What Goes around Comes around-A Comparative Study of the Influence of Chemical Modifications on the Antimicrobial Properties of Small Cyclic Peptides


 and
and
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
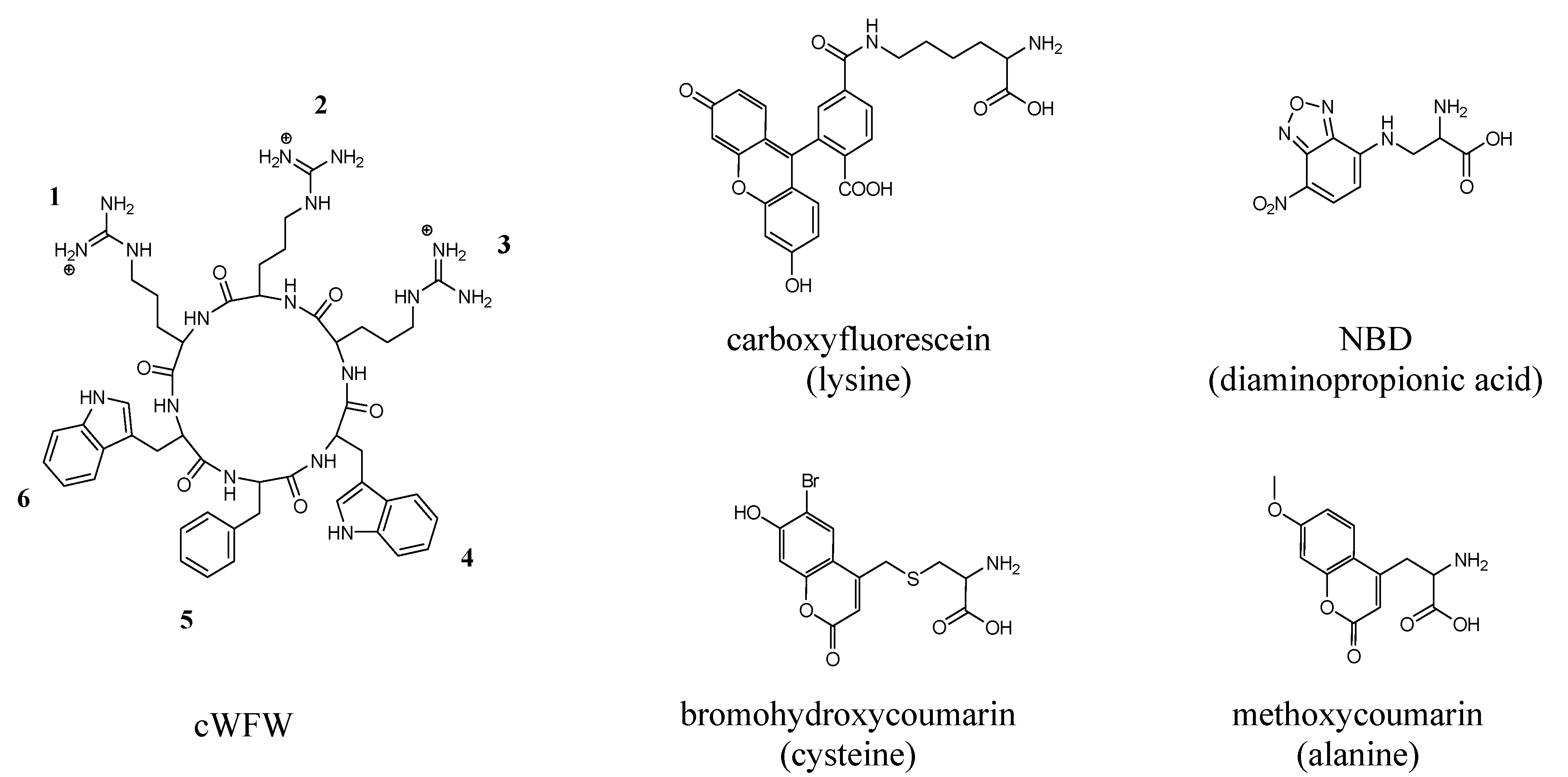
2.2. Peptide Synthesis
2.3. CD Spectroscopy
2.4. Hydrophobicity
2.5. Antimicrobial Activity
2.6. Hemolytic Activity
2.7. Propidium Iodide Influx
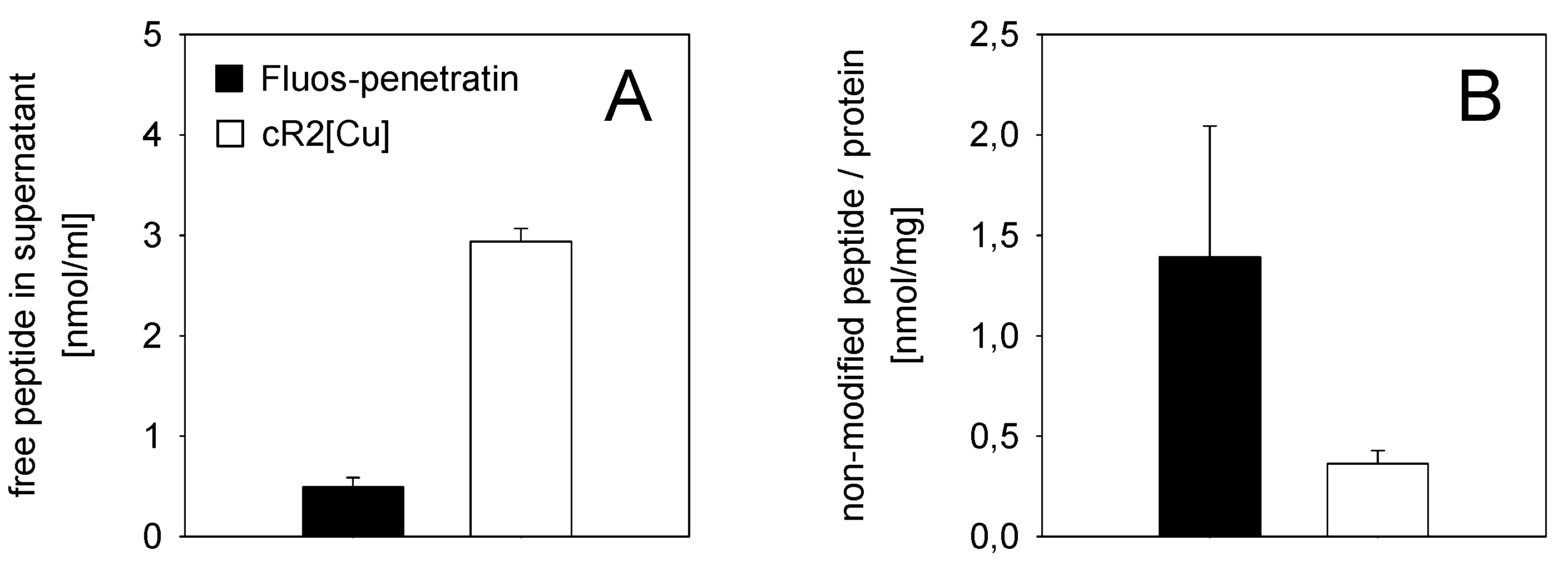
2.8. Cellular Uptake
3. Results and Discussion
3.1. Peptide Modifications and Antimicrobial Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Sequences | MIC [µM] | Hemolysis [%]
at 100 µM | |
|---|---|---|---|---|
| E. coli DH5α | B. subtilis DSM 347 | |||
| cWFW | c-RRRWFW | 3 | 3 | 4 |
| cW2[Fluos] | c-RRRWWK[Fluos] | >100 | >100 | 9 |
| cW3[Fluos] | c-RRWWWK[Fluos] | >100 | >100 | 2 |
| cW[Cu]W | c-RRRW[Ala-Coumarin]W | 25 | 6 | 23 |
| cW[S-Cu]W | c-RRRW[Cys-Br-Coumarin]W | 25 | 6 | 76 |
| cW[NBD]W | c-RRRW[Dap-NBD]W | 50 | 25 | 76 |
| cRKR | c-RKRWFW | 50 | 12 | 4 |
| cRKK | c-RKKWFW | 50 | 25 | 56 |
| cKRK | c-KRKWFW | 12 | 25 | 2 |
| cKKR | c-KKRWFW | 100 | 25 | 3 |
| cKKK | c-KKKWFW | 100 | 50 | 8 |
| cR2[Cu] | c-KRKW[Ala-Coumarin]W | 50 | 6 | 4 |
| cR2[S-Cu] | c-KRKW[Cys-Br-Coumarin]W | 50 | 25 | 5 |
