Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Introduction for Mizoribine
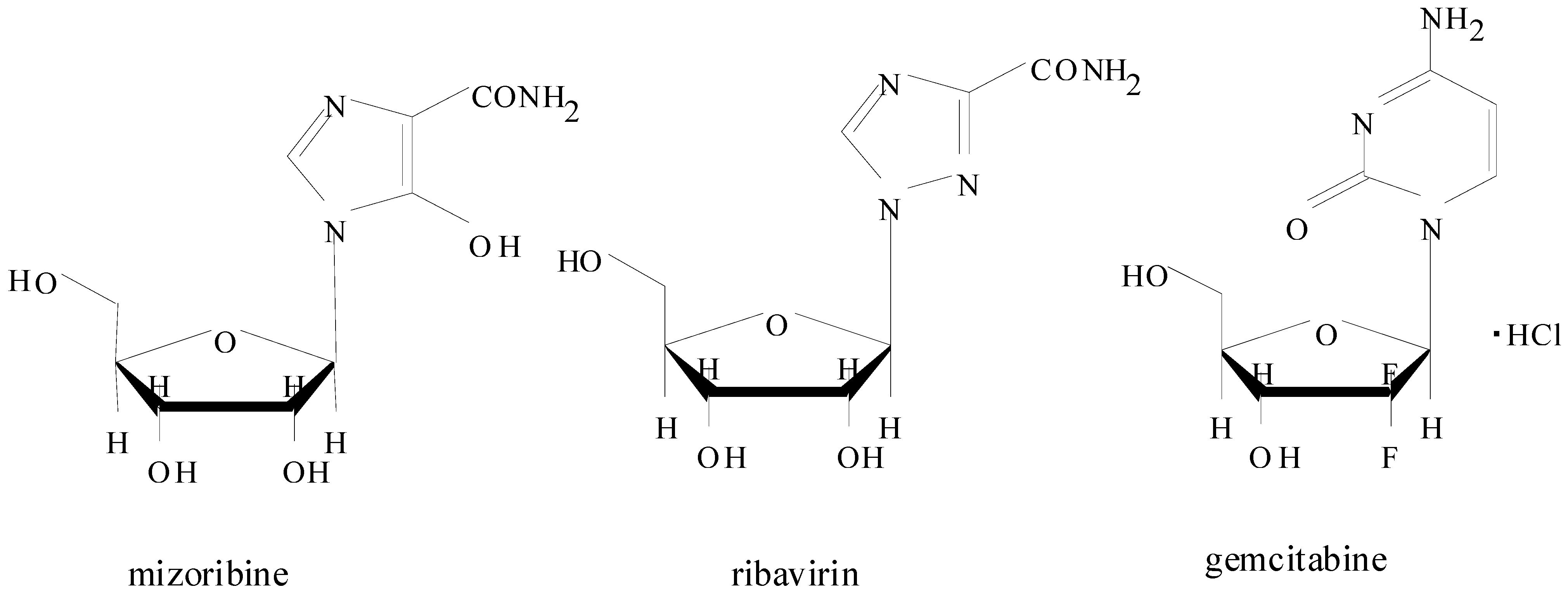
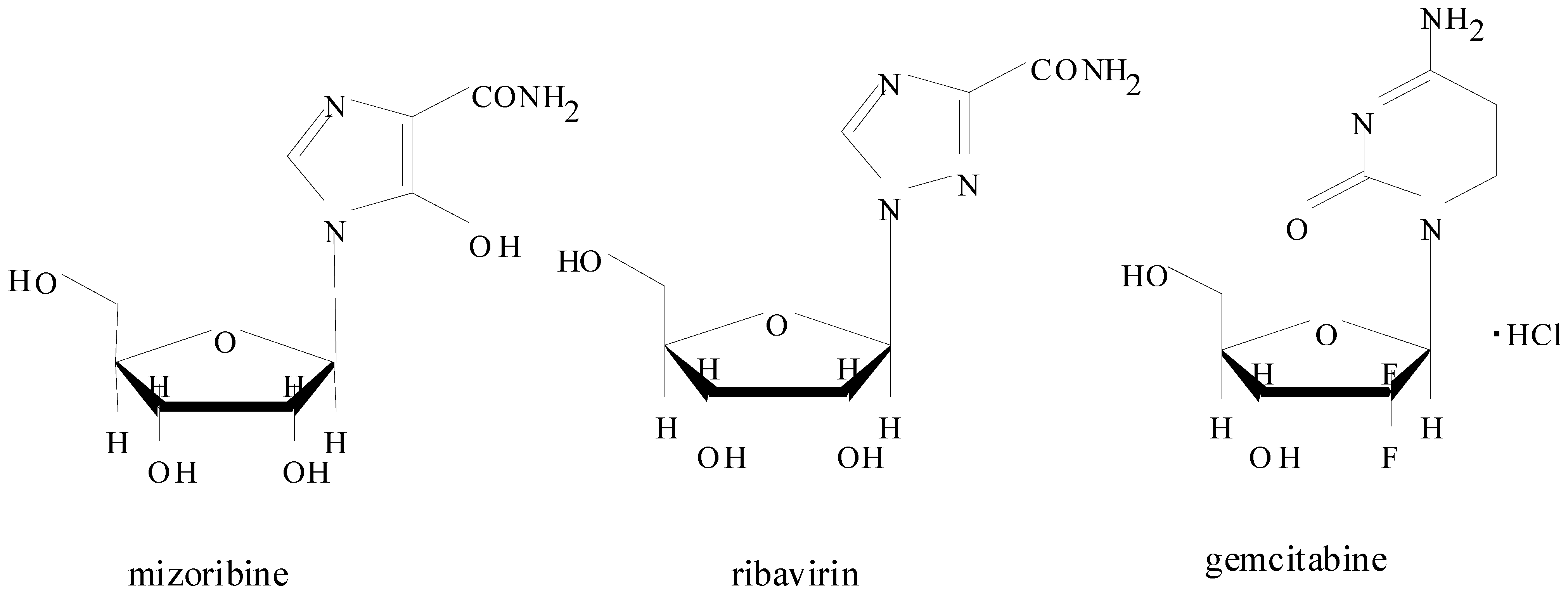
2.1. Pharmacokinetics of Mizoribine in Human Subjects
2.2. Effect of Polymorphisms on Mizoribine Bioavailability
2.3. Pharmacokinetics of Mizoribine in Animal Studies
2.4. Summary for Mizoribine Pharmacokinetics
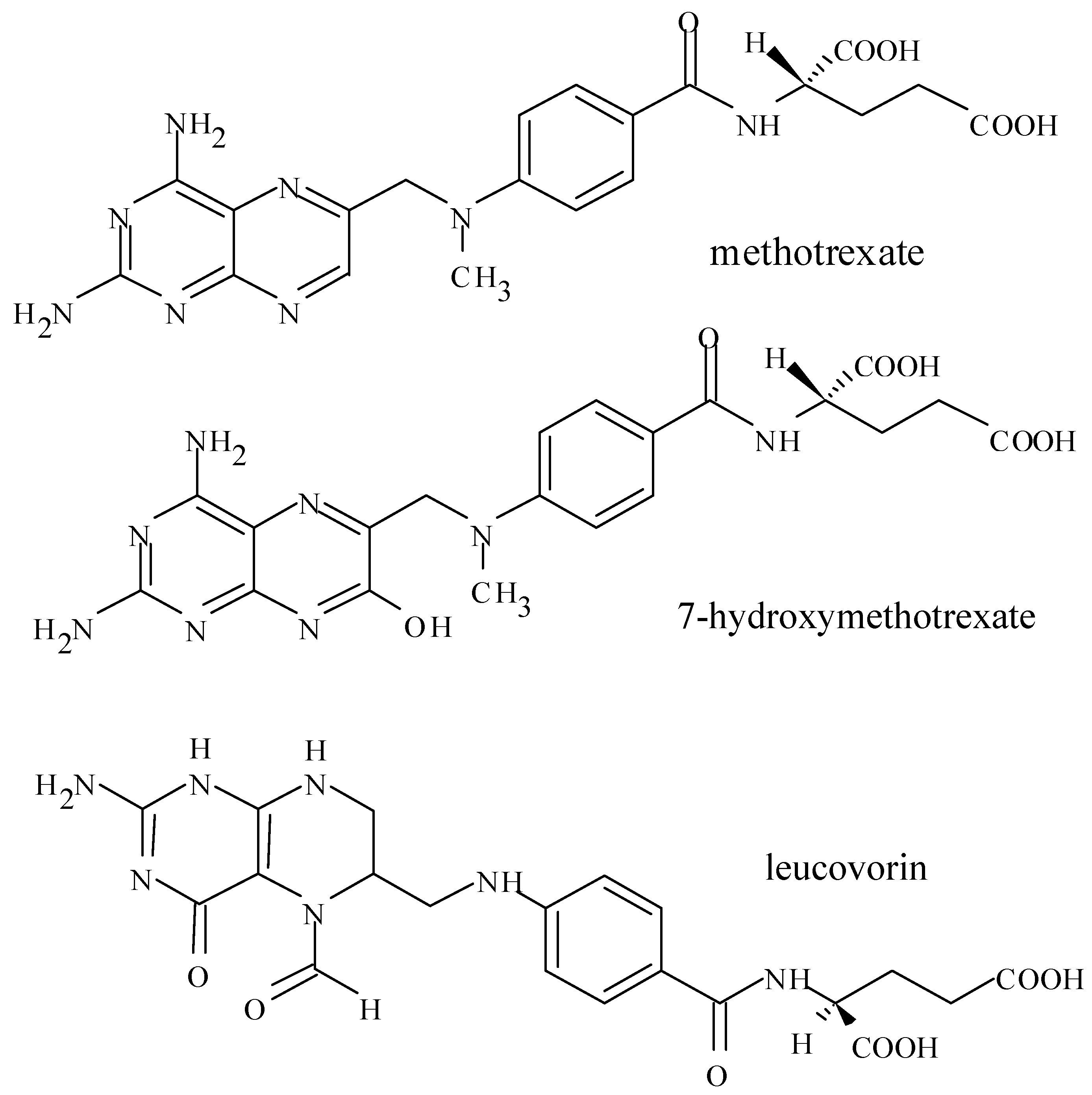
3. Introduction for Methotrexate

3.1. Oral Bioavailability of MTX
3.2. Contribution of Multiple Transporters to Intestinal Absorption of MTX
3.2.1. Site-Specific Contribution of PCFT in Intestinal MTX Absorption
3.2.2. Site-Specific Contribution of ABC transporters in Intestinal MTX Absorption
3.2.3. Effect of Gastric Emptying Rates on MTX Oral Absorption in Rats
3.3. Systemic Clearance of MTX
3.4. Tissue Distribution of MTX
3.5. Formation of MTX Polyglutamate in Cells
3.6. Effect of Polymorphisms on Methotrexate Pharmacokinetics
3.7. Summary for Methotrexate Pharmacokinetics
4. Conclusions
References
- Kasama, T.; Wakabayashi, K.; Odai, T.; Isozaki, T.; Matsunawa, M.; Yajima, N.; Miwa, Y.; Negishi, M.; Ide, H. Effects of low-dose mizoribine pulse therapy in combination with methotrexate in rheumatoid arthritis patients with an insufficient response to methotrexate. Mod. Rheumatol. 2009, 19, 395–400. [Google Scholar]
- Suzuki, D.; Kimoto, O.; Sawada, J.; Shimoyama, K.; Kawashima, M.; Mukai, T.; Ohashi, H.; Yamamura, M.; Ogawa, N. The efficacy and safety of combination therapy with mizoribine and methotrexate for rheumatoid arthritis resistant with methotrexate. Jpn. J. Clin. Immunol. 2011, 34, 149–153. [Google Scholar] [CrossRef]
- Mizuno, K.; Tsujino, M.; Takada, M.; Hayashi, M.; Atsumi, K. Studies on bredinin. I. Isolation, characterization and biological properties. J. Antibiot. (Tokyo) 1974, 27, 775–782. [Google Scholar]
- Kamata, K.; Okubo, M.; Ishigamori, E.; Masaki, Y.; Uchida, H.; Watanabe, K.; Kashiwagi, N. Immunosuppressive effect of bredinin on cell-mediated and humoral immune reactions in experimental animals. Transplantation 1983, 35, 144–149. [Google Scholar] [CrossRef]
- Hughes, S.E.; Gruber, S.A. New immunosuppressive drugs in organ transplantation. J. Clin. Pharmacol. 1996, 36, 1081–1092. [Google Scholar]
- Takei, S. Mizoribine in the treatment of rheumatoid arthritis and juvenile idiopathic arthritis. Pediatr. Int. 2002, 44, 205–209. [Google Scholar] [CrossRef]
- Yokota, S. Mizoribine: Mode of action and effects in clinical use. Pediatr. Int. 2002, 44, 196–198. [Google Scholar] [CrossRef]
- Kawasaki, Y. Mizoribine: A new approach in the treatment of renal disease. Clin. Dev. Immunol. 2009, 681482. [Google Scholar]
- Webster, H.K.; Whaun, J.M. Antimalarial properties of bredinin. Prediction based on identification of differences in human host-parasite purine metabolism. J. Clin. Invest. 1982, 70, 461–469. [Google Scholar]
- Naka, K.; Ikeda, M.; Abe, K.; Dansako, H.; Kato, N. Mizoribine inhibits hepatitis C virus RNA replication: Effect of combination with interferon-alpha. Biochem. Biophys. Res. Commun. 2005, 330, 871–879. [Google Scholar]
- Ghose, A.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef]
- Podgorska, M.; Kocbuch, K.; Pawelczyk, T. Recent advances in studies on biochemical and structural properties of equilibrative and concentrative nucleoside transporters. Acta Biochim. Pol. 2005, 52, 749–758. [Google Scholar]
- Pastor-Anglada, M.; Errasti-Murugarren, E.; Aymerich, I.; Casado, F.J. Concentrative nucleoside transporters (CNTs) in epithelia: From absorption to cell signaling. J. Physiol. Biochem. 2007, 63, 97–110. [Google Scholar] [CrossRef]
- Casado, F.J.; Lostao, M.P.; Aymerich, I.; Larráyoz, I.M.; Duflot, S.; Rodríguez-Mulero, S.; Pastor-Anglada, M. Nucleoside transporters in absorptive epithelia. J. Physiol. Biochem. 2002, 58, 207–216. [Google Scholar] [CrossRef]
- Lu, H.; Chen, C.; Klaassen, C. Tissue distribution of concentrative and equilibrative nucleoside transporters in male and female rats and mice. Drug Metab. Dispos. 2004, 32, 1455–1461. [Google Scholar]
- Ritzel, M.W.; Ng, A.M.; Yao, S.Y.; Graham, K.; Loewen, S.K.; Smith, K.M.; Ritzel, R.G.; Mowles, D.A.; Carpenter, P.; Chen, X.Z.; et al. Molecular identification and characterization of novel human and mouse concentrative Na+-nucleoside cotransporter proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). J. Biol. Chem. 2001, 276, 2914–2927. [Google Scholar]
- Gray, J.H.; Owen, R.P.; Giacomini, K.M. The concentrative nucleoside transporter family, SLC28. Pflugers Arch. Eur. J. Physiol. 2004, 447, 728–734. [Google Scholar] [CrossRef]
- Mori, N.; Yokooji, T.; Kamio, Y.; Murakami, T. Characterization of intestinal absorption of mizoribine mediated by concentrative nucleoside transporters in rats. Eur. J. Pharmacol. 2008, 586, 52–58. [Google Scholar] [CrossRef]
- Mackey, J.R.; Yao, S.Y.; Smith, K.M.; Karpinski, E.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J. Natl. Cancer Inst. 1999, 91, 1876–1881. [Google Scholar] [CrossRef]
- Rauchwerger, D.R.; Firby, P.S.; Hedley, D.W.; Moore, M.J. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res. 2000, 60, 6075–6079. [Google Scholar]
- Okada, M.; Suzuki, K.; Nakashima, M.; Nakanishi, T.; Fujioka, N. The nucleotide derivatives inosine and inosinic acid inhibit intestinal absorption of mizoribine in rats. Eur. J. Pharmacol. 2006, 531, 140–144. [Google Scholar] [CrossRef]
- Tanaka, H.; Tsugawa, K.; Nakahata, T.; Kudo, M.; Suzuki, K.; Ito, E. Implication of the peak serum level of mizoribine for control of the serum anti-dsDNA antibody titer in patients with lupus nephritis. Clin. Nephrol. 2005, 63, 417–422. [Google Scholar]
