
4-Heteroaryl Substituted Amino-3,5-Dicyanopyridines as New Adenosine Receptor Ligands: Novel Insights on Structure-Activity Relationships and Perspectives

 , , , , , , and
, , , , , , and
Abstract
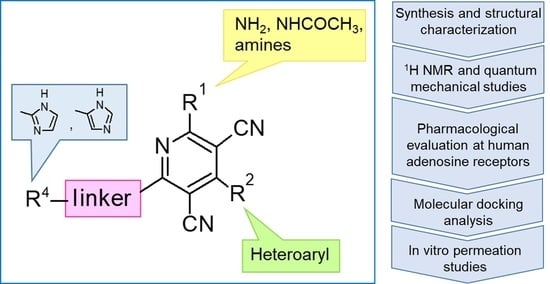
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Pharmacological Assays
2.3. Molecular Docking Studies
2.4. In Vitro Permeation Studies
3. Discussion
3.1. Structure–Activity Relationships

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Binding Experiments a | cAMP Assays | ||||||
| Ki (nM) or I% | EC50 (nM) b Efficacy c (%) | ||||||
| Compd | R1 | R2 | R | hA1 d | hA2A e | hA3 f | hA2B |
| 1 | NH2 |  |  | 9.63 ± 1.61 | 21 ± 2 | 52 ± 5 | 1.4 ± 0.2 (52%) |
| 2 g | NH2 |  |  | 190 ± 16 | 233 ± 19 | 290 ± 23 | >1000 (8%) |
| 3 | NH2 |  |  | 10.1 ± 0.8 | 10.5 ± 0.9 | 279 ± 21 | >1000 1% |
| 4 | NH2 |  |  | 0.77 ± 0.09 | 37 ± 3 | 274 ± 23 | 2.32 ± 0.21 (42%) |
| 5 | NH2 |  |  | 2.50 ± 0.20 | 24 ± 2 | 25 ± 2 | 1.12 ± 0.11 (73%) |
| 6 | NH2 |  |  | 1.01 ± 0.09 | 55 ± 6 | 221 ± 20 | 3.15 ± 0.29 (47%) |
| 7 | NH2 |  |  | 8.85 ± 0.82 | 81 ± 7 | 26% | 12.5 ± 1.3 (36%) |
| 8 | NH2 |  |  | 5.26 ± 0.48 | 463 ± 38 | 19% | 63 ± 5 (58%) |
| 9 | NH2 |  |  | 2.71 ± 0.18 | 377 ± 32 | 798 ± 72 | >1000 (7%) |
| 10 | NH2 |  |  | 149 ± 11 | 613 ± 53 | 23% | >1000 (1%) |
| 11 h | NH2 |  |  | 51 ± 4 | 442 ± 37 | 849 ± 74 | >1000 (4%) |
| 12 | NH2 |  |  | 315 ±28 | 157 ± 14 | 16% | >1000 (3%) |
| 13 | NH2 |  |  | 174 ± 14 | 125 ± 10 | 6% | >1000 (9%) |
| 14 | NH2 |  |  | 87 ± 7 | 817 ± 71 | 33% | >1000 (6%) |
| 15 | NH2 |  |  | 378 ± 26 | 225 ± 18 | 758 ± 66 | >1000 (1%) |
| 16 | NH2 |  |  | 30 ± 3 | 138 ± 11 | 30% | >1000 (1%) |
| 17 | NH2 |  |  | 33 ± 3 | 279 ± 22 | 542 ± 48 | >1000 6% |
| 18 |  |  |  | 393 ± 32 | 138 ± 11 | 279 ± 21 | >1000 2% |
| 19 |  |  |  | 292 ± 26 | 1% | 18% | >1000 (13%) |
| 20 |  |  |  | 41 ± 3 | 89 ± 7 | 118 ± 11 | >1000 (8%) |
| 21 | NHCOCH3 |  |  | 34 ± 3 | 394 ± 27 | 768 ± 63 | >1000 (1%) |
| LUF5833i | NH2 |  |  | 2.4 ± 1 | 28 ± 1 | 171 ± 109 | 19 ± 7 (81%) |
| hA1AR | hA2AAR | hA3AR | ||||
|---|---|---|---|---|---|---|
| Compd | Efficacy, b % (Profile) | EC50 or IC50 (nM) | Efficacy, b % | EC50 (nM) | Efficacy, b % | IC50 (nM) c |
| 1 | 31 ± 3 (Partial Agonist) | 12.2 ± 1.2 c | 39 ± 3 | 15.1 ± 1.3 | 45 ± 4 | 68 ± 6 |
| 5 | 75 ± 5 (Partial Agonist) | 1.95 ± 0.16 c | 40 ± 3 | 11.3 ± 1.0 | 34 ± 3 | 33 ± 2 |
| 11 | 0.82 ± 0.07 (Antagonist) | 125 ± 11 d | NT e | NT | NT | NT |
| 12 | 1.31 ± 0.11 (Antagonist) | 768 ± 67 d | NT | NT | NT | NT |
| 16 | −43 ± 4 (Inverse Agonist) | 59 ± 4 c | NT | NT | NT | NT |
| 17 | 54 ± 4 (Partial Agonist) | 38 ± 3 c | NT | NT | NT | NT |
3.2. Ab Initio Quantum Mechanical Studies
3.3. Molecular Modeling Studies
3.4. In Vitro Permeation Studies
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. General Procedure for the Synthesis of 2-Amino-4-(heteroaryl)-6-[(1H-imidazol-2-ylmethyl)sulfanyl]pyridine-3,5-dicarbonitriles 1, 3