| cR2[NBD] | c-KRKW[Dap-NBD]W | 50 | 12 | 26 |
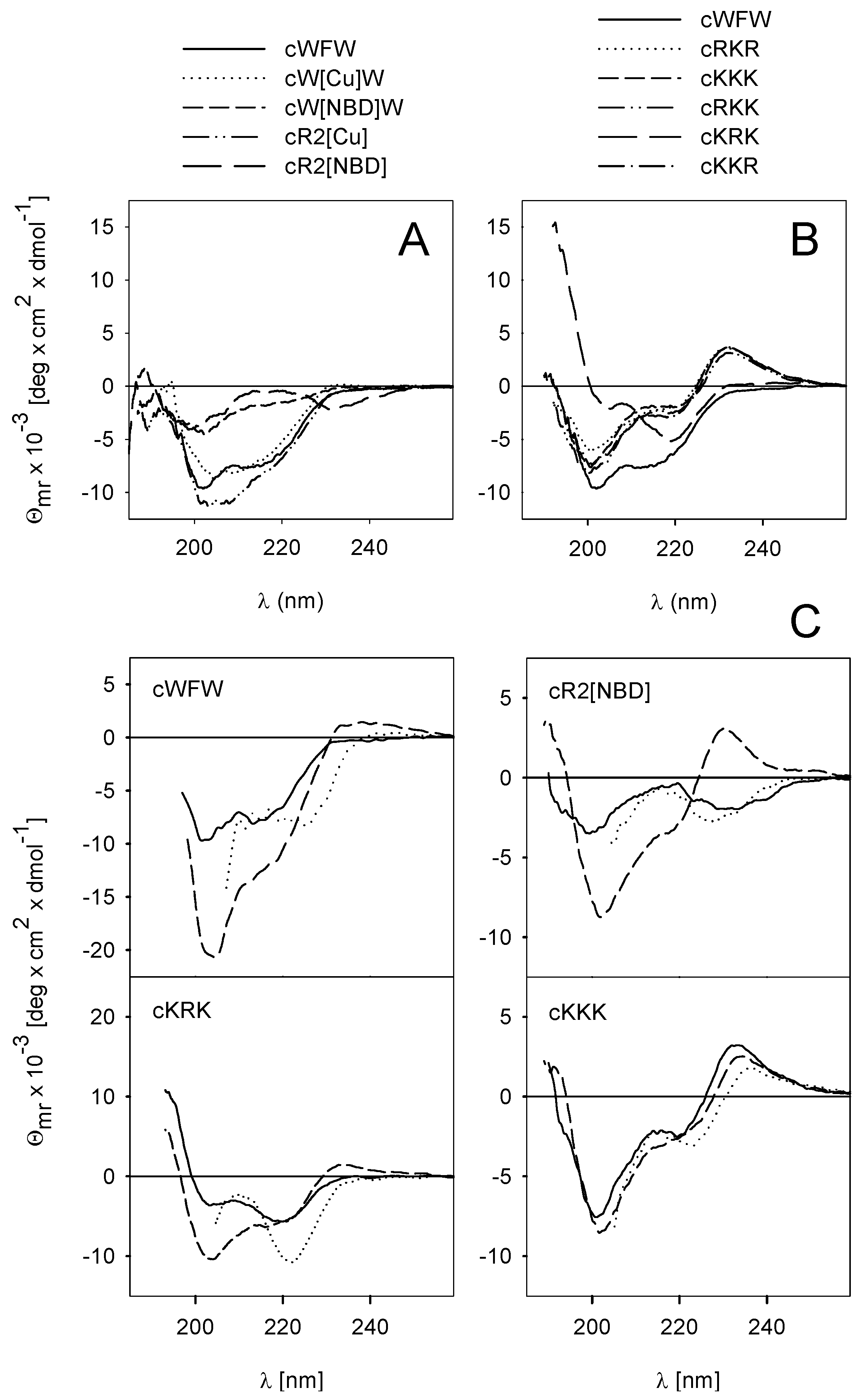
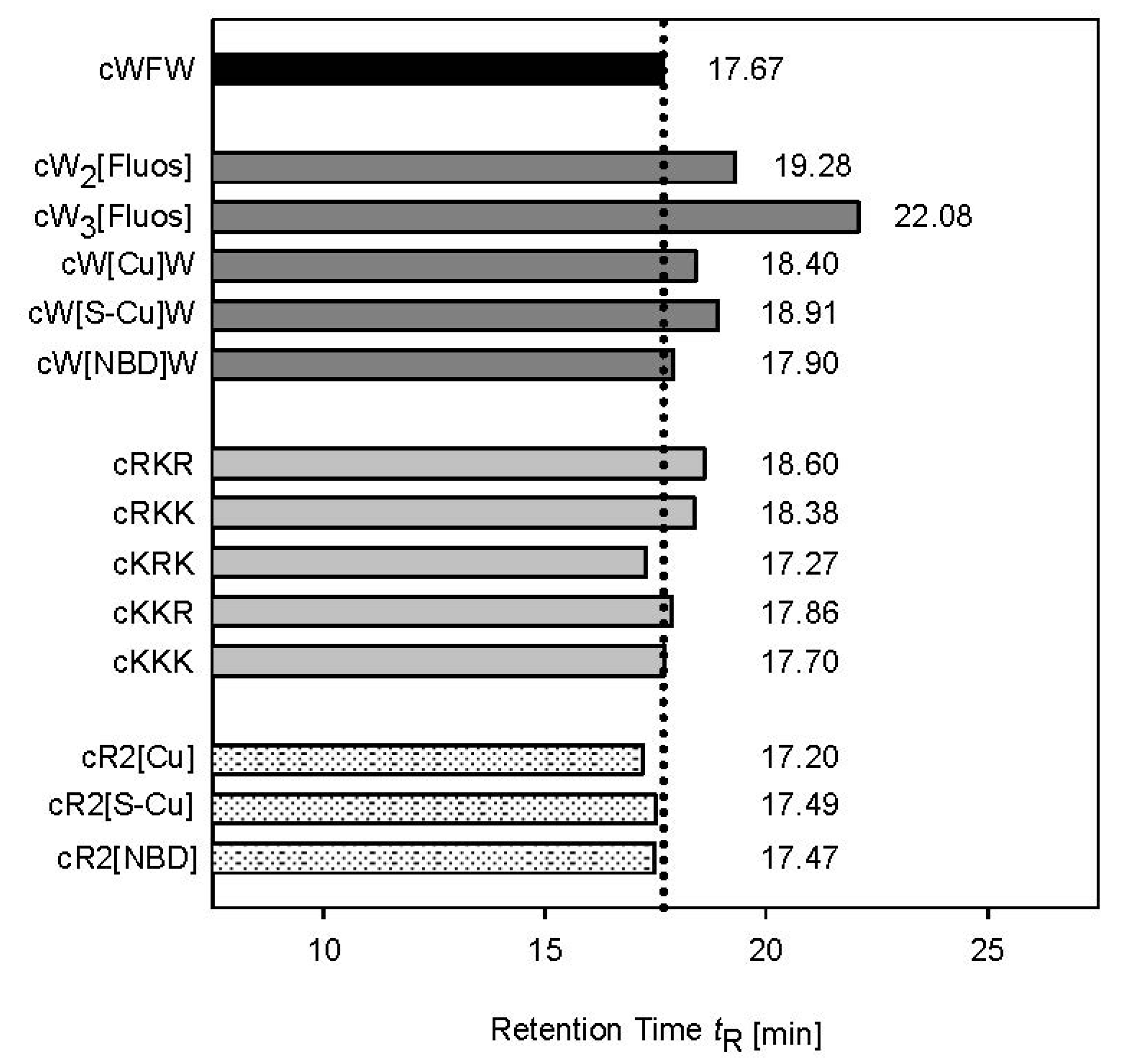
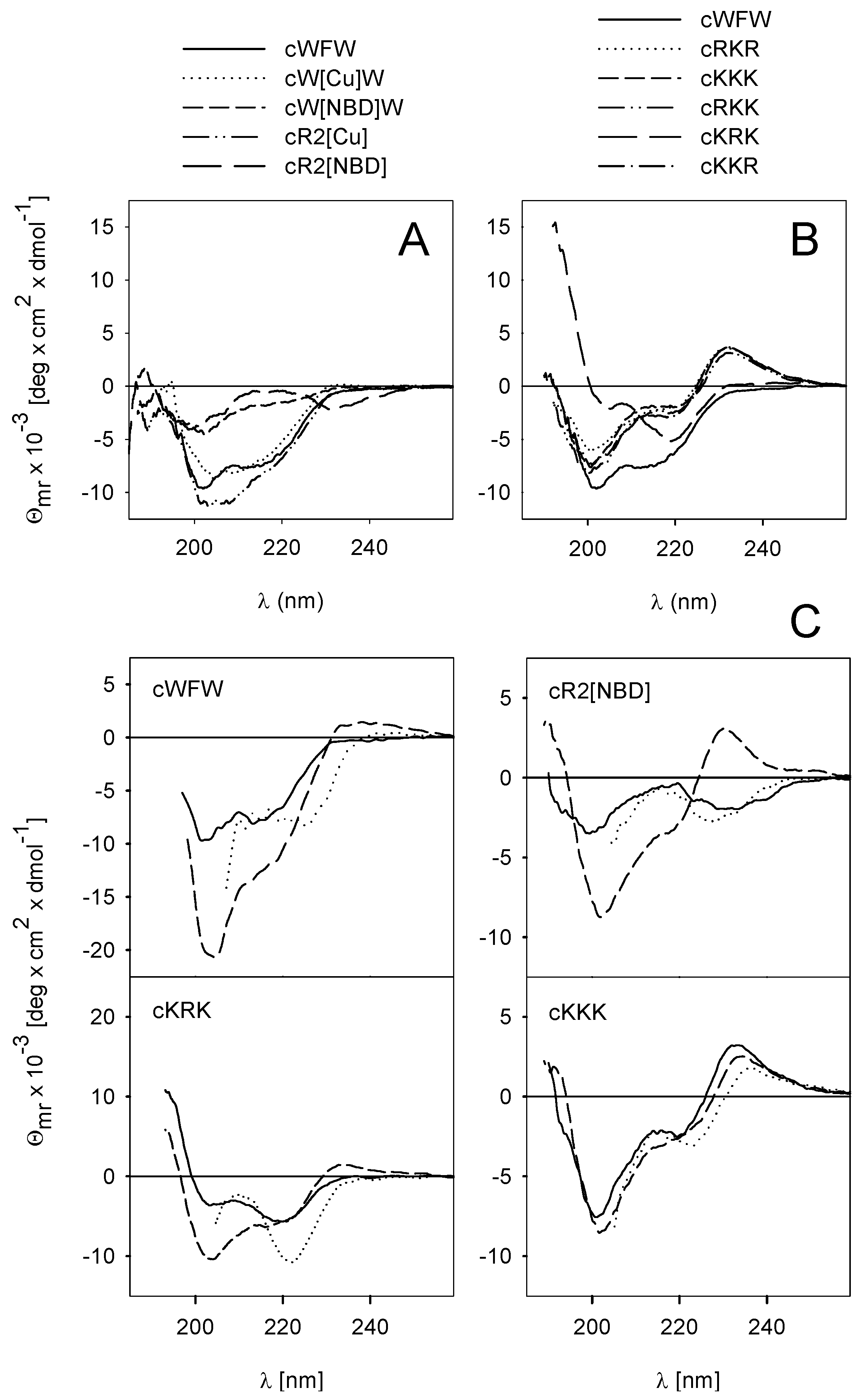
3.2. Peptide Hydrophobicity and Conformation
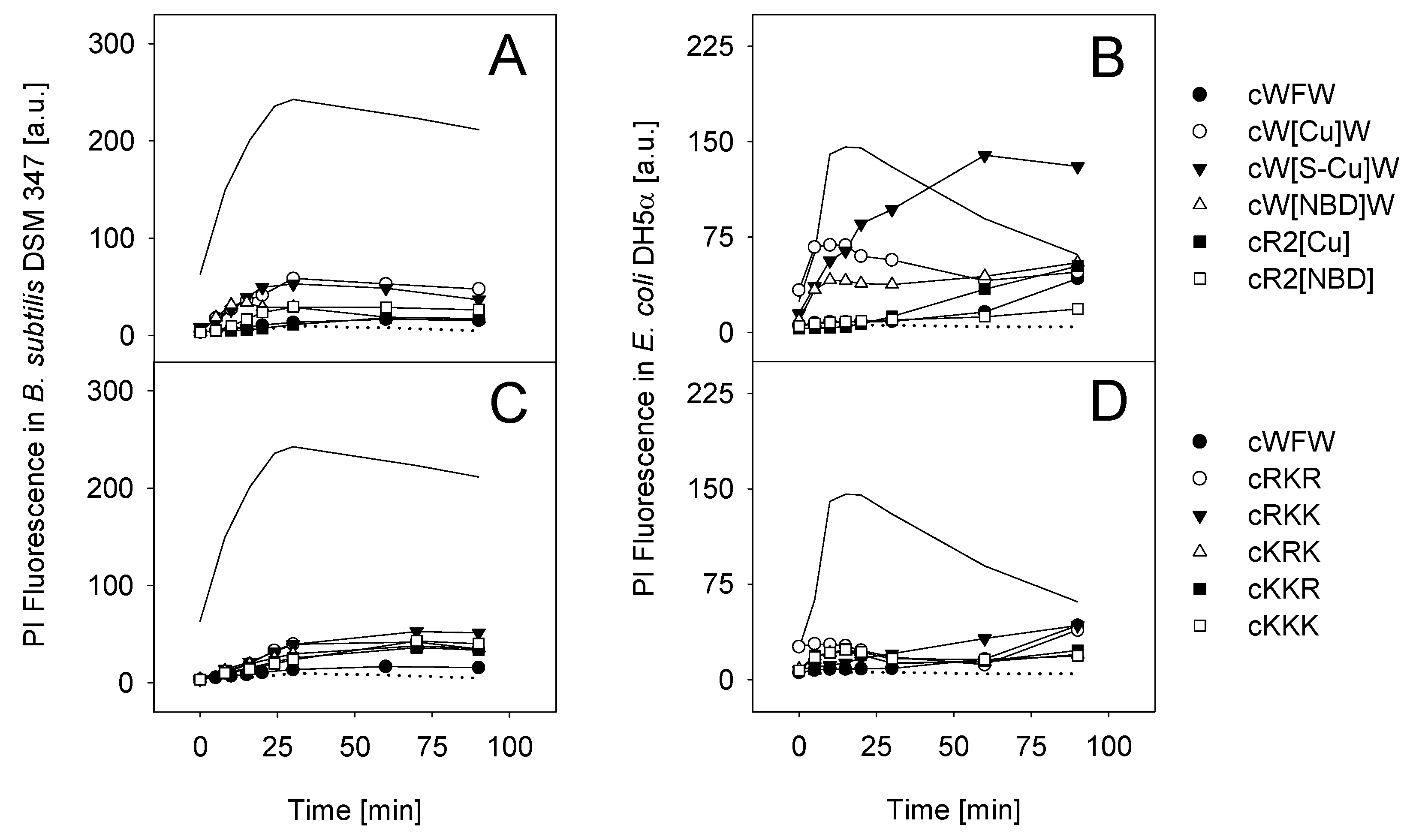
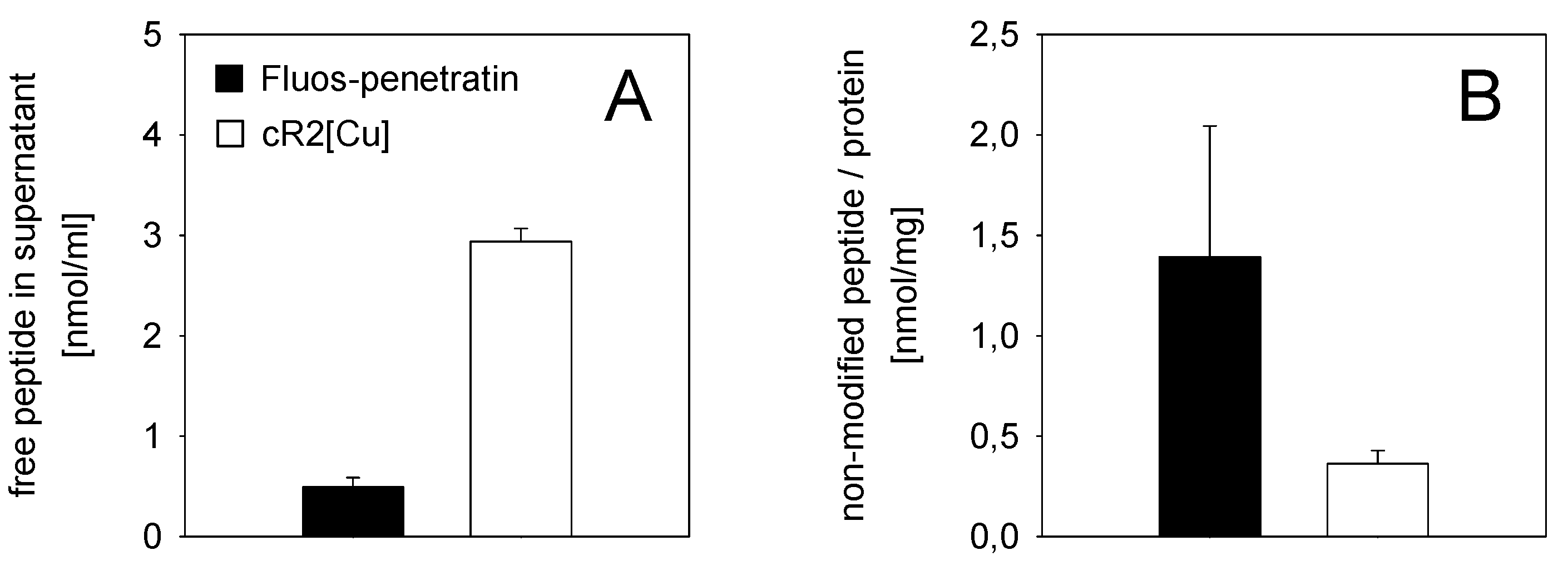
3.3. Mode of Antimicrobial Action

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar] [CrossRef]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef]
- Dathe, M.; Nikolenko, H.; Klose, J.; Bienert, M. Cyclization increases the antimicrobial activity andselectivity of arginine- and tryptophan-containing hexapeptides. Biochemistry 2004, 43, 9140–9150. [Google Scholar] [CrossRef]
- Junkes, C.; Harvey, R.D.; Bruce, K.D.; Dolling, R.; Bagheri, M.; Dathe, M. Cyclic antimicrobial R-, W-rich peptides: The role of peptide structure and E. coli outer and inner membranes in activity and the mode of action. Eur. Biophys. J. 2011, 40, 515–528. [Google Scholar] [CrossRef]
- Wells, S.; Johnson, I. Fluorescent Labels for Confocal Microscopy. In Three-Dimensional Confocal Microscopy: Volume Investigation of Biological Specimens; Academic Press: London, UK, 1994; pp. 101–129. [Google Scholar]