- Liu, D.; Kobayashi, T.; Nagasaki, T.; Yokoyama, I.; Ma, Y.; Miwa, Y.; Kuzuya, T.; Morozumi, K.; Oikawa, T.; Shimano, Y.; Takeuchi, O.; Uchida, K.; Nakao, A. Potential value of high-dose mizoribine as rescue therapy for ongaing acute humoral rejection. Transpl. Int. 2005, 18, 401–407. [Google Scholar]
- Kawasaki, Y.; Takano, K.; Isome, M.; Suzuki, J.; Suyama, K.; Kanno, H.; Fujiki, T.; Suzuki, H.; Hosoya, M. Efficacy of single dose of oral mizoribine pulse therapy two times per week for frequently relapsing nephritic syndrome. J. Nephrol. 2007, 20, 52–56. [Google Scholar]
- Koyama, H.; Tsuji, M. Genetic and biochemical studies on the activation and cytotoxic mechanism of bredinin, a potent inhibitor of purine biosynthesis in mammalian cells. Biochem. Pharmacol. 1983, 32, 3547–3553. [Google Scholar] [CrossRef]
- Kusumi, T.; Tsuda, M.; Katsunuma, T.; Yamamura, M. Dual inhibitory effect of bredinin. Cell Biochem. Funct. 1989, 7, 201–204. [Google Scholar] [CrossRef]
- Stypinski, D.; Obaidi, M.; Combs, M.; Weber, M.; Stewart, A.J.; Ishikaw, H. Safety, tolerability and pharmacokinetics of higher-dose mizoribine in healthy male volunteers. Br. J. Clin. Pharmacol. 2007, 63, 459–468. [Google Scholar] [CrossRef]
- Takada, K.; Asada, S.; Ichikawa, Y.; Sonoda, T.; Takahara, S.; Nagano, S.; Fukunishi, T. Pharmacokinetics of bredinin in renal transplant patients. Eur. J. Clin. Pharmacol. 1983, 24, 457–461. [Google Scholar] [CrossRef]
- Warabi, H.; Abe, S.; Kondo, H.; Ishihara, Y.; Sugawara, S.; Shichikawa, K.; Shiokawa, Y. Consecutive administration of mizoribine in rheumatoid arthritis: Pharmacokinetic study. Rheumatology 1981, 5, 287–300. [Google Scholar]
- Ihara, H.; Shinkuma, D.; Kubo, M.; Miyamoto, I.; Nojima, M.; Koike, H.; Yabumoto, H.; Ikoma, F. Influence of bioavailability on blood level of mizoribine in kidney transplant recipients. Transplant Proc. 1996, 28, 1321–1323. [Google Scholar]
- Abe, Y.; Mikawa, T.; Fuke, T.; Hisano, M.; Tsuji, Y.; Watanabe, S.; Itabashi, K. Pharmacokinetic study of mizoribine in child-onset glomerulonephritis. Pediatr. Int. 2008, 50, 615–619. [Google Scholar]
- Naito, T.; Tokashiki, S.; Mino, Y.; Otsuka, A.; Ozono, S.; Kagawa, Y.; Kawakami, J. Impact of concentrative nucleoside transporter 1 gene polymorphism on oral bioavailability of mizoribine in stable kidney transplant recipients. Basic Clin. Pharmacol. Toxicol. 2010, 106, 310–316. [Google Scholar]
- Fukao, M.; Ishida, K.; Sakamoto, T.; Taguchi, M.; Matsukura, H.; Miyawaki, T.; Hashimoto, Y. Effect of genetic polymorphisms of SLC28A1, ABCG2, and ABCC4 on bioavailability of mizoribine in healthy Japanese males. Drug Metab. Pharmacokinet. 2011, 26, 538–543. [Google Scholar] [CrossRef]
- Murase, J.; Mizuno, K.; Kawai, K.; Nishiumi, S.; Kobayashi, Y.; Hayashi, M.; Morino, T.; Suzuki, T.; Baba, S. Absorption, distribution, metabolism and excretion of bredinin in rats. Pharmacometrics 1978, 15, 829–835. [Google Scholar]
- Uchida, H.; Masaki, Y.; Taira, M.; Maruyama, S.; Yasuda, S. Long-term toxicological studies with mizoribine (Bredinin) in beagle dogs (the first report). Blood chemistries, pharmacokinetics of mizoribine and fertility studies of the male dogs. J. Toxicol. Sci. 1985, 10, 35–49. [Google Scholar]
- To, H.; Xiu, D.R.; Hishikawa, S.; Uchida, H.; Sudoh, T.; Sunaga, K.; Sugimoto, K.; Higuchi, S.; Fujimura, A.; Kobayashi, E. Dosing time-dependent pharmacological effects of anti-metabolites for rat cardiac graft. Res. Commun. Mol. Pathol. Pharmacol. 2001, 110, 319–332. [Google Scholar]
- Takaai, M.; Morishita, H.; Ishida, K.; Taguchi, M.; Hashimoto, Y. Contribution of Na+-independent nucleoside transport to ribavirin uptake in the rat intestine and human epithelial LS180 cells. Eur. J. Pharmacol. 2008, 601, 61–65. [Google Scholar]
- Mori, N.; Yokooji, T.; Kamio, Y.; Murakami, T. Increased intestinal absorption of mizoribine, an immunosuppressive agent, in cholestatic rats. Pharmazie 2010, 65, 457–460. [Google Scholar]
- Mori, N.; Shimomukai, Y.; Yokooji, T.; Ishiguro, M.; Kamio, Y.; Murakami, T. Modulation in concentrative nucleoside transporters-mediated intestinal absorption of mizoribine, an immunosuppressive agent, in lipopolysaccharide-treated rats. Pharmazie 2011, 66, 207–211. [Google Scholar]
- Honda, M.; Itoh, H.; Suzuki, T.; Hashimoto, Y. Population pharmacokinetics of higher-dose mizoribine in healthy male volunteers. Biol. Pharm. Bull. 2006, 29, 2460–2464. [Google Scholar] [CrossRef]
- Yuen, C.W.; Winter, M.E. In Methotrexate (MTX) in Basic clinical pharmacokinetics; Michael, E., Ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, USA, 2010; pp. 304–325, Winter-5th. [Google Scholar]
- Kitamura, S.; Sugihara, K.; Nakatani, K.; Ohta, S.; Ohhara, T.; Ninomiya, S.; Green, C.E.; Tyson, C.A. Variation of hepatic methotrexate 7-hydroxylase activity in animals and humans. IUBMB Life 1999, 48, 607–611. [Google Scholar]
- Dhondt, J.L.; Hayte, J.M.; Millot, F.; Klein, R.; Blais, J.C.; Pfleiderer, W. 2,4-diamino-7-hydroxy-pteridines, a new class of catabolites of methotrexate. Eur. J. Biochem. 1991, 200, 237–244. [Google Scholar] [CrossRef]
- Cairnes, D.A.; Evans, W.E. A simple preparation of the methotrexate metabolites 7-hydroxymethotrexate and 4-deoxy-4-amino-N10-methylpteroic acid. Ther. Drug Monit. 1983, 5, 363–366. [Google Scholar] [CrossRef]
- Fabre, G.; Goldman, I.D. Formation of 7-hydroxymethotrexate polyglutamyl derivatives and their cytotoxicity in human chronic myelogenous leukemia cells, in vitro. Cancer Res. 1985, 45, 80–85. [Google Scholar]