4.1.3. 2-Amino-4-[5-(hydroxymethyl)furan-2-yl]-6-[(1H-imidazol-2-ylmethyl)sulfanyl]pyridine-3,5-dicarbonitrile hydrobromide (2)
4.1.4. General Procedure for the Synthesis of 2-Amino-4-(heteroaryl)-6-[(1H-imidazol-2-ylmethyl)sulfanyl]pyridine-3,5-dicarbonitriles 4–10
4.1.5. 2-Amino-4-(furan-2-yl)-6-[(1H-imidazol-5-ylmethyl)sulfanyl]pyridine-3,5-dicarbonitrile hydrochloride (11)
4.1.6. General Procedure for the Synthesis of 2-Amino-substituted Derivatives 12, 13
4.1.7. General Procedure for the Synthesis of 2-{[6-Amino-3,5-dicyano-4-(furan-2-yl)pyridin-2-yl]sulfanyl}-N-acetamides 14–16
4.1.8. 4-({[6-Amino-3,5-dicyano-4-(furan-2-yl)pyridin-2-yl]sulfanyl}methyl)-N-[2-(1H-imidazol-5-yl)ethyl]-1,3-thiazole-2-carboxamide (17)
4.1.9. General Procedure for the Synthesis of the Target Compounds 18–21
4.1.10. General Procedure for the Synthesis of 2-Amino-4-heteroaryl-6-(phenylsulfanyl)pyridine-3,5-dicarbonitriles 23, 24, 30
4.1.11. The 2-Amino-6-(phenylsulfanyl)-4,4′-bipyridine-3,5-dicarbonitrile (28)
4.1.12. The 2′-Amino-6-hydroxy-6′-(phenylsulfanyl)-3,4′-bipyridine-3′,5′-dicarbonitrile (31)
4.1.13. General Procedure for the Synthesis of 2-Amino-4-(heteroaryl)-6-sulfanylpyridine-3,5-dicarbonitriles 33, 34, 38, 40, 41
4.1.14. {[6-Amino-3,5-dicyano-4-(furan-2-yl)pyridin-2-yl]sulfanyl}acetic acid (42)
4.1.15. 4-({[6-Amino-3,5-dicyano-4-(furan-2-yl)pyridin-2-yl]sulfanyl}methyl)-1,3-thiazole-2-carboxylic acid (43)
4.1.16. 2-Chloro-4-(furan-2-yl)-6-(phenylsulfanyl)pyridine-3,5-dicarbonitrile (44)
4.1.17. General Procedure for the Synthesis of 2(6)-Substituted-4-(furan-2-yl)-6(2)-(phenylsulfanyl)pyridine-3,5-dicarbonitriles 45–47
4.1.18. General Procedure for the Synthesis of 2(6)-Substituted-4-(furan-2-yl)-6(2)-sulfanylpyridine-3,5-dicarbonitriles 48–50
4.1.19. The 4-(Chloromethyl)-1H-imidazole Hydrochloride (53)
4.1.20. The 4-(Azidomethyl)-1H-imidazole (54)
4.1.21. The 1-(1H-Imidazol-4-yl)methanamine (55)
4.1.22. Ethyl 4-(Chloromethyl)-1,3-thiazole-2-carboxylate (56)
4.1.23. The 4-(Chloromethyl)-1,3-thiazole-2-carboxylic acid (57)
4.2. Pharmacological Assays
4.2.1. Cell Culture and Membrane Preparation
4.2.2. Competition Binding Experiments
4.2.3. Cyclic AMP Assays
4.2.4. Data Analysis
4.3. Ab Initio Quantum Mechanical Studies
4.4. Molecular Modeling
4.5. Permeation Studies
4.5.1. HPLC Assay
4.5.2. Evaluation of In Vitro Permeation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rosentreter, U.; Henning, R.; Bauser, M.; Krämer, T.; Vaupel, A.; Hübsch, W.; Dembowsky, K.; Salcher-Schraufstätter, O.; Stasch, J.P.; Krahn, T.; et al. Substituted 2-Thio-3,5-Dicyano-4-Aryl-6-Aminopyridines and the Use Thereof as Adenosine Receptor Ligands. Patent WO2001025210, 19 September 2000. Available online: https://worldwide.espacenet.com/ (accessed on 15 October 2019).