- Lattig-Tunnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta 1998, 1414, 127–139. [Google Scholar] [CrossRef]
- Hagen, V.; Kilic, F.; Schaal, J.; Dekowski, B.; Schmidt, R.; Kotzur, N. [8-[Bis(carboxymethyl)aminomethyl]-6-bromo-7-hydroxycoumarin-4-yl]methyl moieties as photoremovable protecting groups for compounds with COOH, NH2, OH, and C=O functions. J. Org. Chem. 2010, 75, 2790–2797. [Google Scholar] [CrossRef]
- Madani, F.; Abdo, R.; Lindberg, S.; Hirose, H.; Futaki, S.; Langel, U.; Graslund, A. Modeling the endosomal escape of cell-penetrating peptides using a transmembrane pH gradient. Biochim. Biophys. Acta 2013, 1828, 1198–1204. [Google Scholar] [CrossRef]
- Chan, W.; White, P. Fmoc Solid Phase Peptide Synthesis—A Practical Approach; Oxford University Press: Oxford, UK, 1999; p. 370. [Google Scholar]
- Pearson, D.A.; Blanchette, M.; Baker, M.L.; Guindon, C.A. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989, 30, 2739–2742. [Google Scholar] [CrossRef]
- Ehrlich, A.; Heyne, H.U.; Winter, R.; Beyermann, M.; Haber, H.; Carpino, L.A.; Bienert, M. Cyclization of all-l-pentapeptides by means of 1-hydroxy-7-azabenzotriazole-derived uronium and phosphonium reagents. J. Org. Chem. 1996, 61, 8831–8838. [Google Scholar] [CrossRef]
