Epigenetic Mechanisms and Therapeutic Perspectives for Neurodevelopmental Disorders

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
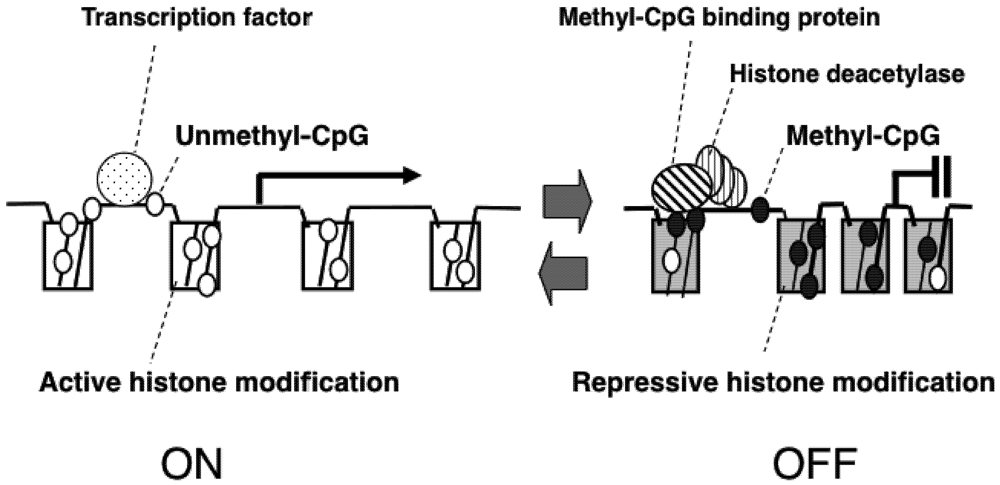
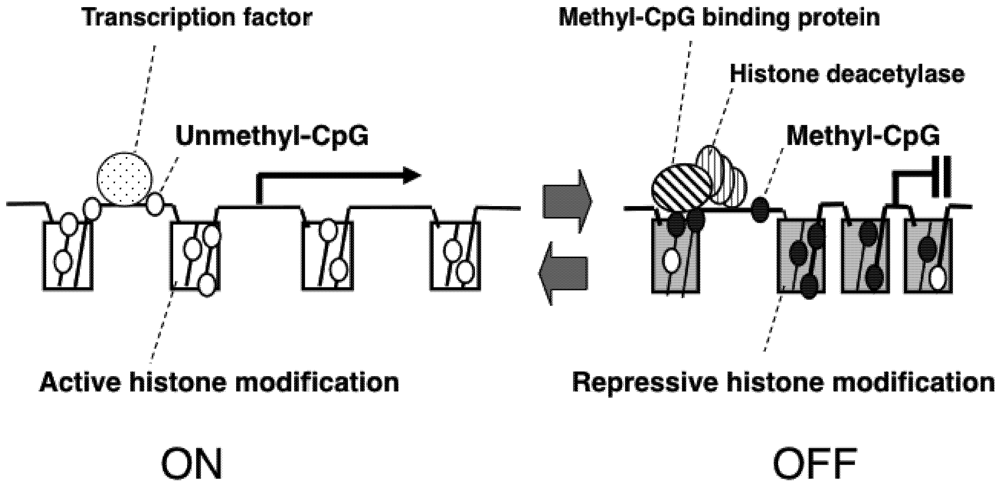
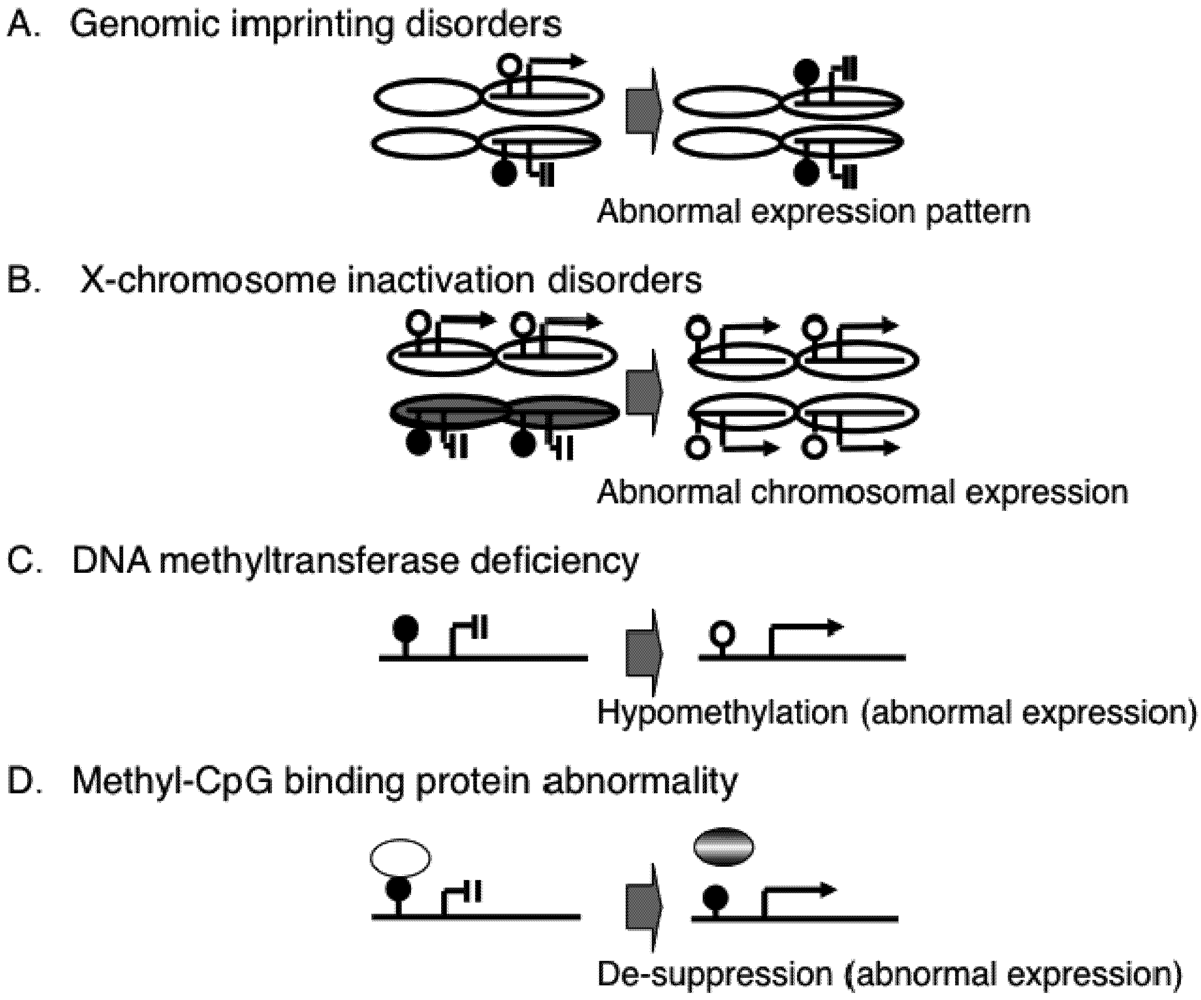
2. Epigenetic Mechanisms of Congenital Neurodevelopmental Disorders
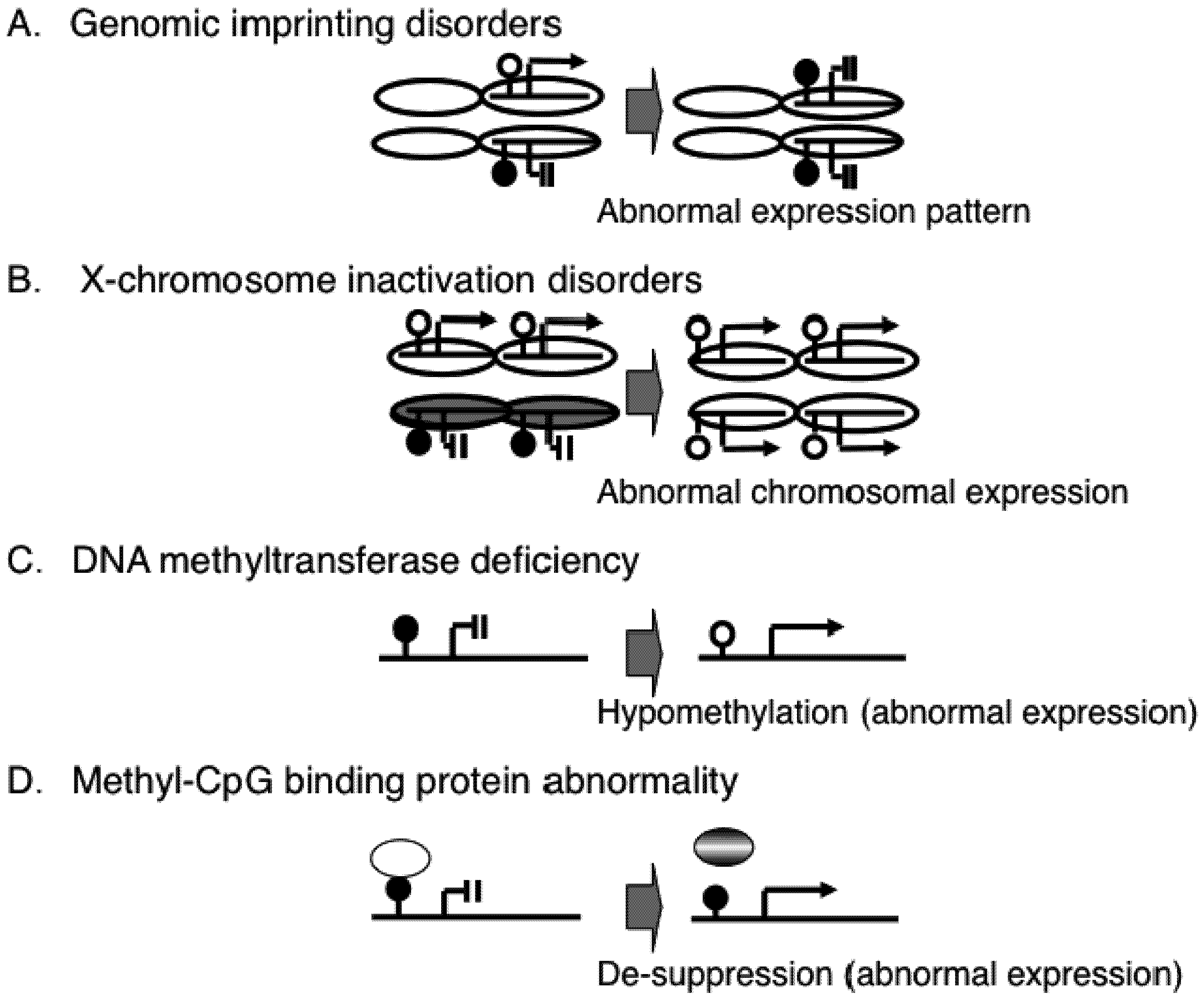
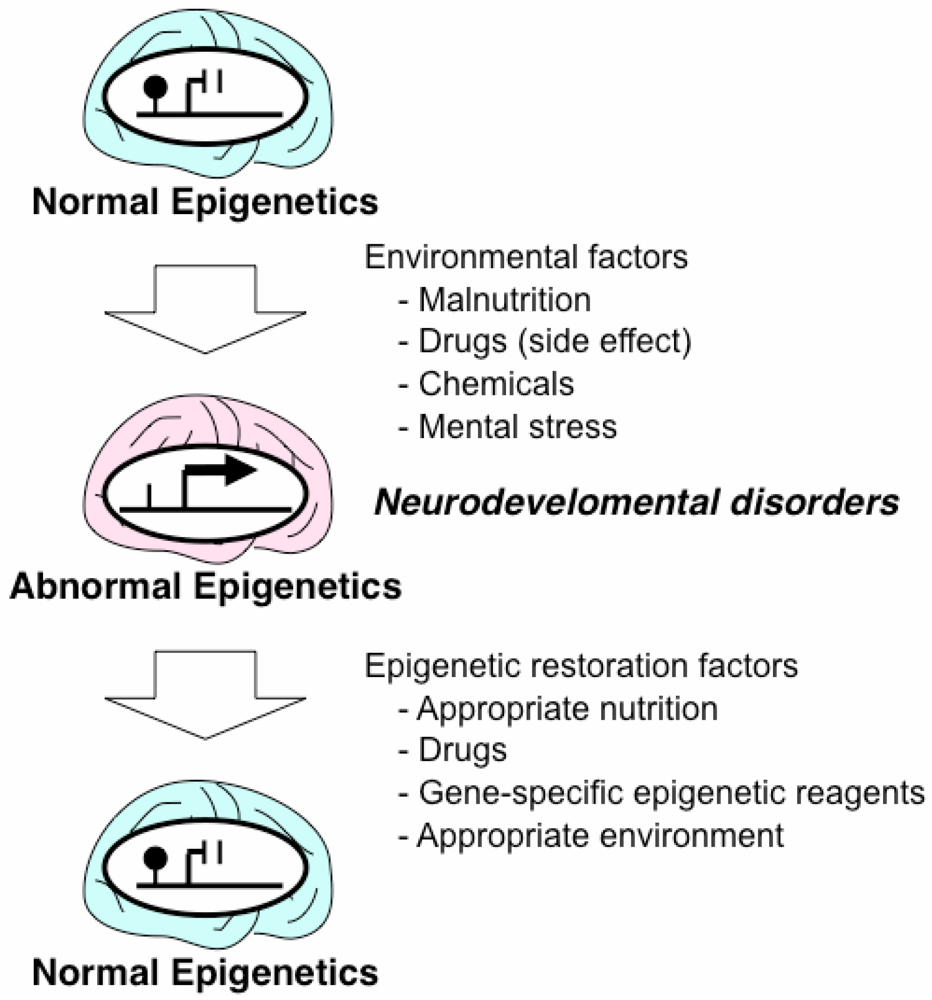
3. Epigenetic Mechanisms of Acquired Neurodevelopmental Disorders
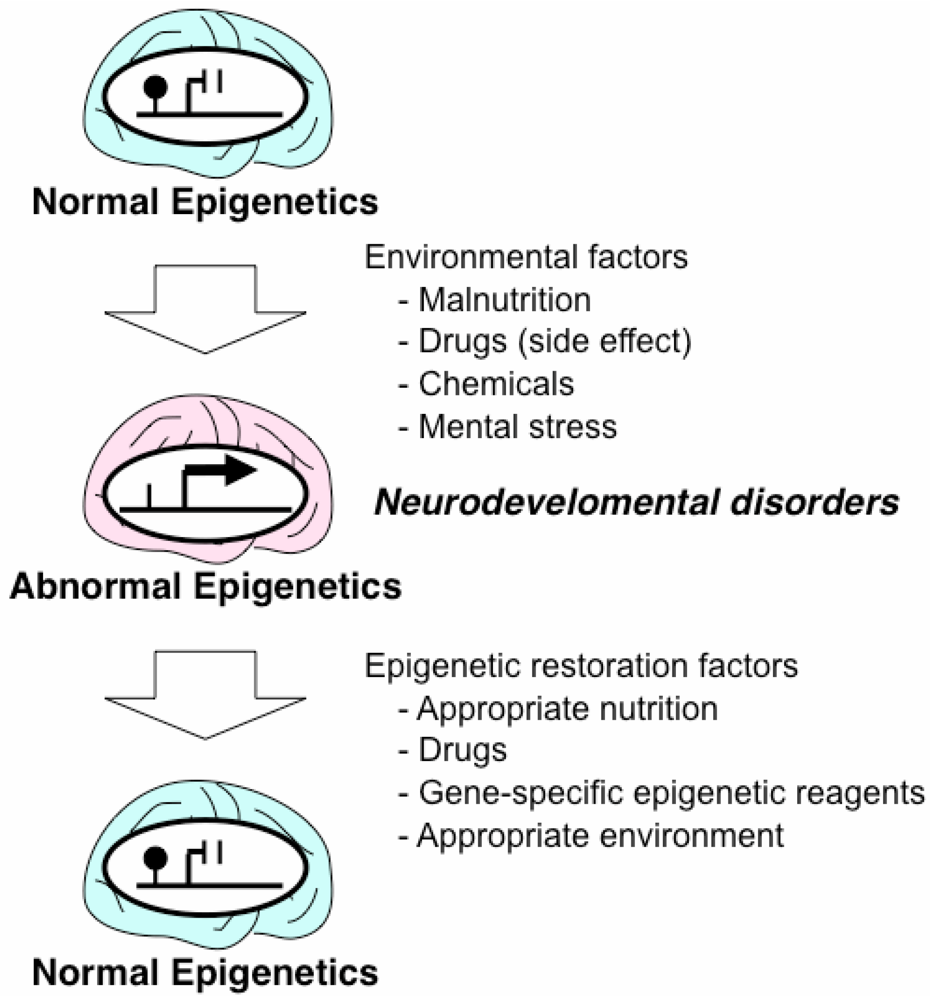
4. Drugs and Nutrition with Epigenetic Effects
5. Epigenetic Therapeutic Strategies for Neurodevelopmental Disorders
6. Conclusions
Acknowledgments
Conflict of Interest
References
- McNairn, A.J.; Gilbert, D.M. Epigenomic replication: Linking epigenetics to DNA replication. Bioessays 2003, 25, 647–656. [Google Scholar]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988, 203, 971–983. [Google Scholar] [CrossRef]
- Waddington, C.H. Epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar]