- Widemann, B.C.; Sung, E.; Anderson, L.; Salzer, W.L.; Balis, F.M.; Monitjo, K.S.; McCully, C.; Hawkins, M.; Adamson, P.C. Pharmacokinetics and metabolism of the methotrexate metabolite 2, 4-diamino-N(10)-methylpteroic acid. J. Pharmacol. Exp. Ther. 2000, 294, 894–901. [Google Scholar]
- Kuo, C.Y.; Wu, H.L.; Kou, H.S.; Chiou, S.S.; Wu, D.C.; Wu, S.M. Simultaneous determination of methotrexate and its eight metabolites in human whole blood by capillary zone electrophoresis. J. Chromatogr. A 2003, 1014, 93–101. [Google Scholar] [CrossRef]
- Panetta, J.C.; Wall, A.; Pui, C.H.; Relling, M.V.; Evans, W.E. Methotrexate intracellular disposition in acute lymphoblastic leukemia: A mathematical model of gamma-glutamyl hydrolase activity. Clin. Cancer Res. 2002, 8, 2423–2429. [Google Scholar]
- Ranganathan, P.; McLeod, H.L. Methotrexate pharmacogenetics: The first step toward individualized therapy in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 1366–1377. [Google Scholar] [CrossRef]
- Seideman, P.; Beck, O.; Eksborg, S.; Wennberg, M. The pharmacokinetics of methotrexate and its 7-hydroxy metabolite in patients with rheumatoid arthritis. Br. J. Clin. Pharmacol. 1993, 35, 409–412. [Google Scholar] [CrossRef]
- Uchiyama, M.; Matsumoto, T.; Matsumoto, T.; Jimi, S.; Takamatsu, Y.; Tamura, K.; Hara, S. Simple and sensitive HPLC method for the fluorometric determination of methotrexate and its major metabolites in human plasma by post-column photochemical reaction. Biomed. Chromatogr. 2012, 26, 76–80. [Google Scholar] [CrossRef]
- Kremer, J.; Genovese, M.; Cannon, G.W.; Caldwell, J.; Cush, J.; Furst, D.E.; Luggen, M.; Keystone, E.; Bathon, J.; Kavanaugh, A.; et al. Combination leflunomide and methotrexate (MTX) therapy for patients with active rheumatoid arthritis failing MTX monotherapy: Open-label extension of a randomized, double-blind, placebo controlled trial. J. Rheumatol. 2004, 31, 1521–1531. [Google Scholar]
- Swierkot, J.; Szechiński, J. Methotrexate in rheumatoid arthritis. Pharmacol. Rep. 2006, 58, 473–492. [Google Scholar]
- Jacobs, S.A.; Stoller, R.G.; Chabner, B.A.; Johns, D.G. Dose-dependent metabolism of methotrexate in man and rhesus monkeys. Cancer Treat. Rep. 1977, 61, 651–656. [Google Scholar]
- Ortiz, Z.; Shea, B.; Suarez-Almazor, M.E.; Moher, D.; Wells, G.A.; Tugwell, P. The efficacy of folic acid and folinic acid in reducing methotrexate gastrointestinal toxicity in rheumatoid arthritis. A metaanalysis of randomized controlled trials. J. Rheumatol. 1998, 25, 36–43. [Google Scholar]
- Ortiz, Z.; Shea, B.; Suarez-Almazor, M.; Moher, D.; Wells, G.; Tugwell, P. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst. Rev. 1999. [Google Scholar] [CrossRef]
- van Ede, A.E.; Laan, R.F.; Rood, M.J.; Huizinga, T.W.; van de Laar, M.A.; van Denderen, C.J.; Westgeest, T.A.; Romme, T.C.; de Rooij, D.J.; Jacobs, M.J.; et al. Effect of folic or folinic acid supplementation on the toxicity and efficacy of methotrexate in rheumatoid arthritis: A forty-eight week, multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2001, 44, 1515–1524. [Google Scholar]
- Widemann, B.C.; Hetherington, M.L.; Murphy, R.F.; Balis, F.M.; Adamson, P.C. Carboxypeptidase-G2 rescue in a patient with high dose methotrexate-induced nephrotoxicity. Cancer 1995, 76, 521–526. [Google Scholar] [CrossRef]
- Widemann, B.C.; Balis, F.M.; Murphy, R.F.; Sorensen, J.M.; Montello, M.J.; O'Brien, M.; Adamson, P.C. Carboxypeptidase-G2, thymidine, and leucovorin rescue in cancer patients with methotrexate-induced renal dysfunction. J. Clin. Oncol. 1997, 15, 2125–2134. [Google Scholar]
- Widemann, B.C.; Balis, F.M.; Kim, A.; Boron, M.; Jayaprakash, N.; Shalabi, A.; ÓBrien, M.; Eby, M.; Cole, D.E.; Murphy, R.F.; et al. Glucarpidase, leucovorin, and thymidine for high-dose methotrexate-induced renal dysfunction: Clinical and pharmacologic factors affecting outcome. J. Clin. Oncol. 2010, 28, 3979–3986. [Google Scholar]
- Visser, K.; van der Heijde, D.M. Risk and management of liver toxicity during methotrexate treatment in rheumatoid and psoriatic arthritis: A systematic review of the literature. Clin. Exp. Rheumatol. 2009, 27, 1017–1025. [Google Scholar]
- Katchamart, W.; Bourré-Tessier, J.; Donka, T.; Drouin, J.; Rohekar, G.; Bykerk, V.P.; Haraoui, B.; Leclerq, S.; Mosher, D.P.; Pope, J.E.; et al. Canadian recommendations for use of methotrexate in patients with rheumatoid arthritis. J. Rheumatol. 2010, 37, 1422–1430. [Google Scholar] [CrossRef]
- Singh, J.A.; Furst, D.E.; Bharat, A.; Curtis, J.R.; Kavanaugh, A.F.; Kremer, J.M.; Moreland, L.W.; ÓDell, J.; Winthrop, K.L.; Beukelman, T.; et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res. 2012, 64, 625–639. [Google Scholar]
- Daikh, D.I.; St Clair, E.W. Updated recommendations for the treatment of rheumatoid arthritis: Another step on a long road. Arthritis Care Res. 2012, 64, 648–651. [Google Scholar] [CrossRef]
- Niehues, T.; Horneff, G.; Michels, H.; Höck, M.S.; Schuchmann, L. Evidence-based use of methotrexate in children with rheumatic diseases: A consensus statement of the Working Groups Pediatric Rheumatology Germany (AGKJR) and Pediatric Rheumatology Austria. Rheumatol. Int. 2005, 25, 169–178. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Breedveld, F.C.; Dougados, M.; Emery, P.; Gaujoux-Viala, C.; Gorter, S.; Knevel, R.; Nam, J.; Schoels, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann. Rheum. Dis. 2010, 69, 964–975. [Google Scholar]
- Henderson, E.S.; Adamson, R.H.; Oliverio, V.T. The metabolic fate of tritiated methotrexate. II. Absorption and excretion in man. Cancer Res. 1965, 25, 1018–1024. [Google Scholar]
- Wan, S.H.; Huffman, D.H.; Azarnoff, D.L.; Stephens, R.; Hoogstraten, B. Effect of route of administration and effusions on methotrexate pharmacokinetics. Cancer Res. 1974, 34, 3487–3491. [Google Scholar]