- Dathe, M.; Schumann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef]
- Dathe, M.; Meyer, J.; Beyermann, M.; Maul, B.; Hoischen, C.; Bienert, M. General aspects of peptide selectivity towards lipid bilayers and cell membranes studied by variation of the structural parameters of amphipathic helical model peptides. Biochim. Biophys. Acta 2002, 1558, 171–186. [Google Scholar] [CrossRef]
- Lindgren, M.; Gallet, X.; Soomets, U.; Hallbrink, M.; Brakenhielm, E.; Pooga, M.; Brasseur, R.; Langel, U. Translocation properties of novel cell penetrating transportan and penetratin analogues. Bioconjugate Chem. 2000, 11, 619–626. [Google Scholar] [CrossRef]
- Oehlke, J.; Savoly, B.; Blasig, I.E. Utilization of endothelial cell monolayers of low tightness for estimation of transcellular transport characteristics of hydrophilic compounds. Eur. J. Pharm. Sci. 1994, 2, 365–372. [Google Scholar] [CrossRef]
- Dansen, T.B.; Pap, E.H.W.; Wanders, R.J.; Wirtz, K.W. Targeted fluorescent probes in peroxisome function. Histochem. J. 2001, 33, 65–69. [Google Scholar] [CrossRef]
- Junkes, C.; Wessolowski, A.; Farnaud, S.; Evans, R.W.; Good, L.; Bienert, M.; Dathe, M. The interaction of arginine- and tryptophan-rich cyclic hexapeptides with Escherichia coli membranes. J. Pept. Sci. 2008, 14, 535–543. [Google Scholar] [CrossRef]
