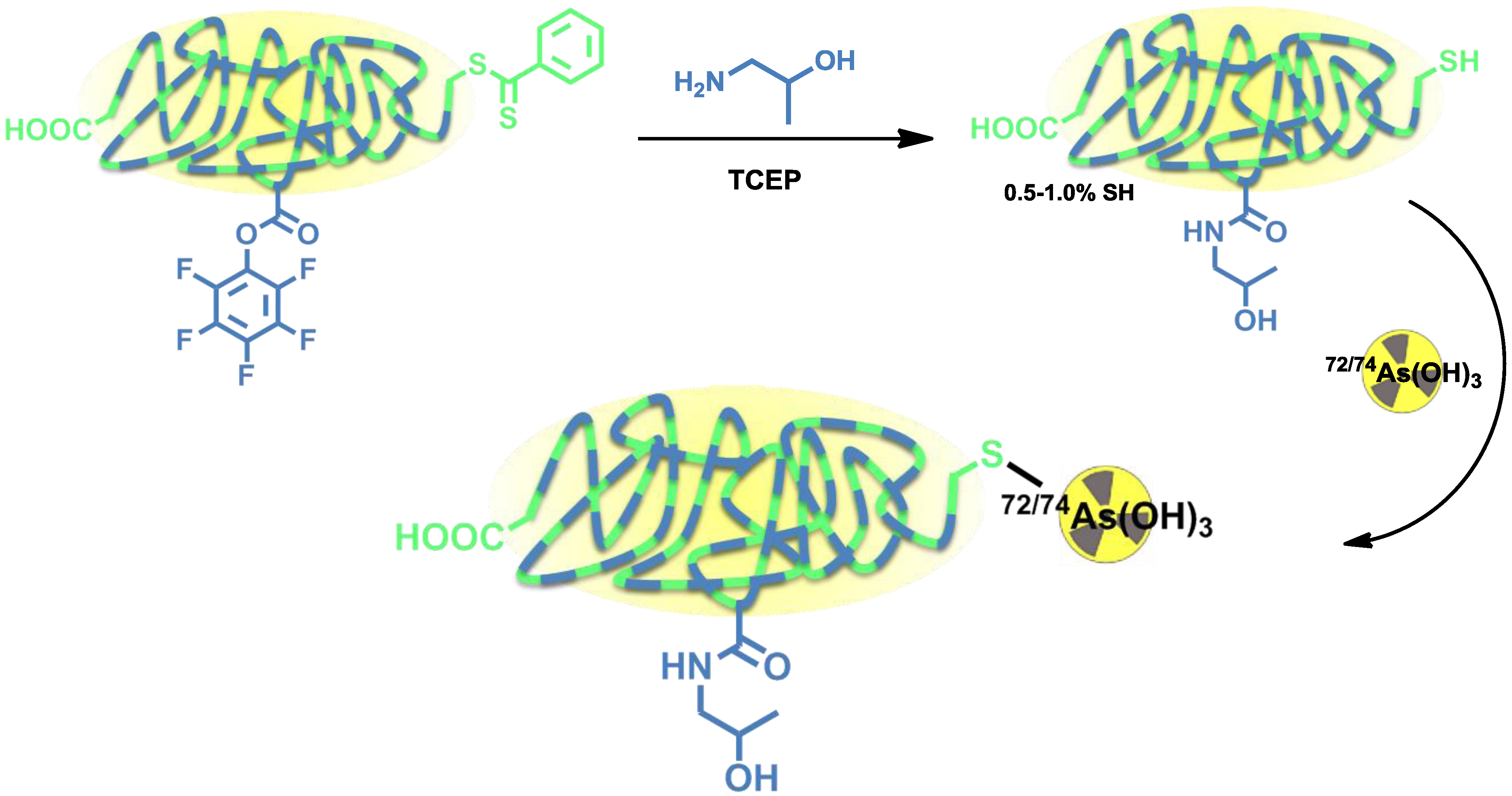
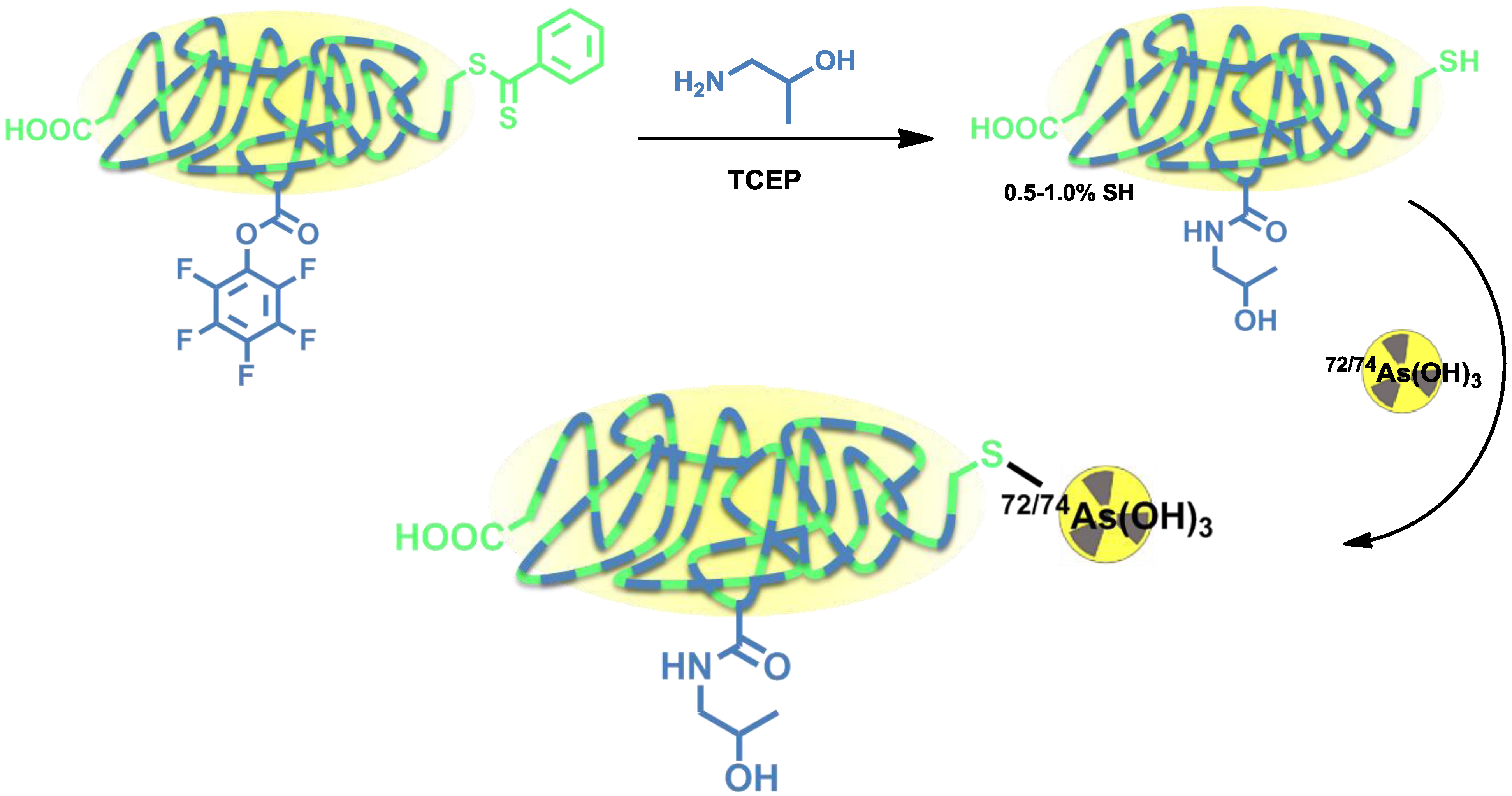
Herth
et al. developed the radiolabeling of HPMA-based polymers for
72/74As (
Figure 7) [
12]. Radioarsenic was achieved from a proton (15 MeV) irradiation of a natural germanium target leading to 4 GBq
72As (30 µA beam current) and 400 MBq
74As (200 µA beam current), respectively. To enable the radiolabeling of four different HPMA-based polymers, the polymers were functionalized with dithiobenzoic ester end groups which can be reduced to free thiol moieties by tris(2-carboxyethyl)phosphine (TCEP) and covalently bind to arsenic (
Figure 7).