- Inoue, K.; Kanai, M.; Tanabe, Y.; Kubota, T.; Kashork, C.D.; Wakui, K.; Fukushima, Y.; Lupski, J.R.; Shaffer, L.G. Prenatal interphase FISH diagnosis of PLP1 duplication associated with Pelizaeus-Merzbacher disease. Prenat. Diagn. 2001, 21, 1133–1136. [Google Scholar]
- Reiner, O.; Carrozzo, R.; Shen, Y.; Wehnert, M.; Faustinella, F.; Dobyns, W.B.; Caskey, C.T.; Ledbetter, D.H. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 1993, 364, 717–721. [Google Scholar]
- Bi, W.; Sapir, T.; Shchelochkov, O.A.; Zhang, F.; Withers, M.A.; Hunter, J.V. ; Levy, T.; Shinder, V.; Peiffer, D.A.; Gunderson, K.L.; et al. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009, 41, 168–177. [Google Scholar]
- Roa, B.B.; Lupski, J.R. Molecular basis of Charcot-Marie-Tooth disease type 1A: Gene dosage as a novel mechanism for a common autosomal dominant condition. Am. J. Med. Sci. 1993, 306, 177–184. [Google Scholar]
- Obi, T.; Nishioka, K.; Ross, O.A.; Terada, T.; Yamazaki, K.; Sugiura, A.; Takanashi, M.; Mizoguchi, K.; Mori, H.; Mizuno, Y.; et al. Clinicopathologic study of a SNCA gene duplication patient with Parkinson disease and dementia. Neurology 2008, 70, 238–241. [Google Scholar]
