3.1. Common Feature Pharmacophore Hypothesis
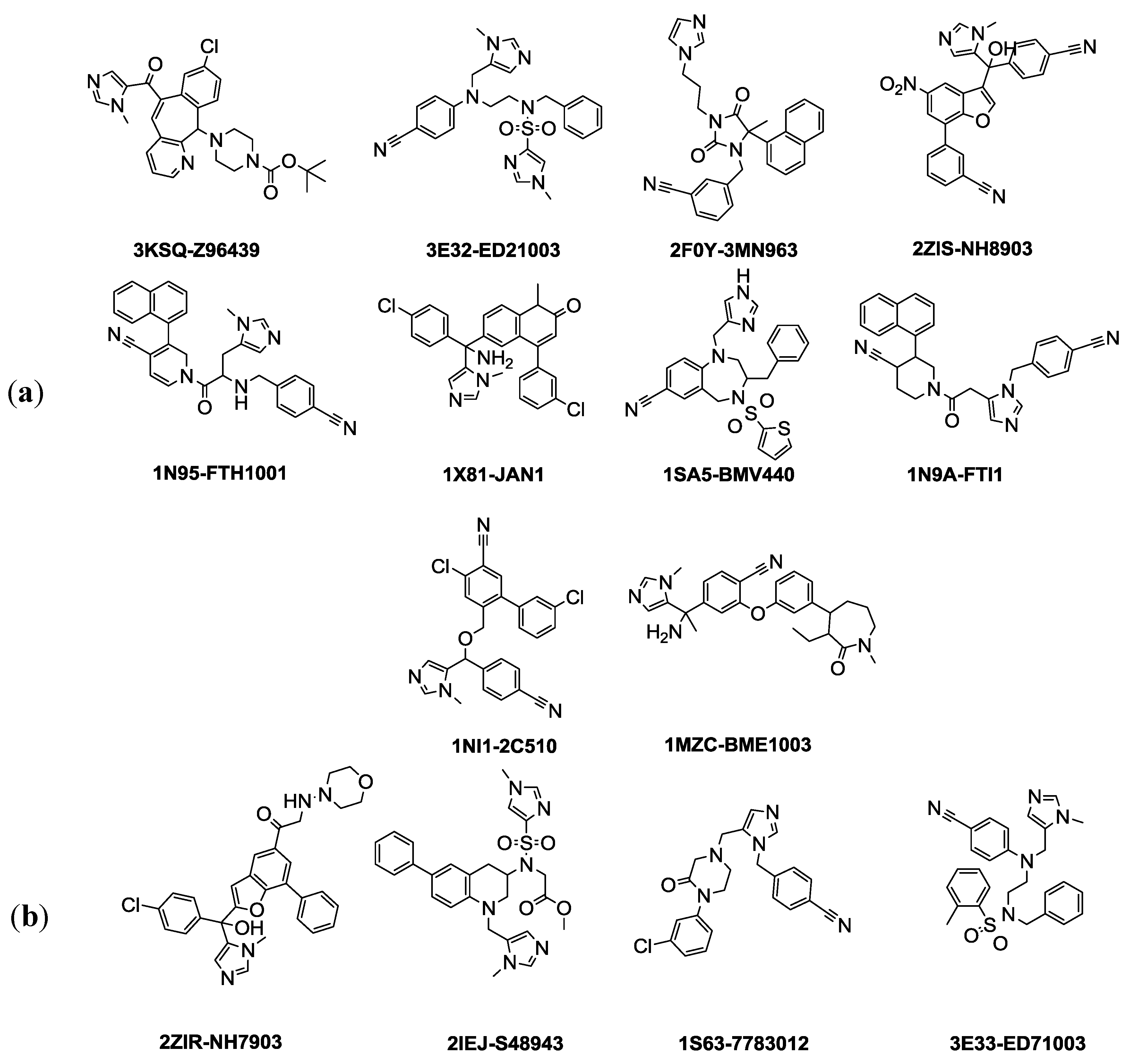
The selection of training and test set compounds for pharmacophore hypotheses generation was performed by subjecting twenty three ligands extracted from PDB of FTase enzyme to both Lipinisks’ rule of five; which limits the selection of molecules of appropriate characteristics with respect to size and molecular weight, number of HBD and HBA, and logP values in addition to Vebers’ rules with respect to the number of rotatable bonds and polar surface area, yielding fourteen candidate compounds (
Figure 1). In this way selecting the compounds will allow building a pharmacophore hypothesis that possesses the essential properties and at the same time avoids chemical compounds that could cause generated pharmacophore hypothesis to deviate from the optimum configuration. The selected fourteen compounds were further divided into training set and test set using the
Generate training and test data protocol at DS 3.1. Splitting of these molecules is based on diversity and it is assumed that 70% of them (10 compounds) are the training set. The crystallized biologically active conformations were mapped without any conformation generation as they were considered to possess the required and optimum conformation for pharmacophore hypothesis generation. In this study, the selected features are
HBA,
HBD,
hydrophobic,
ring aromatic, and
zinc binder. Ten pharmacophore hypotheses based on common features were generated and they are displayed in (
Table 1). According to the table, all the training set compounds fit all the features of the pharmacophore hypotheses and the rank scores ranged from 89.866 to 86.374. Only two of the 10 pharmacophore hypotheses contain Zn
2+ feature and they are ranked in the third and the sixth positions.