Figure 7.
Thiol-functionalization of HPMA-based polymers and radiolabeling strategy for 72/74As-labeled HPMA-based polymers.
Figure 7.
Thiol-functionalization of HPMA-based polymers and radiolabeling strategy for 72/74As-labeled HPMA-based polymers.
Four different HPMA-based polymers were modified, three of them with a thiol content of 0.5%–1.0%. Another HPMA-polymer was especially designed to exhibit a higher thiol content of 10% by introducing additional disulfide sidechains (
Figure 7). For radiolabeling, the radioarsenic was separated from the germanium target by distillation and after further work-up,
72/74As was available in PBS solution (pH 7) [
62]. The different polymers were radiolabeled in aqueous solutions at 70 °C (low thiol) or 30 °C (high-thiol) and gave RCYs of ~20% for low-thiol-content polymers and up to 90% for the high-thiol-content polymer. The
72/74As-labeled HPMA-polymers were tested
in vitro towards their stability. All new
72/74As-labeled HPMA-polymers showed an excellent stability in physiological saline over a time period of 48 h. The radiolabeled polymers were not further investigated or evaluated.
In similarity to McDevitt
et al. [
64], single walled nanotubes (SWNTs) were employed in combination with the longer-lived
89Zr by Ruggiero
et al. [
66]. The SWNTs were amino-functionalized and further derivatized with the chelator DFO for
89Zr-radiolabeling. Moreover, the SWNTs were coupled to an antiVE-cad (vascular endothelial cadherin) antibody (E4G10) for active targeting of neo-vascularization (angiogenesis).
89Zr-Radiolabeling was facilitated using
89Zr-oxalate at pH 5 for 60 min at 60 °C. The
89Zr-SWNTs were isolated via SEC. The new
89Zr-nanotubes were applied to
in vivo PET studies using a human colon adenocarcinoma model (LS174T) in mice. PET imaging was performed daily over one week. The imaging studies revealed a fast blood clearance within one hour and a specific and high uptake in the targeted sites.
Choi
et al. applied
124I to iron oxide NPs [
67]. The magnetic radiolabeled NPs are intended to act as a dual-modality imaging agent for MRI/PET imaging of SLNs. Synthesized magnetic iron oxide NPs were highly monodispersed (σ < 5%) with a size of 15 nm. To facilitate the radioiodination, the surface of the NP was coated with serum albumin, of which the tyrosine groups offer the possibility of direct electrophilic iodination in the
ortho-position. The hydrodynamic size of the albumin-coated particles was 32 nm. The radiolabeling was performed using [
124I]NaI in presence of Iodo-Beads as oxidant. The
124I-labeled NPs were evaluated in Sprague-Dawley rats applying PET and MRI imaging with the focus on SLN detection. Using both modalities for functional and morphological information, a clear visualization of two brachial lymph nodes in rats was achieved. Furthermore, a dissection of the corresponding lymph node ducts in which the radiolabeled NPs were detected, confirmed the imaging results.
5. Conclusions
Radiolabeling approaches for NPs and polymers are essential tools in the development of potential drug delivery systems. In this regard, positron emitters are of special interest as they allow PET imaging. Furthermore, a radiolabeled analog of a drug delivery system for PET enables patient selection, therapy planning and monitoring by molecular imaging.