- Kubota, T.; Das, S.; Christian, S.L.; Baylin, S.B.; Herman, J.G.; Ledbetter, D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997, 16, 16–17. [Google Scholar]
- Kubota, T.; Nonoyama, S.; Tonoki, H.; Masuno, M.; Imaizumi, K.; Kojima, M.; Wakui, K.; Shimadzu, M.; Fukushima, Y. A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum. Genet. 1999, 104, 49–55. [Google Scholar]
- Xue, F.; Tian, X.C.; Du, F.; Kubota, C.; Taneja, M.; Dinnyes, A.; Dai, Y.; Levine, H.; Pereira, L.V.; Yang, X. Aberrant patterns of X chromosome inactivation in bovine clones. Nat. Genet. 2002, 31, 216–220. [Google Scholar]
- Nolen, L.D.; Gao, S.; Han, Z.; Mann, M.R.; Gie, C.Y.; Otte, A.P.; Bartolomei, M.S.; Latham, K.E. X chromosome reactivation and regulation in cloned embryos. Dev. Biol. 2005, 279, 525–540. [Google Scholar] [CrossRef]
- Kubota, T.; Wakui, K.; Nakamura, T.; Ohashi, H.; Watanabe, Y.; Yoshino, M.; Kida, T.; Okamoto, N.; Matsumura, M.; Muroya, K.; et al. The proportion of cells with functional X disomy is associated with the severity of mental retardation in mosaic ring X Turner syndrome females. Cytogenet. Genome Res. 2002, 99, 276–284. [Google Scholar] [CrossRef]
- Rodríguez, L.; Diego-Alvarez, D.; Lorda-Sanchez, I.; Gallardo, F.L.; Martínez-Fernández, M.L.; Arroyo-Muñoz, M.E.; Martínez-Frías, M.L. A small and active ring X chromosome in a female with features of Kabuki syndrome. Am. J. Med. Genet. A 2008, 146A, 2816–2821. [Google Scholar]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 276–257. [Google Scholar]
- Shirohzu, H.; Kubota, T.; Kumazawa, A.; Sado, T.; Chijiwa, T.; Inagaki, K.; Suetake, I.; Tajima, S.; Wakui, K.; Miki, Y.; et al. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 2002, 112, 31–37. [Google Scholar] [CrossRef]
- Kubota, T.; Furuumi, H.; Kamoda, T.; Iwasaki, N.; Tobita, N.; Fujiwara, N.; Goto, Y.; Matsui, A.; Sasaki, H.; Kajii, T. ICF syndrome in a girl with DNA hypomethylation but without detectable DNMT3B mutation. Am. J. Med. Genet. A 2004, 129A, 290–293. [Google Scholar]
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2; encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Chunshu, Y.; Endoh, K.; Soutome, M.; Kawamura, R.; Kubota, T. A patient with classic Rett syndrome with a novel mutation in MECP2 exon 1. Clin. Genet. 2006, 70, 530–531. [Google Scholar]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar]
- Itoh, M.; Ide, S.; Takashima, S.; Kudo, S.; Nomura, Y.; Segawa, M.; Kubota, T.; Mori, H.; Tanaka, S.; Horie, H.; et al. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 2007, 66, 117–123. [Google Scholar] [CrossRef]
- Miyake, K.; Hirasawa, T.; Soutome, M.; Itoh, M.; Goto, Y.; Endoh, K.; Takahashi, K.; Kudo, S.; Nakagawa, T.; Yokoi, S.; et al. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: Implication for pathogenesis of Rett syndrome. BMC Neurosci. 2011, 12, 81. [Google Scholar]
- Basic investigation report for handicapped children 2005 (in Japanese). Available online: http://www.mhlw.go.jp/toukei/saikin/hw/titeki/index.html/ (accessed on 16 February 2012).