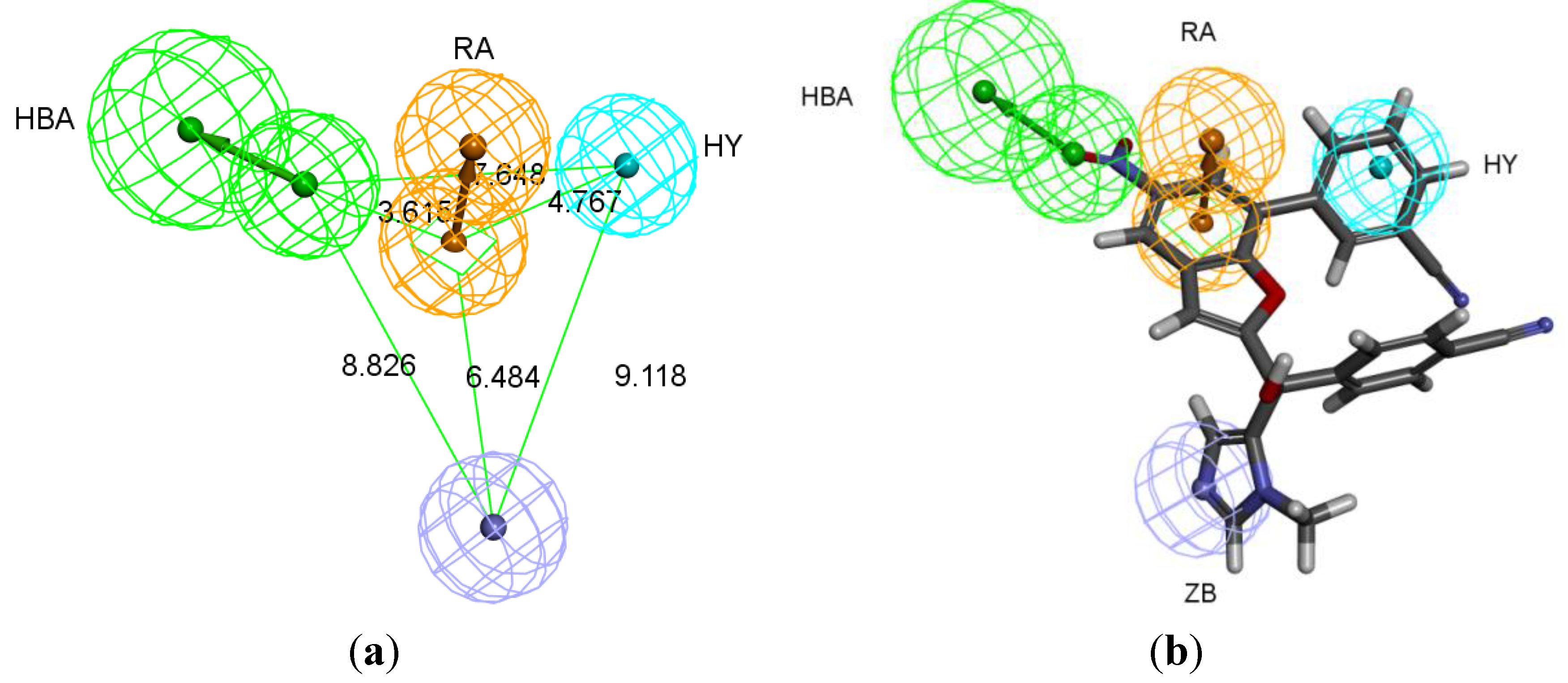
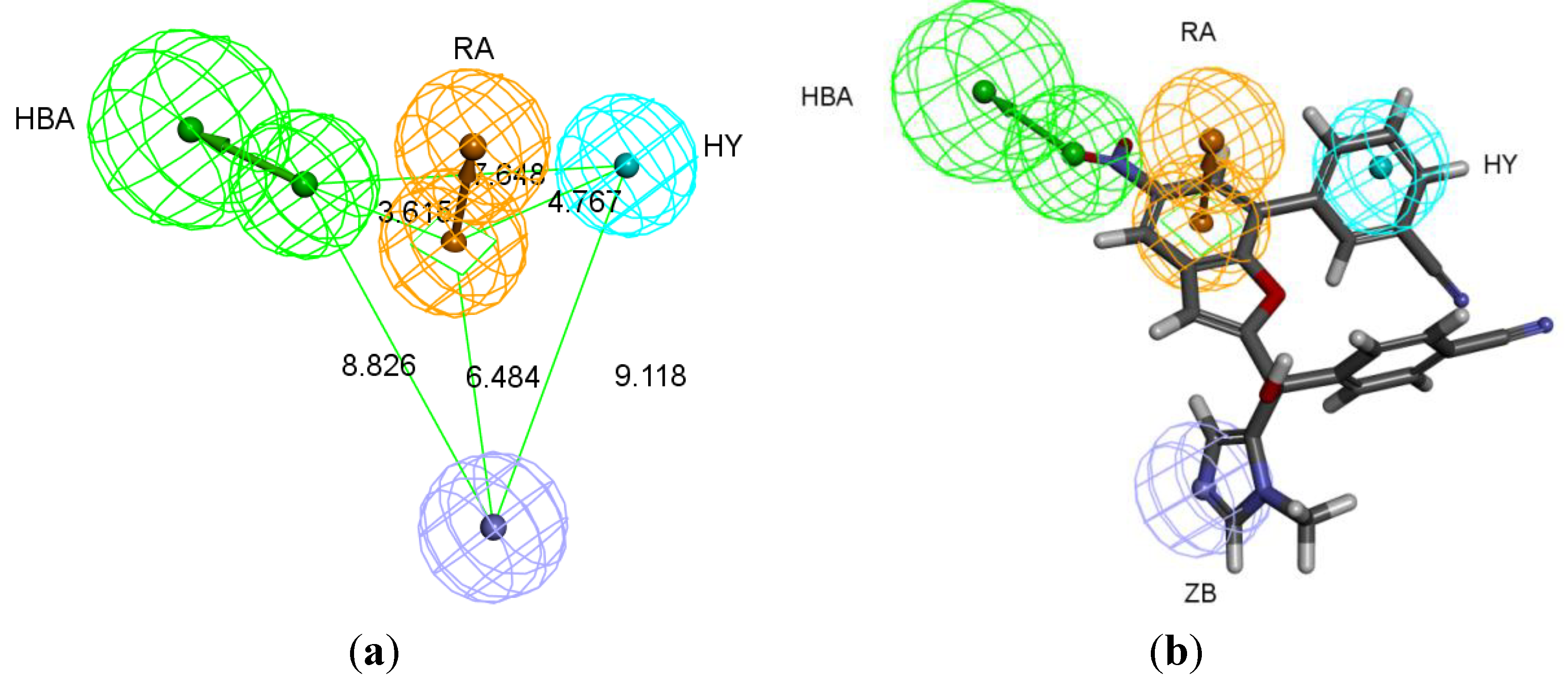
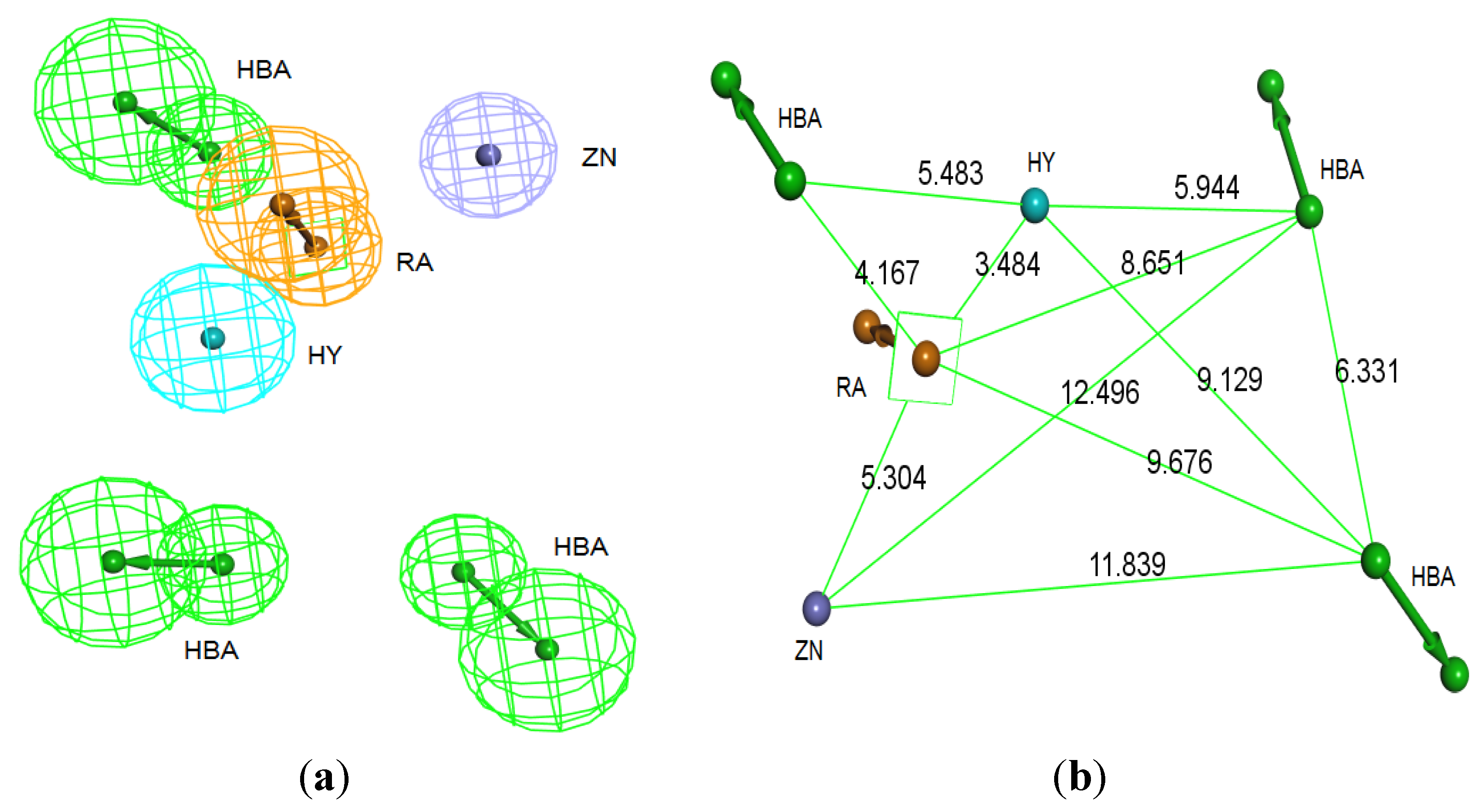
The best pharmacophore hypothesis was chosen based on the fact that the presence of the zinc binding feature is essential. Hypothesis three has showed high values compared to other hypotheses and contains the zinc binding feature, named as Pharm-3A (
Table 2). Pharm-3A comprises one zinc binder, one ring aromatic, one hydrophobic and one HBA feature (
Figure 3a). Each of the training set molecules has mapped all the features in the pharmacophore hypothesis. For instance, compound 2ZIS-NH8903 has the imidazole ring mapped to the Zn
+2 binding feature, whereas the nitro group matched the HBA, in addition, one of the aromatic rings was mapped to the ring aromatic feature while the other one was mapped to the hydrophobic feature (
Figure 3b).