Several radiolabeling strategies with quite a variety of different PET isotopes have been developed and show very promising results. In principle, the very wide range of half-lives (see “
Clock-Of-Nuclides”,
Figure 2) allows tracking and detection of nanodimensional probes from the time point of administration to several days and even weeks p.i. Interestingly, several examples with short-lived isotopes (
i.e.,
18F,
68Ga and even
13N) demonstrate the fundamental importance of the initial few hours. Moreover, short-lived isotopes have been used in pre-targeting concepts, which offer a great flexibility regarding the time point of imaging.
However, there are quite a number of problems one has to deal with during the evaluation of nanoparticular and polymeric systems for PET imaging. It starts with the synthesis of the structures and the decision of what labeling approach should be investigated or is suitable for the biological question asked (half-life; biological target). Subsequently, the radiosynthesis comes to the fore, meaning how to get the NPs or polymers labeled (direct labeling, prosthetic group labeling or in situ generation of the radionuclide) in sufficient RCYs and in the right formulation, like volume activity and/or purification. If all the physicochemical properties can be fulfilled, the in vitro and in vivo testing begins. In vitro tests of some QDs showed an increased toxicity, which could be decreased by using the known concept of PEGylation. It has also been shown that coating NPs with biocompatible/biodegradable structures has a positive effect on the in vitro/in vivo behavior. If the toxicity of the NPs and polymers could be excluded micro-PET studies were conducted.
The next step is the use of the radiolabeling and PET imaging platform for systematic optimization of NPs and polymers towards specific applications. A major aspect, which repeats throughout all in vivo evaluations of NPs or polymeric structures, is their accumulation in either the liver/spleen (large sizes) or the kidneys (small sizes). Noticeable is that not only the size of the particles matters, rather its architecture, which significantly determines the biodistribution. However, as long as new nanomaterials for potential drug delivery systems are developed, there is still a demand for new methods and strategies in radiolabeling of NPs and polymers to provide a suitable (dual)system for the asked biological question. Further research in this exciting field can be expected, ideally with a NP or polymer which accumulates specifically at target sites and can be tracked in vivo, and/or can transport a therapeutic agent.


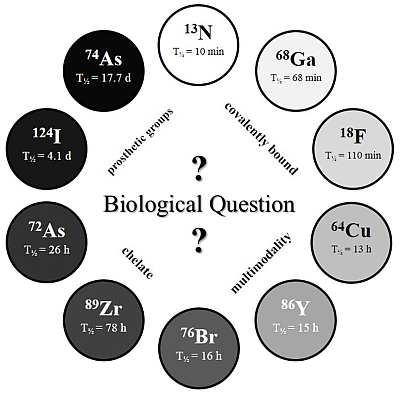


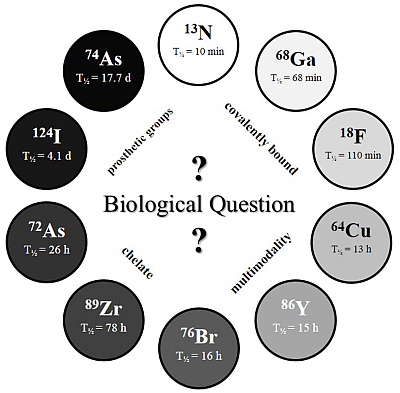{kind=link}
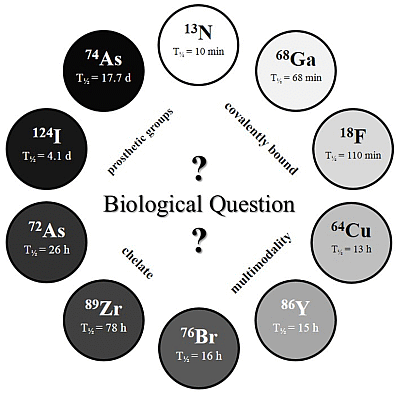{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}