- Yeargin-Allsopp, M.; Rice, C.; Karapurkar, T.; Doernberg, N.; Boyle, C.; Murphy, C. Prevalence of autism in a US metropolitan area. JAMA 2003, 289, 49–55. [Google Scholar]
- Holoden, C. Autism now. Science 2009, 323, 565. [Google Scholar]
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 2009, 65, 591–598. [Google Scholar]
- Kim, Y.S.; Leventhal, B.L.; Koh, Y.J.; Fombonne, E.; Laska, E.; Lim, E.C.; Cheon, K.A.; Kim, S.J.; Kim, Y.K.; Lee, H.; et al. Prevalence of autism spectrum disorders in a total population sample. Am. J. Psychiatry 2011, 168, 904–912. [Google Scholar] [CrossRef]
- Lord, C. Epidemiology: How common is autism? Nature 2011, 474, 166–168. [Google Scholar]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science 2003, 302, 862–830. [Google Scholar] [CrossRef]
- Persico, A.M.; Bourgeron, T. Searching for ways out of the autism maze: Genetic, epigenetic and environmental clues. Trends Neurosci. 2006, 29, 349–358. [Google Scholar]
- Arlt, M.F.; Mulle, J.G.; Schaibley, V.M.; Ragland, R.L.; Durkin, S.G.; Warren, S.T.; Glover, T.W. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009, 84, 339–350. [Google Scholar] [CrossRef]
- Qiu, J. Epigenetics: Unfinished symphony. Nature 2006, 441, 143–145. [Google Scholar]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D'Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar]
- McGowan, P.O.; Sasaki, A.; D'Alessio, A.C.; Dymov, S.; Labonté, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar]
- Claes, B.; Buysschaert, I.; Lambrechts, D. Pharmaco-epigenomics: Discovering therapeutic approaches and biomarkers for cancer therapy. Heredity (Edinb) 2010, 105, 152–160. [Google Scholar]
- Spannhoff, A.; Hauser, A.T.; Heinke, R.; Sippl, W.; Jung, M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem 2009, 4, 1568–1582. [Google Scholar]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar]
- Jessberger, S.; Nakashima, K.; Clemenson, G.D., Jr.; Mejia, E.; Mathews, E.; Ure, K.; Ogawa, S.; Sinton, C.M.; Gage, F.H.; Hsieh, J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 2007, 27, 5967–5975. [Google Scholar]
- Renthal, W.; Nestler, E.J. Epigenetic mechanisms in drug addiction. Trends Mol. Med. 2008, 14, 341–350. [Google Scholar]
- Xu, C.; Spragni, E.; Jacques, V.; Rsche, J.R.; Gottesfeld, J.M. Improved histone deacetylase inhibitors as therapeutics for the neurodegenerative disease Friedreich’s ataxia: A new synthetic route. Pharmaceuticals 2011, 4, 1578–1590. [Google Scholar]
- Sleiman, S.F.; Berlin, J.; Basso, M.; Karuppagounder, S.S.; Rohr, J.; Ratan, R.R. Histone deacetylase inhibitors and Mitramycin A impact a similar neuroprotective pathway at a crossroad between cancer and neurodegeneration. Pharmaceuticals 2011, 4, 1183–1185. [Google Scholar]
- Csoka, A.B.; Szyf, M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. Med. Hypotheses 2009, 73, 770–780. [Google Scholar]
- Watanabe, H.; Fukuoka, H.; Sugiyama, T.; Nagai, Y.; Ogasawara, K.; Yoshiike, N. Dietary folate intake during pregnancy and birth weight in Japan. Eur. J. Nutr. 2008, 47, 341–347. [Google Scholar]
- Hsieh, H.Y.; Chiu, P.H.; Wang, S.C. Epigenetics in traditional chinese pharmacy: A bioinformatic study at pharmacopoeia scale. Evid. Based Complement. Alternat. Med. 2011, 2011, 816714. [Google Scholar]
- Dietrich, M.; Brown, C.J.; Block, G. The effect of folate fortification of cereal-grain products on blood folate status, dietary folate intake, and dietary folate sources among adult non-supplement users in the United States. J. Am. Coll. Nutr. 2005, 24, 266–274. [Google Scholar]