Table 2.
Best-fit values of the training and test set compounds based on common feature pharmacophore technique.
Table 2.
Best-fit values of the training and test set compounds based on common feature pharmacophore technique.
| PDB ID | Inhibitor Name | Fit Value |
|---|
| Pharm-3A |
|---|
| 2ZIS | NH8903 | 4.000 |
| 1N95 | FTH1001 | 3.643 |
| 3E32 | ED21003 | 3.191 |
| 1NI1 | 2C510 | 3.178 |
| 3KSQ | Z96439 | 3.018 |
| 2F0Y | 3MN963 | 2.564 |
| 1SA5 | BMV440 | 2.112 |
| 1X81 | JAN1 | 1.826 |
| 1MZC | BME1003 | 1.164 |
| 1N9A | FTI1 | 0.788 |
| 2ZIR | NH7903 * | 3.585 |
| 3E33 | ED71003 * | 3.096 |
| 2IEJ | S48943 * | 2.748 |
| 1S63 | 7783012 * | 1.784 |
Figure 3.
Pharm-3A and its overlay with a training set compound (a) Chemical features of pharm-3A with its inter-feature distances. (b) 2ZIS-NH8903 overlaid on pharm-3A hypothesis. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB; zinc binder.
Figure 3.
Pharm-3A and its overlay with a training set compound (a) Chemical features of pharm-3A with its inter-feature distances. (b) 2ZIS-NH8903 overlaid on pharm-3A hypothesis. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB; zinc binder.
3.2. Structure-Based Pharmacophore Hypothesis
This pharmacophore was generated in three stages; the first, Receptor-ligand pharmacophore generation protocol was performed for each co-crystallized ligand inside the active site in order to discover the potentially important amino acids that are of strategic contribution to ligand binding. These amino acids will be later considered in the final pharmacophore of the active site. The results collected from running Receptor-ligand pharmacophore generation protocol have showed a series of amino acids that contribute to ligand bindings in the active site. Leu96, Tyr93 and Trp 106 performed hydrophobic interaction with lipophilic groups found in the inhibitors’ structures. Tyr361 and Tyr166 have participated in ligand binding by forming ring aromatic interaction with aromatic rings in the inhibitors’ structures. Moreover, Arg202 and Tyr93 were prominent amino acids by being HBDs to their corresponding acceptors in the inhibitors. All the tested inhibitors have showed covalent interaction between the zinc binding group of the inhibitors and the Zn2+ inside the active site.
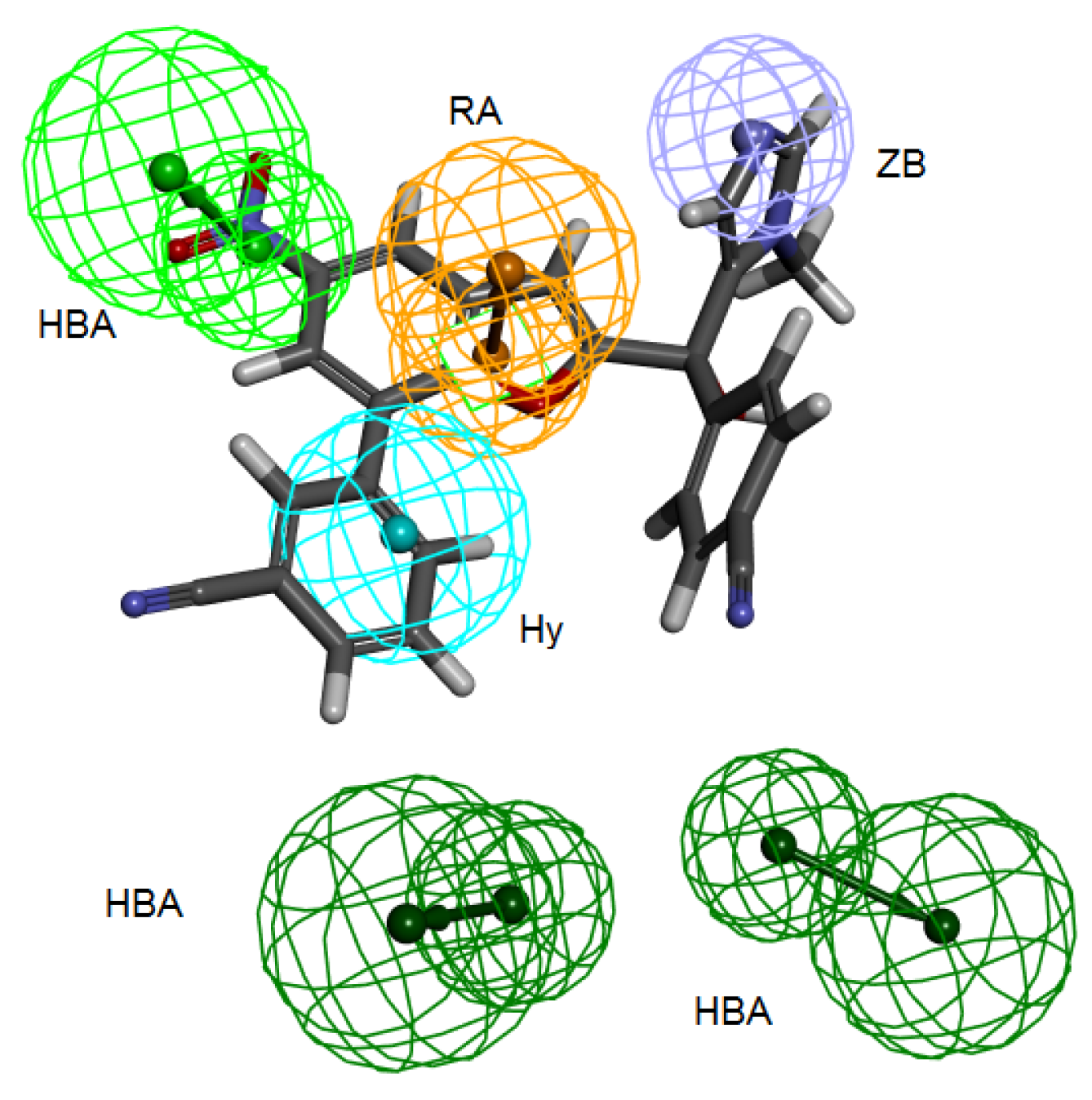
Secondly,
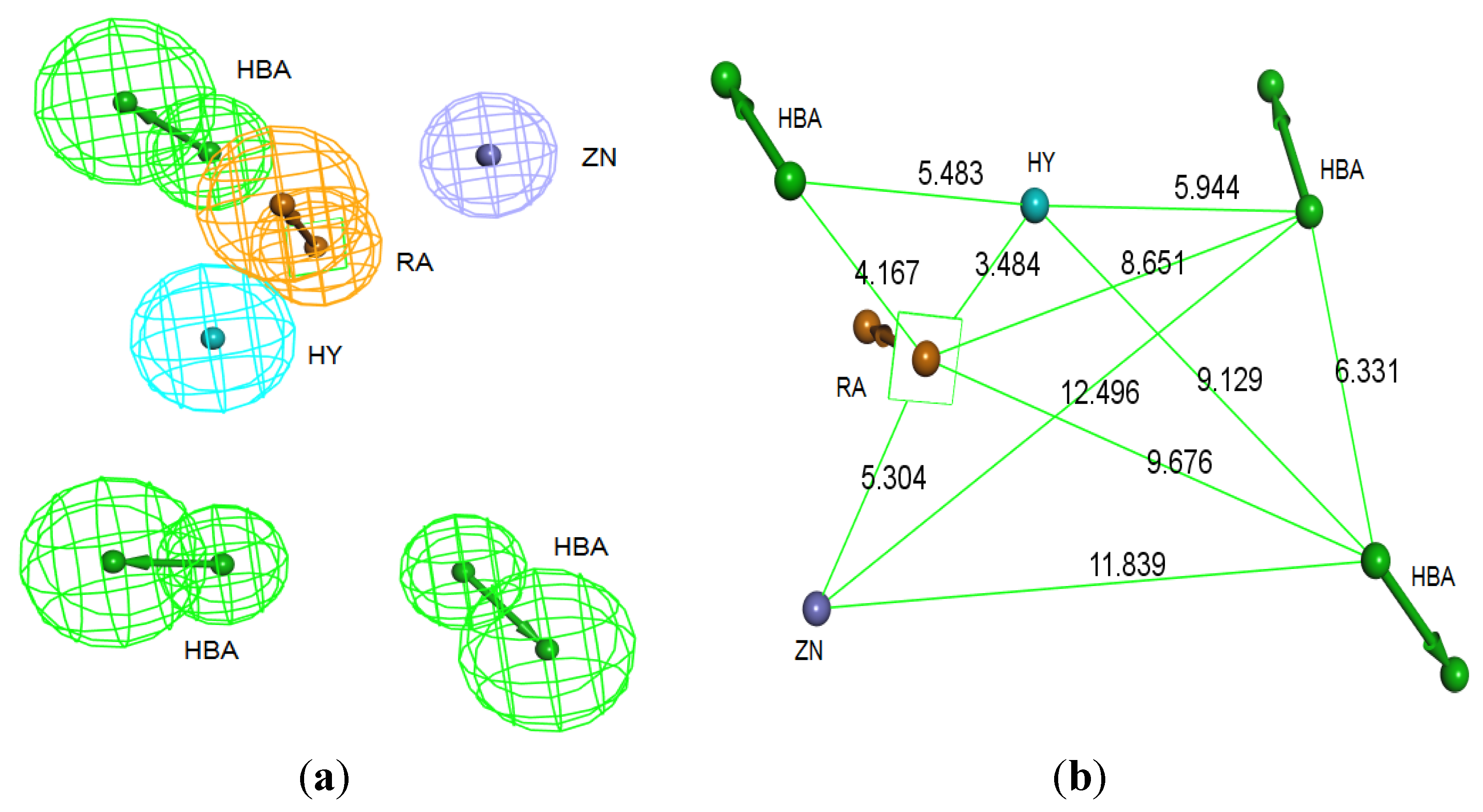
Interaction Generation protocol was generated based on the lipophilic and the hydrophilic regions within the active site, which was later subjected to further cleaning yielding a pharmacophore hypothesis (Pharm-B) containing six features that are complementary to Tyr361, Tyr166, Phe360, Leu96, Tyr93 and the Zn
2+ atom. The third stage involved customization of the pharmacophore by selectively replacing HBA feature pointed to the Zn
2+ atom by zinc feature available on DS 3.1 (
Figure 4). The generated pharmacophore from this step is then compared with pharm-3A by utilizing
Pharmacophore comparison pharmacophore. The results indicated that there was a comparable similarity between the two pharmacophores with RMSD value of 2.52 (
Figure 5).