- Beukers, M.W.; Chang, L.C.W.; von Frijtag Drabbe Kunzel, J.K.; Mulder-Krieger, T.; Spanjersberg, R.F.; Brussee, J.; IJzerman, A.P. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J. Med. Chem. 2004, 47, 3707–3709. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.W.; von Frijtag Drabbe Künzel, J.K.; Mulder-Krieger, T.; Spanjersberg, R.F.; Roerink, S.F.; Van den Hout, G.; Beukers, M.W.; Brussee, J.; IJzerman, A.P. A series of ligand displaying a remarkable agonistic-antagonistic profile at the adenosine A1 receptor. J. Med. Chem. 2005, 48, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Meibom, D.; Albrecht-Küpper, B.; Diedrichs, N.; Hübsch, W.; Kast, R.; Krämer, T.; Krenz, U.; Lerchen, H.-J.; Mittendorf, J.; Nell, P.G.; et al. Neladenoson bialanate hydrochloride: A prodrug of a partial adenosine A1 receptor agonist for the chronic treatment of heart disease. ChemMedChem 2017, 12, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Dal Ben, D.; Lambertucci, C.; Colotta, V. The aminopyridine-3,5-dicarbonitrile core for the design of new non-nucleoside-like agonists of the human adenosine A2B receptor. Eur. J. Med. Chem. 2018, 150, 127–139. [Google Scholar] [CrossRef]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Pasquini, S.; Mannelli, L.D.C.; Ghelardini, C.; Lucarini, E.; et al. Modifications on the amino-3,5-dicyanopyridine core to obtain multifaceted adenosine receptor ligands with antineuropathic activity. J. Med. Chem. 2019, 62, 6894–6912. [Google Scholar] [CrossRef]
- Catarzi, D.; Varano, F.; Varani, K.; Vincenzi, F.; Pasquini, S.; Dal Ben, D.; Volpini, R.; Colotta, V. Amino-3,5-Dicyanopyridines Targeting the Adenosine Receptors. Ranging from Pan Ligands to Combined A1/A2B Partial Agonists. Pharmaceuticals 2019, 12, 159. [Google Scholar] [CrossRef] [Green Version]
- Bennett, K.A.; Tehan, B.; Lebon, G.; Tate, C.G.; Weir, M.; Marshall, F.H.; Langmead, C.J. Pharmacology and structure of isolated conformations of the adenosine A2A receptor define ligand efficacy. Mol. Pharmacol. 2013, 83, 949–958. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Wilder, T.; Cronstein, B.N. Rolofylline, an adenosine A1 receptor antagonist, inhibits osteoclast differentiation as an inverse agonist. Br. J. Pharmacol. 2013, 170, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ye, K.; Blad, C.C.; den Dulk, H.; Brouwer, J.; Ijzerman, A.P.; Beukers, M.W. ZM241385, DPCPX, MRS1706 are inverse agonists with different relative intrinsic efficacies on constitutively active mutants of the human adenosine A2B receptor. J. Pharmacol. Exp. Ther. 2007, 320, 637–645. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, A.M.; Ribeiro, J.A. Fine-tuning neuromodulation by adenosine. Trends Pharmacol. Sci. 2000, 21, 341–346. [Google Scholar] [CrossRef]
- De Mendoncca, A.; Ribeiro, J.A. Adenosine and synaptic plasticity. Drug Dev. Res. 2001, 52, 283–290. [Google Scholar] [CrossRef]
- Cunha, R.A. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem. Int. 2001, 38, 107–125. [Google Scholar] [CrossRef]
- Ferreira, J.M.; Paes-de-Carvalho, R. Long-term activation of adenosine A(2a) receptors blocks glutamate excitotoxicity in cultures of avian retinal neurons. Brain Res. 2001, 900, 169–176. [Google Scholar] [CrossRef]