- Ichi, S.; Costa, F.F.; Bischof, J.M.; Nakazaki, H.; Shen, Y.W.; Boshnjaku, V.; Sharma, S.; Mania-Farnell, B.; McLone, D.G.; Tomita, T.; et al. Folic acid remodels chromatin on Hes1 and Neurog2 promoters during caudal neural tube development. J. Biol. Chem. 2010, 285, 36922–36932. [Google Scholar]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Invest. 2008, 118, 2316–2324. [Google Scholar]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar]
- Lillycrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modification. Br. J. Nutr. 2007, 97, 1064–1073. [Google Scholar]
- Burdge, G.C.; Lillycrop, K.A.; Phillips, E.S.; Slater-Jefferies, J.L.; Jackson, A.A.; Hanson, M.A. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J. Nutr. 2009, 139, 1054–1060. [Google Scholar]
- Rimland, B. Controversies in the treatment of autistic children: Vitamin and drug therapy. J. Child. Neurol. 1988, 3, S68–S72. [Google Scholar]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 20, 1611–1617. [Google Scholar]
- Moretti, P.; Sahoo, T.; Hyland, K.; Bottiglieri, T.; Peters, S.; del Gaudio, D.; Roa, B.; Curry, S.; Zhu, H.; Finnell, R.H.; et al. Cebral folate deficiency with developmental delay; autism; and response to folinic acid. Neurology 2005, 64, 1088–1090. [Google Scholar]
- Kucharski, R.; Maleszka, J.; Foret, S.; Maleszka, R. Nutritional control of reproductive status in haneybees via DNA methylation. Science 2008, 319, 1827–1830. [Google Scholar]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar]
- Xin, Z.; Tachibana, M.; Guggiari, M.; Heard, E.; Shinkai, Y.; Wagstaff, J. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J. Biol. Chem. 2003, 278, 14996–15000. [Google Scholar]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar]
- Qi, H.H.; Sarkissian, M.; Hu, G.Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar]
- Huang, H.S.; Allen, J.A.; Mabb, A.M.; King, I.F.; Miriyala, J.; Taylor-Blake, B.; Sciaky, N.; Dutton, J.W., Jr.; Lee, H.M.; Chen, X.; et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar]
- Matsuda, H.; Fukuda, N.; Ueno, T.; Katakawa, M.; Wang, X.; Watanabe, T.; Matsui, S.; Aoyama, T.; Saito, K.; Bando, T.; et al. Transcriptional inhibition of progressive renal disease by gene silencing pyrrole-imidazole polyamide targeting of the transforming growth factor-β1 promoter. Kidney Int. 2011, 79, 46–56. [Google Scholar] [CrossRef]
- Ohtsuki, A.; Kimura, M.T.; Minoshima, M.; Suzuki, T.; Ikeda, M.; Bando, T.; Nagase, H.; Shinohara, K.; Sugiyama, H. Synthesis and properties of PI polyamide-SAHA conjugate. Tetrahedron Lett. 2009, 50, 7288–7292. [Google Scholar]
- Kondo, M.; Gray, L.J.; Pelka, G.J.; Christodoulou, J.; Tam, P.P.; Hannan, A.J. Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome—Mecp2 gene dosage effects and BDNF expression. Eur. J. Neurosci. 2008, 27, 3342–3350. [Google Scholar]
- Nag, N.; Moriuchi, J.M.; Peitzman, C.G.; Ward, B.C.; Kolodny, N.H.; Berger-Sweeney, J.E. Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp2 1lox mice. Behav. Brain Res. 2009, 196, 44–48. [Google Scholar]
- Kerr, B.; Silva, P.A.; Walz, K.; Young, J.I. Unconventional transcriptional response to environmental enrichment in a mouse model of Rett syndrome. PLoS One 2010, 5, e11534. [Google Scholar]
- Lonetti, G.; Angelucci, A.; Morando, L.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T. Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol. Psychiatry 2010, 67, 657–665. [Google Scholar]
- Woods, R.; Vallero, R.O.; Golub, M.S.; Suarez, J.K.; Ta, T.A.; Yasui, D.H.; Chi, L.H.; Kostyniak, P.J.; Pessah, I.N.; Berman, R.F.; et al. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2 308 mutation. Hum. Mol. Genet. 2012. [Google Scholar] [CrossRef]