The fourteen compounds used to generate the common feature pharmacophore Pharm-3A were used to check whether Pharm-B can accommodate these compounds and map them. The fit values were extracted for these compounds which showed good fitting values (
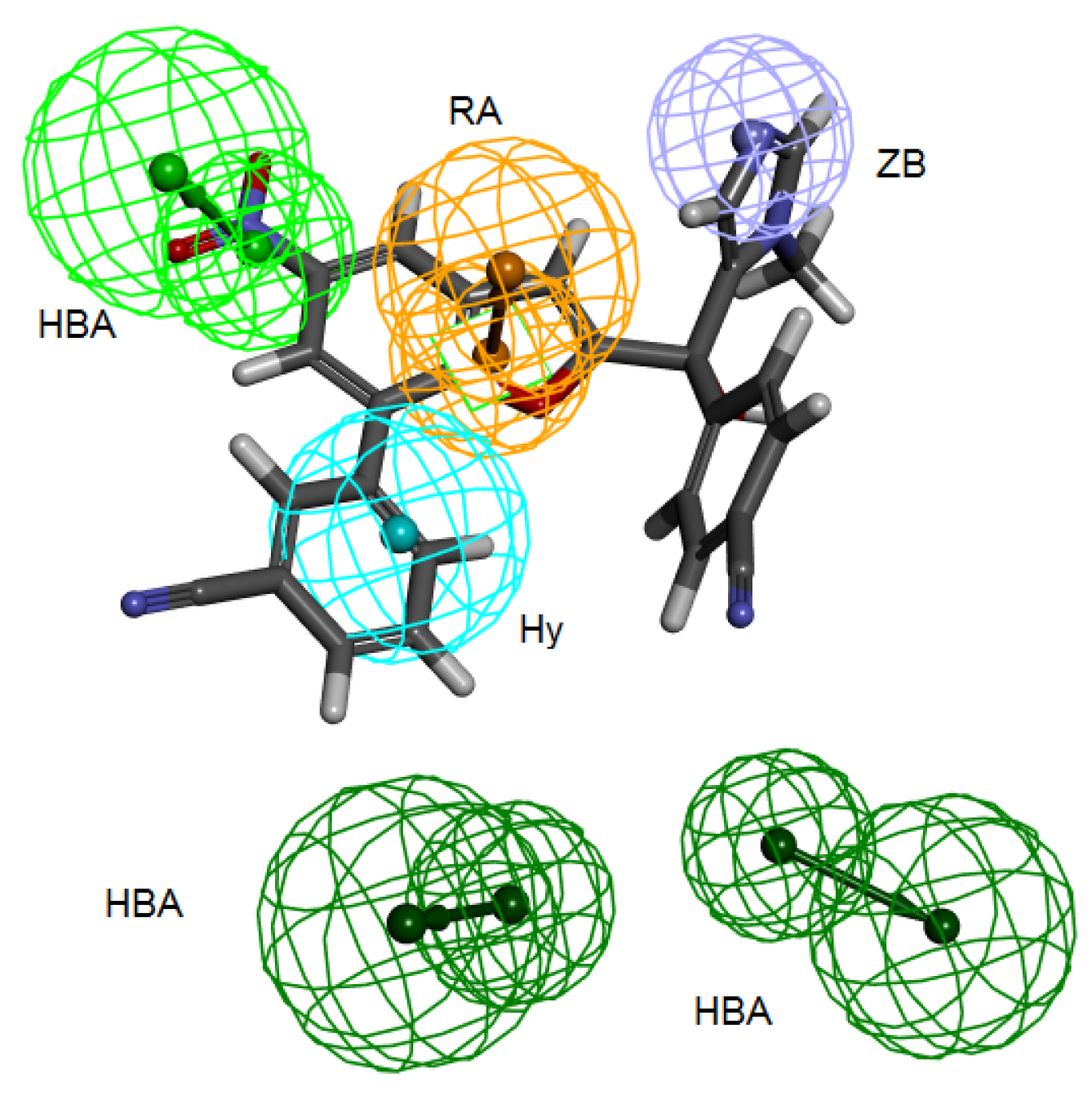
Table 3) and all the fourteen compounds mapped at least four features of the six pharmacophoric features of the structure-based hypothesis and every time the zinc binding feature was occupied with zinc binding group. Compound 2ZIS-NH8903 mapping on Pharm-B is depicted in (
Figure 6).
Figure 4.
Structure-based pharmacophore hypothesis, Pharm-B. (a) Arrangement of pharmacophoric features of Pharm-B. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB; zinc binder. (b) Pharmacophoric features are displayed with inter-feature distances. Tolerance spheres were removed for simplification purposes.
Figure 4.
Structure-based pharmacophore hypothesis, Pharm-B. (a) Arrangement of pharmacophoric features of Pharm-B. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB; zinc binder. (b) Pharmacophoric features are displayed with inter-feature distances. Tolerance spheres were removed for simplification purposes.
Figure 5.
Superimposition of pharm-3A over Pharm-B showing the degree of similarity between the two pharmacophores of RMSD value of 2.52. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB; zinc binder.
Figure 5.
Superimposition of pharm-3A over Pharm-B showing the degree of similarity between the two pharmacophores of RMSD value of 2.52. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB; zinc binder.
Table 3.
Best-fit values of the training and test set compounds based on structure based pharmacophore techniques.
Table 3.
Best-fit values of the training and test set compounds based on structure based pharmacophore techniques.
| PDB ID | Inhibitor Name | Fit Value |
|---|
| Pharm-B |
|---|
| 2ZIS | NH8903 | 5.088 |
| 1N95 | FTH1001 | 4.979 |
| 3E32 | ED21003 | 5.666 |
| 1NI1 | 2C510 | 4.713 |
| 3KSQ | Z96439 | 4.324 |
| 2F0Y | 3MN963 | 5.457 |
| 1SA5 | BMV440 | 4.956 |
| 1X81 | JAN1 | 4.627 |
| 1MZC | BME1003 | 4.778 |
| 1N9A | FTI1 | 4.962 |
| 2ZIR | NH7903 * | 4.933 |
| 3E33 | ED71003 * | 5.266 |
| 2IEJ | S48943 * | 5.579 |
| 1S63 | 7783012 * | 4.167 |
Figure 6.
2ZIS-NH8903 overlaid on pharm-B hypothesis. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB zinc binder.
Figure 6.
2ZIS-NH8903 overlaid on pharm-B hypothesis. HY; hydrophobic, RA; ring aromatic, HBA; hydrogen bond acceptor, ZB zinc binder.
3.3. Validation of Pharmacophore Hypotheses
Ligand Pharmacophore Mapping Protocol was used to perform this step. The four test compounds were used to score their fitting to the generated pharmacophore taking into consideration that there is no conformation generation accompanied with rigid fitting technique. The four compounds were found to completely match Pharm-3A with fitting values ranging from 3.58–1.78 (
Table 2).
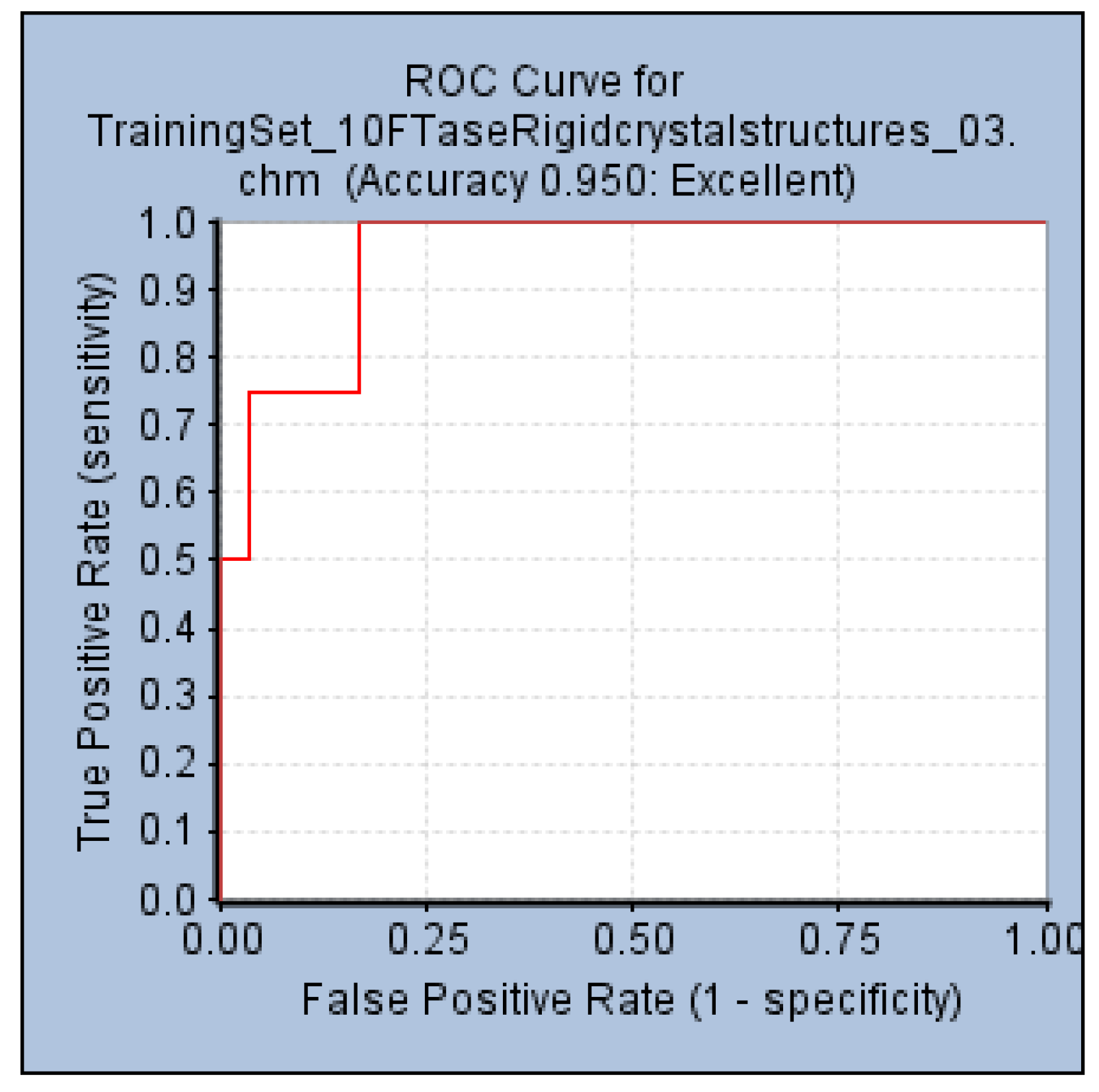
An internal validation step performed by generating a ROC curve was also used to confirm the ability of Pharm-3A to be able to distinguish between active and inactive molecules. Within the
Common Feature Pharmacophore Generation protocol, a validation step was used by providing the active compounds which were the four test set compounds (
Figure 1b), and 85 inactive compounds collected from literature. The AUC, accuracy and specificity of Pharm-3A were shown to be the best among all the pharmacophore hypotheses predicted by the training set compounds by recording 93%, 95% and 55% respectively (
Figure 7).