- Balis, F.M.; Savitch, J.L.; Bleyer, W.A. Pharmacokinetics of oral methotrexate in children. Cancer Res. 1983, 43, 2342–2345. [Google Scholar]
- Lebbe, C.; Beyeler, C.; Gerber, N.J.; Reichen, J. Intraindividual variability of the bioavailability of low dose methotrexate after oral administration in rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 475–477. [Google Scholar]
- Kurnik, D.; Loebstein, R.; Fishbein, E.; Almog, S.; Halkin, H.; Bar-Meir, S.; Chowers, Y. Bioavailability of oral vs. subcutaneous low-dose methotrexate in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2003, 18, 57–63. [Google Scholar]
- Hoekstra, M.; Haagsma, C.; Neef, C.; Proost, J.; Knuif, A.; van de Laar, M. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J. Rheumatol. 2004, 31, 645–648. [Google Scholar]
- Moshkowitz, M.; Oren, R.; Tishler, M.; Konikoff, F.M.; Graff, E.; Brill, S.; Yaron, M.; Gilat, T. The absorption of low-dose methotrexate in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 1997, 11, 569–573. [Google Scholar]
- Balis, F.M.; Holcenberg, J.S.; Poplack, D.G.; Ge, J.; Sather, H.N.; Murphy, R.F.; Ames, M.M.; Waskerwitz, M.J.; Tubergen, D.G.; Zimm, S.; et al. Pharmacokinetics and pharmacodynamics of oral methotrexate and mercaptopurine in children with lower risk acute lymphoblastic leukemia: A joint children’s cancer group and pediatric oncology branch study. Blood 1998, 92, 3569–3577. [Google Scholar]
- Herfarth, H.H.; Osterman, M.T.; Isaacs, K.L.; Lewis, J.D.; Sands, B.E. Efficacy of methotrexate in ulcerative colitis: Failure or promise. Inflamm. Bowel Dis. 2010, 16, 1421–1430. [Google Scholar]
- Christophidis, N.; Vajda, F.J.; Lucas, I.; Moon, W.J.; Louis, W.J. Comparison of intravenous and oral high-dose methotrexate in treatment of solid tumours. Br. Med. J. 1979, 1, 298–300. [Google Scholar]
- Hoekstra, M.; Haagsma, C.; Neef, C.; Proost, J.; Knuif, A.; van de Laar, M. Splitting high-dose oral methotrexate improves bioavailability: A pharmacokinetic study in patients with rheumatoid arthritis. J. Rheumatol. 2006, 33, 481–485. [Google Scholar]
- Hamilton, R.A.; Kremer, J.M. The effects of food on methotrexate absorption. J. Rheumatol. 1995, 22, 630–632. [Google Scholar]
- Murry, D.J.; Synold, T.W.; Pui, C.H.; Rodman, J.H. Renal function and methotrexate clearance in children with newly diagnosed leukemia. Pharmacotherapy 1995, 15, 144–149. [Google Scholar]
- Zhao, R.; Assaraf, Y.G.; Goldman, I.D. A reduced folate carrier mutation produces substrate-dependent alterations in carrier mobility in murine leukemia cells and methotrexate resistance with conservation of growth in 5-formyltetrahydrofolate. J. Biol. Chem. 1998, 273, 7873–7879. [Google Scholar]
- Nozaki, Y.; Kusuhara, H.; Endou, H.; Sugiyama, Y. Quantitative evaluation of the drug-drug interactions between methotrexate and nonsteroidal anti-inflammatory drugs in the renal uptake process based on the contribution of organic anion transporters and reduced folate carrier. J. Pharmacol. Exp. Ther. 2004, 309, 226–234. [Google Scholar] [CrossRef]
- Badagnani, I.; Castro, R.A.; Taylor, T.R.; Brett, C.M.; Huang, C.C.; Stryke, D.; Kawamoto, M.; Johns, S.J.; Ferrin, T.E.; Carlson, E.J.; Burchard, E.G.; Giacomini, K.M. Interaction of methotrexate with organic-anion transporting polypeptide 1A2 and its genetic variants. J. Pharmacol. Exp. Ther. 2006, 318, 521–529. [Google Scholar] [CrossRef]
- Shibayama, Y.; Ushinohama, K.; Ikeda, R.; Yoshikawa, Y.; Motoya, T.; Takeda, Y.; Yamada, K. Effect of methotrexate treatment on expression levels of multidrug resistance protein 2, breast cancer resistance protein and organic anion transporters Oat1, Oat2 and Oat3 in rats. Cancer Sci. 2006, 97, 1260–1266. [Google Scholar] [CrossRef]
- Inoue, K.; Nakai, Y.; Ueda, S.; Kamigaso, S.; Ohta, K.Y.; Hatakeyama, M.; Hayashi, Y.; Otagiri, M.; Yuasa, H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G660–G668. [Google Scholar] [CrossRef]
- VanWert, A.L.; Sweet, D.H. Impaired clearance of methotrexate in organic anion transporter 3 (Slc22a8) knockout mice: A gender specific impact of reduced folates. Pharm. Res. 2008, 25, 453–462. [Google Scholar] [CrossRef]
- Urquhart, B.L.; Gregor, J.C.; Chande, N.; Knauer, M.J.; Tirona, R.G.; Kim, R.B. The human proton-coupled folate transporter (hPCFT): Modulation of intestinal expression and function by drugs. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, 248–254. [Google Scholar] [CrossRef]
- Subramanian, V.S.; Marchant, J.S.; Said, H.M. Apical membrane targeting and trafficking of the human proton-coupled transporter in polarized epithelia. Am. J. Physiol. Cell Physiol. 2008, 294, C233–C240. [Google Scholar] [CrossRef]
- Collins, J.F. Novel insights into intestinal and renal folate transport. Focus on “Apical membrane targeting and trafficking of the human proton-coupled folate transporter in polarized epithelia”. Am. J. Physiol. Cell Physiol. 2008, 294, C381–C382. [Google Scholar]
- Yuasa, H.; Inoue, K.; Hayashi, Y. Molecular and functional characteristics of proton-coupled folate transporter. J. Pharm. Sci. 2009, 98, 1608–1616. [Google Scholar]
- Wang, Y.; Zhao, R.; Russell, R.G.; Goldman, I.D. Localization of the murine reduced folate carrier as assessed by immunohistochemical analysis. Biochim. Biophys. Acta 2001, 1513, 49–54. [Google Scholar] [CrossRef]
- Balamurugan, K.; Said, H.M. Ontogenic regulation of folate transport across rat jejunal brush-border membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G1068–G1073. [Google Scholar]
- Chiao, J.H.; Roy, K.; Tolner, B.; Yang, C.H.; Sirotnak, F.M. RFC-1 gene expression regulates folate absorption in mouse small intestine. J. Biol. Chem. 1997, 272, 11165–11170. [Google Scholar]