- Hasko, G.; Pacher, P. A2A receptors in inflammation and injury: Lessons learned from transgenic animals. J. Leukoc. Biol. 2008, 83, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Fenton, R.A.; Wheeler, H.B.; Powell, C.C.; Peyton, B.D.; Cutler, B.S.; Dobson, J.G.J. Adenosine A2a receptors increase arterial endothelial cell nitric oxide. J. Surg. Res. 1998, 80, 357–364. [Google Scholar] [CrossRef]
- Bouma, M.G.; Stad, R.K.; van den Wildenberg, F.A.; Buurman, W.A. Differential regulatory effects of adenosine on cytokine release by activated human monocytes. J. Immunol. 1994, 153, 4159–4168. [Google Scholar]
- Van der Graaf, P.H.; Van Schaick, E.A.; Visser, S.A.; De Greef, H.J.; Ijzerman, A.P.; Danhof, M. Mechanism-based pharmacokinetic-pharmacodynamic modeling of antilipolytic effects of adenosine A(1) receptor agonists in rats: Prediction of tissue-dependent efficacy in vivo. J. Pharmacol. Exp. Ther. 1999, 290, 702–709. [Google Scholar]
- Borea, P.A.; Gassi, S.; Merighi, S.; Varani, K. Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert protective effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Feoktistov, I.; Biaggioni, I.; Cronstein, B.N. Adenosine receptors in wound healing, fibrosis and angiogenesis. Handb. Exp. Pharmacol. 2009, 193, 383–397. [Google Scholar]
- Flohr, C.; Hay, R. Putting the burden of skin diseases on the global map. Br. J. Dermatol. 2021, 184, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Valls, M.D.; Cronstein, B.N.; Montesinos, M.C. Adenosine receptor agonists for promotion of dermal wound healing. Biochem. Pharmacol. 2009, 77, 1117–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonyanian, Z.; Walker, M.; Du Toit, E.; Rose’Meyer, R.B. Multiple adenosine receptor subtypes stimulate wound healing in human EA.hy926 endothelial cells. Purinergic Signal 2019, 15, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands ad their potential as novel drugs. Biochim. Biophys. Acta 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [Green Version]
- Eissa, A.M.F.; Ezz El-Arab, E.M.; Farag, A.M.; Moharram, H.H. Synthesis and biological evaluation of pyrido [2,3.d]pyrimidine as antitumor effect. Egypt. J. Chem. 2006, 49, 761–774. [Google Scholar]
- Brandt, W.; Mologni, L.; Preu, L.; Lemcke, T.; Gambacorti-Passerini, C.; Kunick, C. Inhibitors of the RET tyrosine based on a 2-(alkylsulfanyl)-4-(3-thienyl)nicotinonitrile scaffold. Eur. J. Med. Chem. 2010, 45, 2919–2927. [Google Scholar] [CrossRef]
- Attaby, F.A.; Elghandour, A.H.H.; Ali, M.A.; Ibrahem, Y.M. Synthesis, reactions, and antiviral activity of 6′-amino-2′-thioxo-1′,2-dihydro-3,4′-bipyridine-3′-5′-dicarbonitrile. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 695–709. [Google Scholar] [CrossRef]
- Sridhar, M.; Ramanaiah, B.C.; Narsaiah, C.; Mahesh, B.; Kumaraswamy, M.; Mallu, K.K.R.; Ankathi, V.M.; Shanthan Rao, P. Novel ZnCl2-catalyzed one-pot multicomponent synthesis of 2-amino-3,5-dicarbonitrile-6-thio-pyridines. Tetrahedron Lett. 2009, 50, 3897–3900. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Kireev, A.S.; Yakovenko, A.A.; Antipin, M.Y.; Magedov, I.V.; Kornienko, A. One-step synthesis of heterocyclic privileged medicinal scaffolds by a multicomponent reaction of malononitrile with aldehydes and thiols. J. Org. Chem. 2007, 72, 3443–3453. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Magedov, I.V.; Kireev, A.S.; Kornienko, A. One-step, three-component synthesis of pyridines and 1,4-dihydropyridines with manifold medicinal utility. Org. Lett. 2006, 8, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Maiti, P.K. First report of multiple metal ions containing glass-ceramic material as a heterogeneous ditopic catalyst for the chromatography free synthesis of 2-amino-3,5-dicarbonitrile-6-arylthio-pyridines in water. Tetrahedron Lett. 2015, 56, 5094–5098. [Google Scholar] [CrossRef]