- Wessolowski, A.; Bienert, M.; Dathe, M. Antimicrobial activity of arginine- and tryptophan-rich hexapeptides: The effects of aromatic clusters, D-amino acid substitution and cyclization. J. Pept. Res. 2004, 64, 159–169. [Google Scholar] [CrossRef]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1996, 1462, 71–87. [Google Scholar]
- Kondejewski, L.H.; Jelokhani-Niaraki, M.; Farmer, S.W.; Lix, B.; Kay, C.M.; Sykes, B.D.; Hancock, R.E.; Hodges, R.S. Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J. Biol. Chem. 1999, 274, 13181–13192. [Google Scholar] [CrossRef]
- Bagheri, M.; Keller, S.; Dathe, M. Interaction of W-substituted analogs of cyclo-RRRWFW with bacterial lipopolysaccharides: The role of the aromatic cluster in antimicrobial activity. Antimicrob. Agents Chemother. 2011, 55, 788–797. [Google Scholar] [CrossRef]
- Radchenko, D.S.; Michurin, O.M.; Grygorenko, O.O.; Scheinpflug, K.; Dathe, M.; Komarov, I.V. Confining the χ space of basic natural amino acids: Cyclobutane-derived χ1,χ2-constrained analogues of arginine, lysine and ornithine. Tetrahedron 2013, 69, 505–511. [Google Scholar] [CrossRef]