- Wang, Y.; Zhao, R.; Goldman, I.D. Characterization of a folate transporter in HeLa cells with a low pH optimum and high affinity for pemetrexed distinct from the reduced folate carrier. Clin. Cancer Res. 2004, 10, 6256–6264. [Google Scholar] [CrossRef]
- Wang, Y.; Rajgopal, A.; Goldman, I.D.; Zhao, R. Preservation of folate transport activity with a low-pH optimum in rat IEC-6 intestinal epithelial cell lines that lack reduced folate carrier function. Am. J. Physiol. Cell Physiol. 2005, 288, C65–C71. [Google Scholar]
- Dudeja, P.K.; Kode, A.; Alnounou, M.; Tyagi, S.; Torania, S.; Subramanian, V.S.; Said, H.M. Mechanism of folate transport across the human colonic basolateral membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G54–G60. [Google Scholar]
- Marchant, J.S.; Subramanian, V.S.; Parker, I.; Said, H.M. Intracellular trafficking and membrane targeting mechanisms of the human reduced folate carrier in Mammalian epithelial cells. J. Biol. Chem. 2002, 277, 33325–33333. [Google Scholar]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar]
- Nakai, Y.; Inoue, K.; Abe, N.; Hatakeyama, M.; Ohta, K.Y.; Otagiri, M.; Hayashi, Y.; Yuasa, H. Functional characterization of human proton-coupled folate transporter/heme carrier protein 1 heterologously expressed in mammalian cells as a folate transporter. J. Pharmacol. Exp. Ther. 2007, 322, 469–476. [Google Scholar] [CrossRef]
- Yokooji, T.; Mori, N.; Murakami, T. Site-Specific contribution of proton-coupled folate transporter/haem carrier protein 1 in the intestinal absorption of methotrexate in rats. J. Pharm. Pharmacol. 2009, 61, 911–918. [Google Scholar]
- Kitamura, Y.; Hirouchi, M.; Kusuhara, H.; Schuetz, J.D.; Sugiyama, Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3). J. Pharmacol. Exp. Ther. 2008, 327, 465–473. [Google Scholar] [CrossRef]
- Strum, W.B. A pH-dependent, carrier-mediated transport system for the folate analog, amethopterin, in rat jejunum. J. Pharmacol. Exp. Ther. 1977, 203, 640–645. [Google Scholar]
- Selhub, J.; Rosenberg, I.H. Folate transport in isolated brush border membrane vesicles from rat intestine. J. Biol. Chem. 1981, 256, 4489–4493. [Google Scholar]
- Masuda, M.; Íizuka, Y.; Yamazaki, M.; Nishigaki, R.; Kato, Y.; Níinuma, K.; Suzuki, H.; Sugiyama, Y. Methotrexate is excreted into the bile by canalicular multispecific organic anion transporter in rats. Cancer Res. 1997, 57, 3506–3510. [Google Scholar]
- Hirohashi, T.; Suzuki, H.; Sugiyama, Y. Characterization of the transport properties of cloned rat multidrug resistance-associated protein 3 (MRP3). J. Biol. Chem. 1999, 274, 15181–15185. [Google Scholar]
- Breedveld, P.; Pluim, D.; Cipriani, G.; Dahlhaus, F.; van Eijndhoven, M.A.; de Wolf, C.J.; Kuil, A.; Beijnen, J.H.; Scheffer, G.L.; Jansen, G.; et al. The effect of low pH on breast cancer resistance protein (ABCG2)-mediated transport of methotrexate, 7-hydroxymethotrexate, methotrexate diglutamate, folic acid, mitoxantrone, topotecan, and resveratrol in in vitro drug transport models. Mol. Pharmacol. 2007, 71, 240–249. [Google Scholar]
- Takano, M.; Yumoto, R.; Murakami, T. Expression and function of efflux drug transporters in the intestine. Pharmacol. Ther. 2006, 109, 137–161. [Google Scholar] [CrossRef]
- Murakami, T.; Takano, M. Intestinal efflux transporters and drug absorption. Expert Opin. Drug Metab. Toxicol. 2008, 4, 923–939. [Google Scholar] [CrossRef]
- Suzuki, H.; Sugiyama, Y. Role of metabolic enzymes and efflux transporters in the absorption of drugs from the small intestine. Eur. J. Pharm. Sci. 2000, 12, 3–12. [Google Scholar] [CrossRef]
- Tanaka, Y.; Slitt, A.L.; Leazer, T.M.; Maher, J.M.; Klaassen, C.D. Tissue distribution and hormonal regulation of the breast cancer resistance protein (Bcrp/Abcg2) in rats and mice. Biochem. Biophys. Res. Commun. 2005, 326, 181–187. [Google Scholar]
- Prime-Chapman, H.M.; Fearn, R.A.; Cooper, A.E.; Moore, V.; Hirst, BH. Differential multidrug resistance-associated protein 1 through 6 isoform expression and function in human intestinal epithelial Caco-2 cells. J. Pharmacol. Exp. Ther. 2004, 311, 476–484. [Google Scholar] [CrossRef]
- Zimmermann, C.; Gutmann, H.; Hruz, P.; Gutzwiller, J.P.; Beglinger, C.; Drewe, J. Mapping of multidrug resistance gene 1 and multidrug resistance-associated protein isoform 1 to 5 mRNA expression along the human intestinal tract. Drug Metab. Dispos. 2005, 33, 219–224. [Google Scholar]
- Johnson, B.M.; Zhang, P.; Schuetz, J.D.; Brouwer, K.L. Characterization of transport protein expression in multidrug resistance-associated protein (Mrp) 2-deficient rats. Drug Metab. Dispos. 2006, 34, 556–562. [Google Scholar] [CrossRef]
- Maher, J.M.; Slitt, A.L.; Callaghan, T.N.; Cheng, X.; Cheung, C.; Gonzalez, F.J.; Klaassen, C.D. Alterations in transporter expression in liver, kidney, and duodenum after targeted disruption of the transcription factor HNF1alph. Biochem. Pharmacol. 2006, 72, 512–522. [Google Scholar]
- Mottino, A.D.; Hoffman, T.; Jennes, L.; Cao, J.; Vore, M. Expression of multidrug resistance-associated protein 2 in small intestine from pregnant and postpartum rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1261–G1273. [Google Scholar]
- Rost, D.; Mahner, S.; Sugiyama, Y.; Stremmel, W. Expression and localization of the multidrug resistance-associated protein 3 in rat small and large intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G720–726. [Google Scholar]