- Horsthemke, B. Heritable germline epimutations in humans. Nat. Genet. 2007, 39, 573–574. [Google Scholar]
- Daxinger, L.; Whitelaw, E. Transgenerational epigenetic inheritance: More questions than answers. Genome. Res. 2010, 20, 1623–1628. [Google Scholar]
- Seong, K.H.; Li, D.; Shimizu, H.; Nakamura, R.; Ishii, S. Inheritance of stress-induced; ATF-2-dependent epigenetic change. Cell 2011, 145, 1049–1061. [Google Scholar] [CrossRef]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar]
- Yaoi, T.; Itoh, K.; Nakamura, K.; Ogi, H.; Fujiwara, Y.; Fushiki, S. Genome-wide analysis of epigenomic alterations in fetal mouse forebrain after exposure to low doses of bisphenol A. Biochem. Biophys. Res. Commun. 2008, 376, 563–567. [Google Scholar]
- Gore, A.C.; Walker, D.M.; Zama, A.M.; Armenti, A.E.; Uzumcu, M. Early life exposure to endocrine-disrupting chemicals causes lifelong molecular reprogramming of the hypothalamus and premature reproductive aging. Mol. Endocrinol. 2011, 25, 2157–2168. [Google Scholar]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.; Truong, H.T.; Russo, S.J.; Laplant, Q.; Sasaki, T.S.; Whistler, K.N.; et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef]
- Pascual, M.; Boix, J.; Felipo, V.; Guerri, C. Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J. Neurochem. 2009, 108, 920–931. [Google Scholar]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 2010, 68, 408–415. [Google Scholar]
- Thayer, Z.M.; Kuzawa, C.W. Biological memories of past environments: Epigenetic pathways to health disparities. Epigenetics 2011, 6, 798–803. [Google Scholar]
- Arai, J.A.; Feig, L.A. Long-lasting and transgenerational effects of an environmental enrichment on memory formation. Brain Res. Bull. 2011, 85, 30–35. [Google Scholar]
- Sakazume, S.; Ohashi, H.; Sasaki, Y.; Harada, N.; Nakanishi, K.; Sato, H.; Emi, M.; Endoh, K.; Sohma, R.; Kido, Y.; et al. Spread of X-chromosome inactivation into chromosome 15 is associated with Prader-Willi syndrome phenotype in a boy with a t(X;15)(p21.1;q11.2) translocation. Hum. Genet. 2012, 131, 121–130. [Google Scholar] [CrossRef]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K Discovery and replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar]
- Poon, L.L.; Leung, T.N.; Lau, T.K.; Chow, K.C.; Lo, Y.M. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin. Chem. 2002, 48, 35–41. [Google Scholar]
- Papageorgiou, E.A.; Karagrigoriou, A.; Tsaliki, E.; Velissariou, V.; Carter, N.P.; Patsalis, P.C. Fetal-specific DNA methylation ratio permits noninvasive prenatal diagnosis of trisomy 21. Nat. Med. 2011, 17, 510–513. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kubota, T.; Takae, H.; Miyake, K. Epigenetic Mechanisms and Therapeutic Perspectives for Neurodevelopmental Disorders. Pharmaceuticals 2012, 5, 369-383. https://doi.org/10.3390/ph5040369
Kubota T, Takae H, Miyake K. Epigenetic Mechanisms and Therapeutic Perspectives for Neurodevelopmental Disorders. Pharmaceuticals. 2012; 5(4):369-383. https://doi.org/10.3390/ph5040369
Chicago/Turabian StyleKubota, Takeo, Hirasawa Takae, and Kunio Miyake. 2012. "Epigenetic Mechanisms and Therapeutic Perspectives for Neurodevelopmental Disorders" Pharmaceuticals 5, no. 4: 369-383. https://doi.org/10.3390/ph5040369
APA StyleKubota, T., Takae, H., & Miyake, K. (2012). Epigenetic Mechanisms and Therapeutic Perspectives for Neurodevelopmental Disorders. Pharmaceuticals, 5(4), 369-383. https://doi.org/10.3390/ph5040369