- Dennison, S.R.; Wallace, J.; Harris, F.; Phoenix, D.A. Amphiphilic alpha-helical antimicrobial peptides and their structure/function relationships. Protein Pept. Lett. 2005, 12, 31–39. [Google Scholar] [CrossRef]
- Spathelf, B.M.; Rautenbach, M. Anti-listerial activity and structure–activity relationships of the six major tyrocidines, cyclic decapeptides from Bacillus aneurinolyticus. Bioorg. Med. Chem. 2009, 17, 5541–5548. [Google Scholar] [CrossRef]
- Marques, M.A.; Citron, D.M.; Wang, C.C. Development of Tyrocidine A analogues with improved antibacterial activity. Bioorg. Med. Chem. 2007, 15, 6667–6677. [Google Scholar] [CrossRef]
- Appelt, C.; Wessolowski, A.; Soderhall, J.A.; Dathe, M.; Schmieder, P. Structure of the antimicrobial, cationic hexapeptide cyclo(RRWWRF) and its analogues in solution and bound to detergent micelles. Chembiochem 2005, 6, 1654–1662. [Google Scholar] [CrossRef]
- Woody, R.W. Contributions of tryptophan side chains to the far-ultraviolet circular dichroism of proteins. Eur. Biophys. J. 1994, 23, 253–262. [Google Scholar] [CrossRef]
- Appelt, C.; Wessolowski, A.; Dathe, M.; Schmieder, P. Structures of cyclic, antimicrobial peptides in a membrane-mimicking environment define requirements for activity. J. Pept. Sci. 2008, 14, 524–527. [Google Scholar] [CrossRef]