- Yokooji, T.; Murakami, T.; Yumoto, R.; Nagai, J.; Takano, M. Site-specific bidirectional efflux of 2,4-dinitrophenyl-S-glutathione, a substrate of multidrug resistance-associated proteins, in rat intestine and Caco-2 cells. J. Pharm. Pharmacol. 2007, 59, 513–520. [Google Scholar]
- Yokooji, T.; Murakami, T.; Yumoto, R.; Nagai, J.; Takano, M. Role of intestinal efflux transporters in the intestinal absorption of methotrexate in rats. J. Pharm. Pharmacol. 2007, 59, 1263–1270. [Google Scholar]
- Evans, D.F.; Pye, G.; Bramley, R.; Clark, A.G.; Dyson, T.J.; Hardcastle, J.D. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef]
- Murakami, T.; Yokooji, T.; Mori, N. Study on absorption sites of quinidine and methotrexate in rat intestine. Pharmazie 2010, 65, 440–447. [Google Scholar]
- Levy, G.; Jusko, W.J. Factors affecting the absorption of riboflavin in man. J. Pharm. Sci. 1966, 55, 285–289. [Google Scholar] [CrossRef]
- Hewitt, R.R.; Levy, G. Effect of viscosity on thiamine and riboflavin absorption in man. J. Pharm. Sci. 1971, 60, 784–786. [Google Scholar] [CrossRef]
- Hamdani, J.; Goole, J.; Moës, A.J.; Amighi, K. In vitro and in vivo evaluation of floating riboflavin pellets developed using the melt pelletization process. Int. J. Pharm. 2006, 323, 86–92. [Google Scholar] [CrossRef]
- Kagan, L.; Lapidot, N.; Afargan, M.; Kirmayer, D.; Moor, E.; Mardor, Y.; Friedman, M.; Hoffman, A. Gastroretentive Accordion Pill: Enhancement of riboflavin bioavailability in humans. J. Control Release. 2006, 113, 208–215. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Ayres, J.W. Bioavailability of riboflavin from a gastric retention formulation. Int. J. Pharm. 2007, 330, 146–154. [Google Scholar] [CrossRef]
- Sklan, D.; Trostler, N. Site and extent of thiamin absorption in the rat. J. Nutr. 1977, 107, 353–356. [Google Scholar]
- McClintock, C.; Shiau, Y.F. Jejunum is more important than terminal ileum for taurocholate absorption in rats. Am. J. Physiol. 1983, 244, G507–G514. [Google Scholar]
- Nakashima, T.; Seto, Y.; Nakajima, T.; Shima, T.; Sakamoto, Y.; Sano, A.; Takino, T. Distribution of tissue bile acids in the human alimentary tract and colon polyps. Jpn. J. Med. 1989, 28, 25–29. [Google Scholar] [CrossRef]
- Kramer, W.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Jouvenal, K.; Müller, G.; Tripier, D.; Wess, G. Intestinal bile acid absorption. Na(+)-dependent bile acid transport activity in rabbit small intestine correlates with the coexpression of an integral 93-kDa and a peripheral 14-kDa bile acid-binding membrane protein along the duodenum-ileum axis. J. Biol. Chem. 1993, 268, 18035–18046. [Google Scholar]
- Breedveld, P.; Zelcer, N.; Pluim, D.; Sönmezer, O.; Tibben, M.M.; Beijnen, J.H.; Schinkel, A.H.; van Tellingen, O.; Borst, P.; Schellens, J.H. Mechanism of the pharmacokinetic interaction between methotrexate and benzimidazoles: Potential role for breast cancer resistance protein in clinical drug-drug interactions. Cancer Res. 2004, 64, 5804–5811. [Google Scholar] [CrossRef]
- Jansen, G.; van der Heijden, J.; Oerlemans, R.; Lems, W.F.; Ifergan, I.; Scheper, R.J.; Assaraf, Y.G.; Dijkmans, B.A. Sulfasalazine is a potent inhibitor of the reduced folate carrier: Implications for combination therapies with methotrexate in rheumatoid arthritis. Arthritis Rheum. 2004, 50, 2130–2139. [Google Scholar]
- Pratt, CB.; Roberts, D.; Shanks, E.C.; Warmath, E.L. Clinical trials and pharmacokinetics of intermittent high-dose methotrexate-“leucovorin rescue” for children with malignant tumors. Cancer Res. 1974, 34, 3326–3331. [Google Scholar]
- Wall, A.M.; Gajjar, A.; Link, A.; Mahmoud, H.; Pui, C.H.; Relling, M.V. Individualized methotrexate dosing in children with relapsed acute lymphoblastic leukemia. Leukemia 2000, 14, 221–225. [Google Scholar] [CrossRef]
- Schrøder, H.; Jensen, K.B.; Brandsborg, M. Lack of correlation between methotrexate concentrations in serum, saliva and sweat after 24 h methotrexate infusions. Br. J. Clin. Pharmacol. 1987, 24, 537–541. [Google Scholar] [CrossRef]
- Egan, L.J.; Sandborn, W.J.; Tremaine, W.J.; Leighton, J.A.; Mays, D.C.; Pike, M.G.; Zinsmeister, A.R.; Lipsky, J.J. A randomized dose-response and pharmacokinetic study of methotrexate for refractory inflammatory Crohn's disease and ulcerative colitis. Aliment. Pharmacol. Ther. 1999, 13, 1597–1604. [Google Scholar] [CrossRef]
- Joerger, M.; Huitema, A.D.; van den Bongard, H.J.; Baas, P.; Schornagel, J.H.; Schellens, J.H.M.; Beijnen, J.H. Determinants of the elimination of methotrexate and 7-hydroxy-methotrexate following high-dose infusional therapy to cancer patients. Br. J. Clin. Pharmacol. 2006, 62, 71–80. [Google Scholar] [CrossRef]
- Yokooji, T.; Mori, N.; Murakami, T. Modulated pharmacokinetics and increased small intestinal toxicity of methotrexate in bilirubin-treated rats. J. Pharm. Pharmacol. 2011, 63, 206–213. [Google Scholar]
- van de Steeg, E.; van der Kruijssen, C.M.; Wagenaar, E.; Burggraaff, J.E.; Mesman, E.; Kenworthy, K.E.; Schinkel, A.H. Methotrexate pharmacokinetics in transgenic mice with liver-specific expression of human organic anion-transporting polypeptide 1B1 (SLCO1B1). Drug Metab. Dispos. 2009, 37, 277–281. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Slørdal, L.; Wist, E.; Aarbakke, J. Dose-dependent pharmacokinetics of methotrexate and 7-hydroxymethotrexate in the rat in vivo. Cancer Res. 1989, 49, 6359–6364. [Google Scholar]
- Moriyasu, A.; Sugihara, K.; Nakatani, K.; Ohta, S.; Kitamura, S. In vivo-in vitro relationship of methotrexate 7-hydroxylation by aldehyde oxidase in four different strain rats. Drug Metab. Pharmacok. 2006, 21, 485–491. [Google Scholar] [CrossRef]
- Hosoyamada, M.; Sekine, T.; Kanai, Y.; Endou, H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am. J. Physiol. 1999, 276, F122–F128. [Google Scholar]