Figure 7.
ROC curve of hypothesis Pharm-3A.
Figure 7.
ROC curve of hypothesis Pharm-3A.
In another validation step, a database of 269 compounds of both active and inactive compounds was collected from the literature and drawn using Chemsketch 12.0 and then converted to .sd format file. Of the 269 compounds, 184 were considered active compounds based on their activity against FTase enzyme of more than 500 nM. Using Ligand Pharmacophore Mapping Protocol, the 184 compounds were used to investigate the ability of the generated pharmacophore hypothesis to map ligands that are known inhibitors to this enzyme. Of 184 compounds, 153 compounds were mapped on all the features of the common feature pharmacophore hypothesis with a hit rate of 83.15%. And the same compounds were mapped on the structure-based pharmacophore and here, 151 of 184 compounds were mapped on all the features of structure-based pharmacophore with a hit rate of 82.06%.
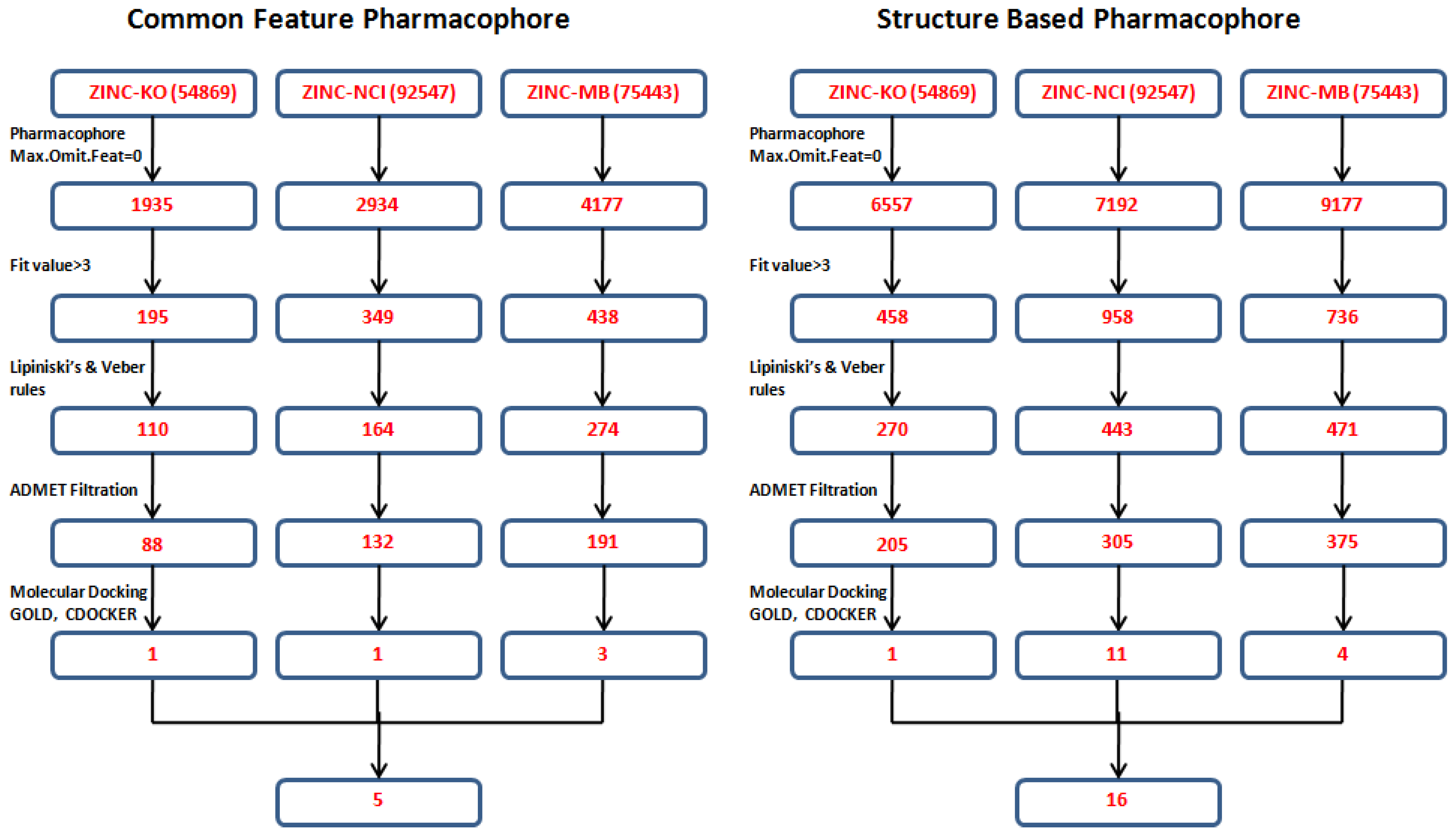
3.4. Database Screening
Both the common feature pharmacophore and the structure-based pharmacophore hypotheses that were generated previously were subjected to database screening process utilizing
Ligand Pharmacophore Mapping Protocol using
Best Flexible search option. These databases are ZINC-NCI_(92547), ZINC-MayBridge_(75443), and ZINC-Key Organics_(54869) [
21] which were prepared previously by our group for the screening process. For Pharm-3A,
Ligand Pharmacophore Mapping protocol was used with
MaxOmitFeat value of zero as the four features of the Pharm-3A should be fulfilled. A compound will be determined as a hit if it maps all the four features that are included in the pharmacophore hypothesis.On the other hand, a relaxed criteria was used for Pharm-B where the hit is accepted if it can map five features out of the six and hence the
Ligand Pharmacophore Mapping protocol which allows some of the features to be missed was used. The extracted compounds were then subjected to further screening processes relying on their fitness value, drug likeness according to Lipinski's rule of five and Vebers’ rule. A compound is determined positive according to
Lipiniski and
Veber if (i) less than five hydrogen bond donor groups; (ii) less than 10 hydrogen bond acceptor groups; (iii) a molecular weight less than 500; (iv) A LogP value less than 5; (v) number of rotatable bonds less than 10; polar surface area less than 140 Å
2; and hydrogen bond donors and acceptors less than 12. A subsequent level of filtration to achieve drug like compounds is to perform ADMET screening techniques in DS 3.1 in which the databases are filtered according to their solubility, intestinal absorption and Blood Brain Barrier penetration. A total of 1,296 compounds (885 from the structure-based design and 411 from the common feature pharmacophore) were found to satisfy the assigned filters for the molecular docking step (
Figure 8).