- Scheller, A.; Oehlke, J.; Wiesner, B.; Dathe, M.; Krause, E.; Beyermann, M.; Melzig, M.; Bienert, M. Structural requirements for cellular uptake of alpha-helical amphipathic peptides. J. Pept. Sci. 1999, 5, 185–194. [Google Scholar] [CrossRef]
- Mercier, R.; Domínguez-Cuevas, P.; Errington, J. Crucial role for membrane fluidity in proliferation of primitive cells. Cell Rep. 2012, 1, 417–423. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 289–294. [Google Scholar] [CrossRef]
- Eun, Y.J.; Zhou, M.; Kiekebusch, D.; Schlimpert, S.; Trivedi, R.R.; Bakshi, S.; Zhong, Z.; Wahlig, T.A.; Thanbichler, M.; Weibel, D.B. Divin: A small molecule inhibitor of bacterial divisome assembly. J. Am. Chem. Soc. 2013, 135, 9768–9776. [Google Scholar] [CrossRef]
- Thanbichler, M. Synchronization of chromosome dynamics and cell division in bacteria. Cold Spring Harb. Perspect. Biol. 2010, 2, a000331. [Google Scholar] [CrossRef]
- Ferullo, D.J.; Cooper, D.L.; Moore, H.R.; Lovett, S.T. Cell cycle synchronization of Escherichia coli using the stringent response, with fluorescence labeling assays for DNA content and replication. Methods 2009, 48, 8–13. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Scheinpflug, K.; Nikolenko, H.; Komarov, I.V.; Rautenbach, M.; Dathe, M. What Goes around Comes around-A Comparative Study of the Influence of Chemical Modifications on the Antimicrobial Properties of Small Cyclic Peptides. Pharmaceuticals 2013, 6, 1130-1144. https://doi.org/10.3390/ph6091130
Scheinpflug K, Nikolenko H, Komarov IV, Rautenbach M, Dathe M. What Goes around Comes around-A Comparative Study of the Influence of Chemical Modifications on the Antimicrobial Properties of Small Cyclic Peptides. Pharmaceuticals. 2013; 6(9):1130-1144. https://doi.org/10.3390/ph6091130
Chicago/Turabian StyleScheinpflug, Kathi, Heike Nikolenko, Igor V. Komarov, Marina Rautenbach, and Margitta Dathe. 2013. "What Goes around Comes around-A Comparative Study of the Influence of Chemical Modifications on the Antimicrobial Properties of Small Cyclic Peptides" Pharmaceuticals 6, no. 9: 1130-1144. https://doi.org/10.3390/ph6091130
APA StyleScheinpflug, K., Nikolenko, H., Komarov, I. V., Rautenbach, M., & Dathe, M. (2013). What Goes around Comes around-A Comparative Study of the Influence of Chemical Modifications on the Antimicrobial Properties of Small Cyclic Peptides. Pharmaceuticals, 6(9), 1130-1144. https://doi.org/10.3390/ph6091130