- Cha, S.H.; Sekine, T.; Fukushima, J.I.; Kanai, Y.; Kobayashi, Y.; Goya, T.; Endou, H. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol. Pharmacol. 2001, 59, 1277–1286. [Google Scholar]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302, 666–671. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Hamilton, K.F.; Seth-Smith, M.L.; Cass, C.E.; Sawyer, M.B. Characterization of binding of folates and antifolates to brush-border membrane vesicles isolated from human kidney. Mol. Pharmacol. 2005, 67, 453–459. [Google Scholar]
- Tracy, T.S.; Worster, T.; Bradley, J.D.; Greene, P.K.; Brater, D.C. Methotrexate disposition following concomitant administration of ketoprofen, piroxicam and flurbiprofen in patients with rheumatoid arthritis. Br. J. Clin. Pharmacol. 1994, 37, 453–456. [Google Scholar] [CrossRef]
- El-Sheikh, A.A.; van den Heuvel, J.J.; Koenderink, J.B.; Russel, F.G. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J. Pharmacol. Exp. Ther. 2007, 320, 229–235. [Google Scholar]
- Shibayama, Y.; Takeda, Y.; Yamada, K. Effect of methotrexate treatment on expression levels of organic anion transporter polypeptide 2, P-glycoprotein and bile salt export pump in rats. Biol. Pharm. Bull. 2009, 32, 493–496. [Google Scholar] [CrossRef]
- Zhao, R.; Qiu, A.; Tsai, E.; Jansen, M.; Akabas, M.H.; Goldman, I.D. The proton-coupled folate transporter: Impact on pemetrexed transport and on antifolates activities compared with the reduced folate carrier. Mol. Pharmacol. 2008, 74, 854–862. [Google Scholar] [CrossRef]
- Smith, S.B.; Kekuda, R.; Gu, X.; Chancy, C.; Conway, S.J.; Ganapathy, V. Expression of folate receptor alpha in the mammalian retinol pigmented epithelium and retina. Invest. Ophthalmol. Vis. Sci. 1999, 40, 840–848. [Google Scholar]
- Spinella, M.J.; Brigle, K.E.; Sierra, E.E.; Goldman, I.D. Distinguishing between folate receptor-alpha-mediated transport and reduced folate carrier-mediated transport in L1210 leukemia cells. J. Biol. Chem. 1995, 270, 7842–7849. [Google Scholar] [CrossRef]
- Henderson, G.B.; Strauss, B.P. Characteristics of a novel transport system for folate compounds in wild-type and methotrexate-resistant L1210 cells. Cancer Res. 1990, 50, 1709–1714. [Google Scholar]
- Deng, Y.; Zhou, X.; Kugel Desmoulin, S.; Wu, J.; Cherian, C.; Hou, Z.; Matherly, L.H.; Gangjee, A. Synthesis and biological activity of a novel series of 6-substituted thieno[2,3-d]pyrimidine antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors over the reduced folate carrier and proton-coupled folate transporter for cellular entry. J. Med. Chem. 2009, 52, 2940–2951. [Google Scholar] [CrossRef]
- Hinken, M.; Halwachs, S.; Kneuer, C.; Honscha, W. Subcellular localization and distribution of the reduced folate carrier in normal rat tissues. Eur. J. Histochem. 2011, 55. [Google Scholar] [CrossRef]
- Hosoya, K.; Fujita, K.; Tachikawa, M. Involvement of reduced folate carrier 1 in the inner blood-retinal barrier transport of methyltetrahydrofolate. Drug Metab. Pharmacok. 2008, 23, 285–292. [Google Scholar] [CrossRef]
- Samuels, L.L.; Feinberg, A.; Moccio, D.M.; Sirotnak, F.M.; Rosen, G. Detection by high-performance liquid chromatography of methotrexate and its metabolites in tumor tissue from osteosarcoma patients treated with high-dose methotrexate/leucovorin rescue. Biochem. Pharmacol. 1984, 33, 2711–2714. [Google Scholar] [CrossRef]
- Koizumi, S.; Curt, G.A.; Fine, R.L.; Griffin, J.D.; Chabner, B.A. Formation of methotrexate polyglutamates in purified myeloid precursor cells from normal human bone marrow. J. Clin. Invest. 1985, 75, 1008–1014. [Google Scholar] [CrossRef]
- Fabre, I.; Fabre, G.; Goldman, I.D. Polyglutamylation, an important element in methotrexate cytotoxicity and selectivity in tumor versus murine granulocytic progenitor cells in vitro. Cancer Res. 1984, 44, 3190–3195. [Google Scholar]
- Jolivet, J.; Chabner, B.A. Intracellular pharmacokinetics of methotrexate polyglutamates in human breast cancer cells. Selective retention and less dissociable binding of 4-NH2-10-CH3- pteroylglutamate4 and 4-NH2-10-CH3-pteroylglutamate5 to dihydrofolate reductase. J. Clin. Invest. 1983, 72, 773–778. [Google Scholar]
- Synold, T.W.; Relling, M.V.; Boyett, J.M.; Rivera, G.K.; Sandlund, J.T.; Mahmoud, H.; Crist, W.M.; Pui, C.H.; Evans, W.E. Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J. Clin. Invest. 1994, 94, 1996–2001. [Google Scholar] [CrossRef]
- Masson, E.; Relling, M.V.; Synold, T.W.; Liu, Q.; Schuetz, J.D.; Sandlund, J.T.; Pui, C.H.; Evans, W.E. Accumulation of methotrexate polyglutamates in lymphoblasts is a determinant of antileukemic effects in vivo. A rationale for high-dose methotrexate. J. Clin. Invest. 1996, 97, 73–80. [Google Scholar]
- Dalrymple, J.M.; Stamp, L.K.; O'Donnell, J.L.; Chapman, P.T.; Zhang, M.; Barclay, M.L. Pharmacokinetics of oral methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2008, 58, 3299–3308. [Google Scholar] [CrossRef]
- Stamp, L.K.; O'Donnell, J.L.; Chapman, P.T.; Zhang, M.; Frampton, C.; James, J.; Barclay, M.L. Determinants of red blood cell methotrexate polyglutamate concentrations in rheumatoid arthritis patients receiving long-term methotrexate treatment. Arthritis Rheum. 2009, 60, 2248–2256. [Google Scholar]
- Stamp, L.K; O'Donnell, J.L.; Chapman, P.T.; Zhang, M.; James, J.; Frampton, C.; Barclay, M.L. Methotrexate polyglutamate concentrations are not associated with disease control in rheumatoid arthritis patients receiving long-term methotrexate therapy. Arthritis Rheum. 2010, 62, 359–368. [Google Scholar]