Figure 8.
Database screening of three databases employing pharm-3A and pharm-B hypotheses.
Figure 8.
Database screening of three databases employing pharm-3A and pharm-B hypotheses.
3.5. Molecular Docking
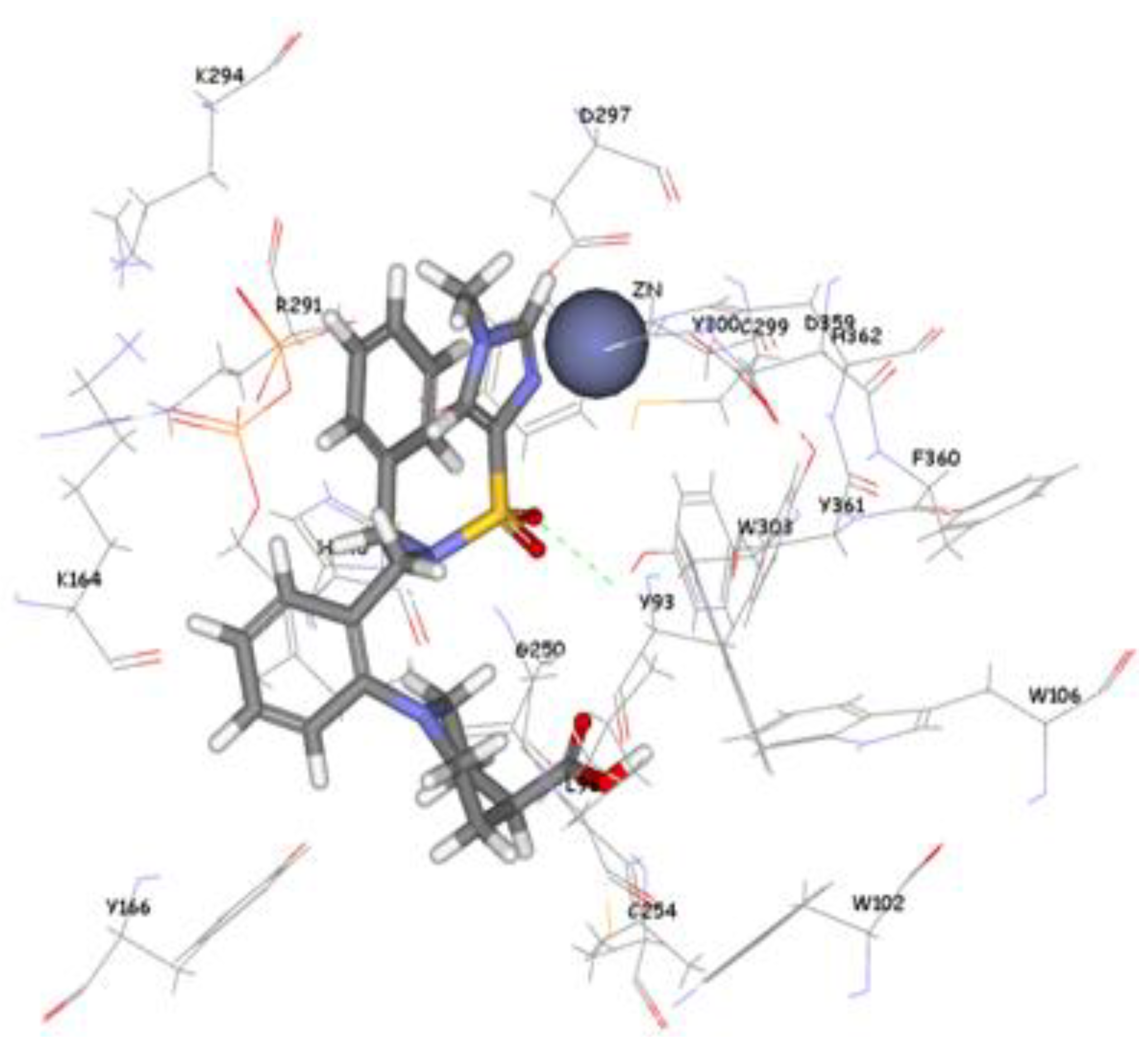
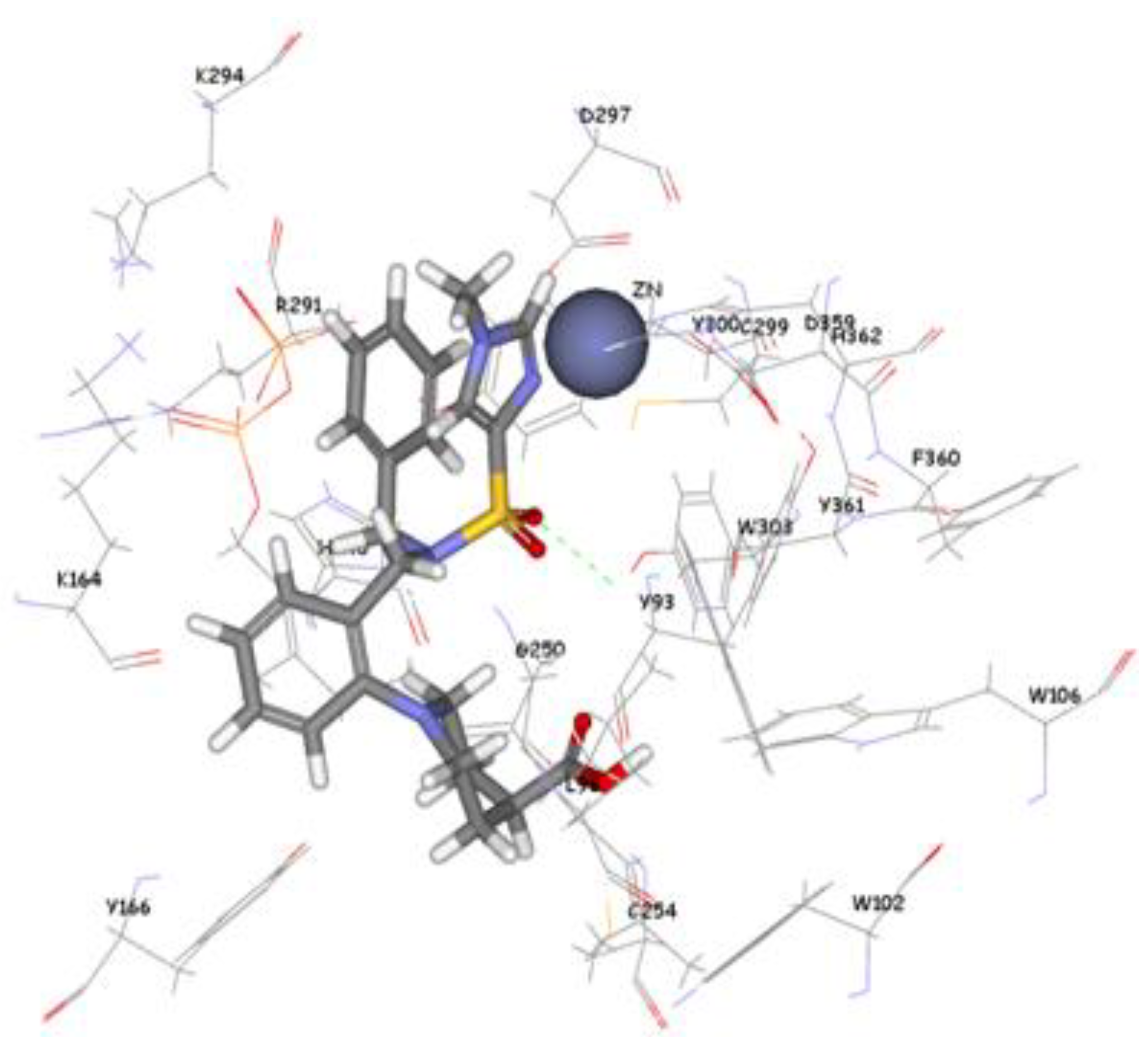
The 1,296 compounds emerged from both structure and ligand-based pharmacophores conjointly with the training set were submitted to docking programs CDOCKER and GOLD. Consensus scores of the candidate compounds from both CDOCKER and GOLD were performed, eventually compounds enumerated the highest value from score summation of the two docking programs were chosen to be candidate molecules. These scores of the candidate compounds were contrasted by scores obtained for the training set compounds (the highest consensus score for the training set compound is 147.4) and only compounds that scored equally or higher than the training set compounds were fully investigated by studying their binding mode. Of the 1,296 compounds, twenty one compounds had shown scores higher than training set scores. Based on molecular fitting mode inside the active site and their proper interaction with zinc atom, two compounds were chosen as potential virtual leads namely ZINC39323901, and ZINC01034774. Compound ZINC01034774 which was identified from structure-based pharmacophore has a fit value, CDOCKER, and GOLD fitness scores of 3.28, 64.65, and 98.11 respectively. The imidazole ring is supposed to form a coordinate bond with zinc cation which has a perfect pose when the pyridine-like