- Dervieux, T.; Zablocki, R.; Kremer, J. Red blood cell methotrexate polyglutamates emerge as a function of dosage intensity and route of administration during pulse methotrexate therapy in rheumatoid arthritis. Rheumatology (Oxford) 2010, 49, 2337–2345. [Google Scholar]
- Becker, M.L.; van Haandel, L.; Gaedigk, R.; Lasky, A.; Hoeltzel, M.; Stobaugh, J.; Leeder, J.S. Analysis of intracellular methotrexate polyglutamates in patients with juvenile idiopathic arthritis: Effect of route of administration on variability in intracellular methotrexate polyglutamate concentrations. Arthritis Rheum. 2010, 62, 1803–1812. [Google Scholar]
- Becker, M.L.; Gaedigk, R.; van Haandel, L.; Thomas, B.; Lasky, A.; Hoeltzel, M.; Dai, H.; Stobaugh, J.; Leeder, J.S. The effect of genotype on methotrexate polyglutamate variability in juvenile idiopathic arthritis and association with drug response. Arthritis Rheum. 2011, 63, 276–285. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, Z.S.; Belinsky, M.G.; Rea, P.A.; Kruh, G.D. Transport of methotrexate (MTX) and folates by multidrug resistance protein (MRP) 3 and MRP1: Effect of polyglutamylation on MTX transport. Cancer Res. 2001, 61, 7225–7232. [Google Scholar]
- Chen, Z.S.; Lee, K.; Walther, S.; Raftogianis, R.B.; Kuwano, M.; Zeng, H.; Kruh, G.D. Analysis of methotrexate and folate transport by multidrug resistance protein 4 (ABCC4): MRP4 is a component of the methotrexate efflux system. Cancer Res. 2002, 62, 3144–3150. [Google Scholar]
- Wielinga, P.R.; van der Heijden, I.; Reid, G.; Beijnen, J.H.; Wijnholds, J.; Borst, P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J. Biol. Chem. 2003, 278, 17664–17671. [Google Scholar]
- Wielinga, P.; Hooijberg, J.H.; Gunnarsdottir, S.; Kathmann, I.; Reid, G.; Zelcer, N.; van der Born, K.; de Haas, M.; van der Heijden, I.; Kaspers, G.; Wijnholds, J.; Jansen, G.; Peters, G.; Borst, P. The human multidrug resistance protein MRP5 transports folates and can mediate cellular resistance against antifolates. Cancer Res. 2005, 65, 4425–4430. [Google Scholar]
- Volk, E.L.; Farley, K.M.; Wu, Y.; Li, F.; Robey, R.W.; Schneider, E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002, 62, 5035–5040. [Google Scholar]
- Ifergan, I.; Shafran, A.; Jansen, G.; Hooijberg, J.H.; Scheffer, G.L.; Assaraf, Y.G. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression. A role for BCRP in cellular folate homeostasis. J. Biol. Chem. 2004, 279, 25527–25534. [Google Scholar]
- Chen, Z.S.; Robey, R.W.; Belinsky, M.G.; Shchaveleva, I.; Ren, X.Q.; Sugimoto, Y.; Ross, D.D.; Bates, S.E.; Kruh, G.D. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: Effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003, 63, 4048–4054. [Google Scholar]
- Volk, E.L.; Schneider, E. Wild-type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res. 2003, 63, 5538–5543. [Google Scholar]
- Rhee, M.S.; Schneider, E. Lack of an effect of breast cancer resistance protein (BCRP/ABCG2) overexpression on methotrexate polyglutamate export and folate accumulation in a human breast cancer cell line. Biochem. Pharmacol. 2005, 69, 123–132. [Google Scholar]
- Sharp, L.; Little, J. Polymorphisms in genes involved in folate metabolism and colorectal neoplasia: A HuGE review. Am. J. Epidemiol. 2004, 159, 423–443. [Google Scholar] [CrossRef]
- Gemmati, D.; Ongaro, A.; Scapoli, G.L.; Della Porta, M.; Tognazzo, S.; Serino, M.L.; Di Bona, E.; Rodeghiero, F.; Gilli, G.; Reverberi, R.; et al. Common gene polymorphisms in the metabolic folate and methylation pathway and the risk of acute lymphoblastic leukemia and non-Hodgkin's lymphoma in adults. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 787–794. [Google Scholar]
- de Jonge, R.; Hooijberg, J.H.; van Zelst, B.D.; Jansen, G.; van Zantwijk, C.H.; Kaspers, G.J.; Peters, G.J.; Ravindranath, Y.; Pieters, R.; Lindemans, J. Effect of polymorphisms in folate-related genes on in vitro methotrexate sensitivity in pediatric acute lymphoblastic leukemia. Blood 2005, 106, 717–720. [Google Scholar]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef]
- Treviño, L.R.; Shimasaki, N.; Yang, W.; Panetta, J.C.; Cheng, C.; Pei, D.; Chan, D.; Sparreboom, A.; Giacomini, K.M.; Pui, C.H.; Evans, W.E.; Relling, M.V. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J. Clin. Oncol. 2009, 27, 5972–5978. [Google Scholar]
- Lopez-Lopez, E.; Martin-Guerrero, I.; Ballesteros, J.; Piñan, M.A.; Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Polymorphisms of the SLCO1B1 gene predict methotrexate-related toxicity in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer. 2011, 57, 612–619. [Google Scholar] [CrossRef]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Treviño, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012, 22, 1–8. [Google Scholar]
- Hon, Y.Y.; Evans, W.E. Making TDM work to optimize cancer chemotherapy: A multidisciplinary team approach. Clin. Chem. 1998, 44, 388–400. [Google Scholar]
- Lennard, L. Therapeutic drug monitoring of cytotoxic drugs. Br. J. Clin. Pharmacol. 2001, 52, 75S–87S. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Murakami, T.; Mori, N. Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics. Pharmaceuticals 2012, 5, 802-836. https://doi.org/10.3390/ph5080802
Murakami T, Mori N. Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics. Pharmaceuticals. 2012; 5(8):802-836. https://doi.org/10.3390/ph5080802
Chicago/Turabian StyleMurakami, Teruo, and Nobuhiro Mori. 2012. "Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics" Pharmaceuticals 5, no. 8: 802-836. https://doi.org/10.3390/ph5080802
APA StyleMurakami, T., & Mori, N. (2012). Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics. Pharmaceuticals, 5(8), 802-836. https://doi.org/10.3390/ph5080802