nitrogen points directly at zinc cation of the active site with optimum distance of 1.76 Å. In addition, internal π-π interaction is shown between the imidazole ring and one of the benzene rings of the compound. The other benzene ring is positioned in a hydrophobic pocket within the active site formed from the hydrophobic tail of farnesyl pyrophosphate (FPP), Tyr166, and Lys164. The sulphonamide moiety interacts via hydrogen bond with Tyr361 phenol side chain (
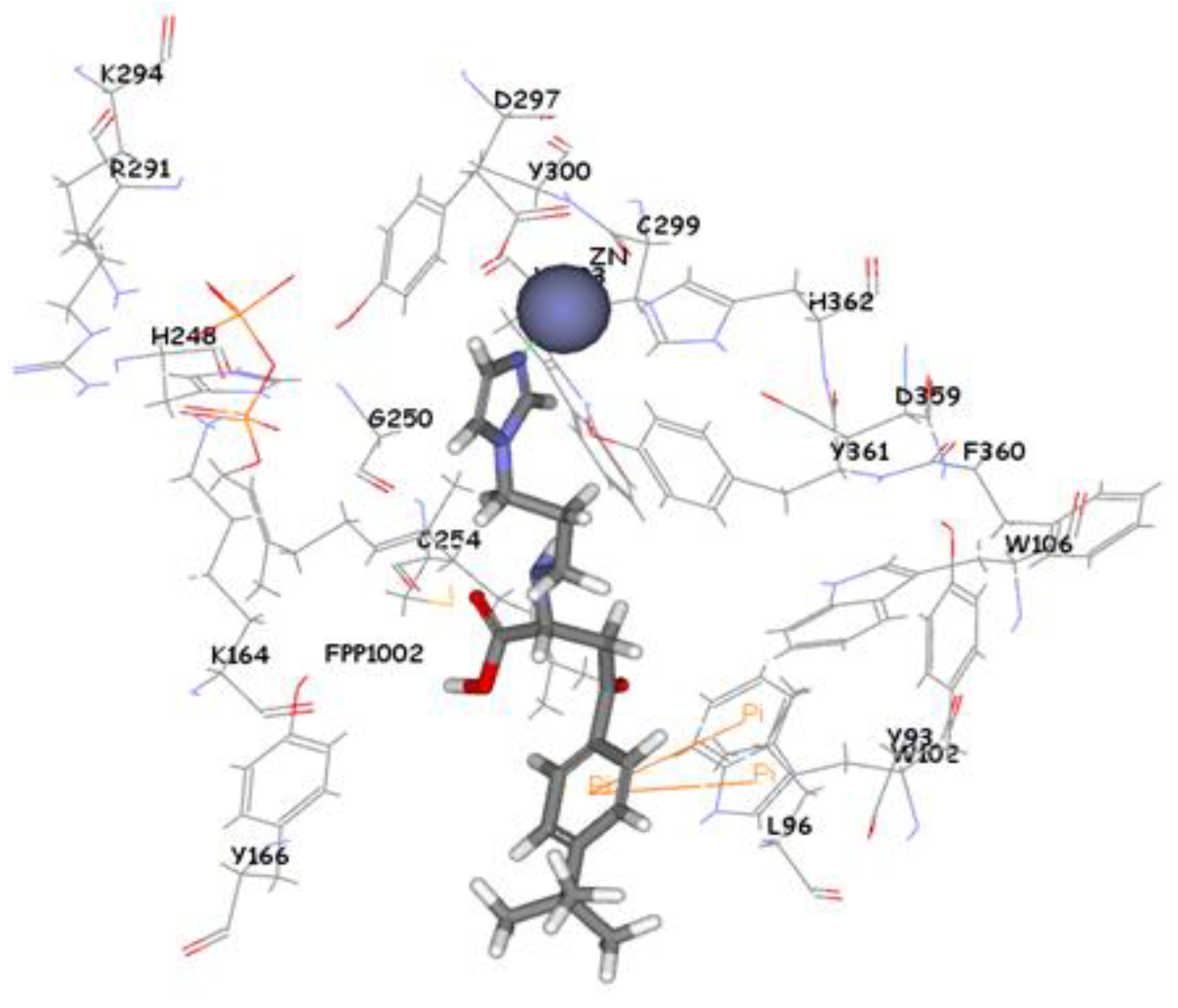
Figure 9). Compound ZINC39323901 which was extracted from common feature pharmacophore has a fit value, CDOCKER, and GOLD fitness scores of 3.64, 65.05, and 95.63 respectively. The imidazole ring is in proximity to the zinc cation with a distance of 1.70 Å which is expected to form coordination bond, and the benzene ring is expected to form π-π interactions with indole ring of Trp102 side chain (
Figure 10).
Figure 9.
Molecular docking result of ZINC01034774 displayed in stick format, zinc atom CPK and active site amino acids presented as line format.
Figure 9.
Molecular docking result of ZINC01034774 displayed in stick format, zinc atom CPK and active site amino acids presented as line format.
Figure 10.
Molecular docking result of ZINC39323901 displayed in stick format, zinc atom CPK and active site amino acids presented as line format.
Figure 10.
Molecular docking result of ZINC39323901 displayed in stick format, zinc atom CPK and active site amino acids presented as line format.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}