- Bartels-Keith, J.R.; Boggs, R.A.; Puttick, A.J.; Sofen, N.M. Photographic Reagent Tetrazoles. U.S. Patent US4847383, 11 July 1989. [Google Scholar]
- Inoue, T.; Tojo, T.; Morita, M. Thiazole Derivatives as VAP-1 Inhibitors, Their Preparation, Pharmaceutical Compositions, and Use in Therapy. Patent WO2006028269A2, 16 March 2006. [Google Scholar]
- De Roulet, D.; Devita, R. Composition and Methods Using the Same for Treatment of Neurodegenerative and Mitochondrial Disease. Patent WO2015123365, 20 August 2015. [Google Scholar]
- Available online: https://www.msg.chem.iastate.edu/gamess/download.html (accessed on 1 September 2021).
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment; Chemical Computing Group: Montreal, QC, Canada, 2022.
- Bartosova, L.; Bajgar, J. Transdermal drug delivery in vitro using diffusion cells. Curr. Med. Chem. 2012, 19, 4671–4677. [Google Scholar] [CrossRef]
- Rosentreter, U.; Kramer, T.; Shimada, M.; Hebsch, W.; Diedrichs, N.; Krahn, T.; Henninger, K.; Stasch, J.-P.; Wischnat, R. New 2-Substituted Methylthio-Dicyanopyridine Derivatives, Useful for Treating or Preventing e.g., Cardiovascular Disease and Inflammation, Are Adenosine A1 Receptor Agonists. Patent WO2003053441A1, 21 August 2022. [Google Scholar]
- Vakalopoulos, A.; Meibom, D.; Albrecht-Kepper, B.; Zimmermann, K.; Keldenich, J.; Lerchen, H.-G.; Nell, P.; Sessmeier, F.; Krenz, U. Preparation of Alkylaminodicyanopyridines as Adenosine Receptor Antagonists. Patent WO2010086101A1, 19 January 2010. [Google Scholar]
- Dal Ben, D.; Lambertucci, C.; Buccioni, M.; Marti Navia, A.; Marucci, G.; Spinaci, A.; Volpini, R. Non-Nucleoside Agonists of the Adenosine Receptors: An Overview. Pharmaceuticals 2019, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, D.; Gao, Z.G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular docking screening using agonist-bound GPCR structures: Probing the A2A adenosine receptor. J. Chem. Inf. Model 2015, 55, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Thomas, A.; Volpini, R. Simulation and Comparative Analysis of Different Binding Modes of Non-nucleoside Agonists at the A2A Adenosine Receptor. Mol. Inform. 2016, 35, 403–413. [Google Scholar] [CrossRef]
- Available online: http://biosig.unimelb.edu.au/pkcsm/ (accessed on 10 April 2015).
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a potent A1 adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Massara, A.; Vincenzi, F.; Tosi, A.; Padovan, M.; Trotta, F.; Borea, P.A. Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum. 2009, 60, 2880–2891. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Merighi, S.; Gessi, S.; Klotz, K.-N.; Leung, E.; Baraldi, P.G.; Cacciari, B.; Romagnoli, R.; Spalluto, G.; Borea, P.A. [3H]MRE 3008F20: A novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol. Pharmacol. 2000, 57, 968–975. [Google Scholar]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and Function of A2A and A3 Adenosine Receptors in Patients with Ankylosing Spondylitis, Psoriatic Arthritis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry: The First Forty Years; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Dougan, L.; Koti, A.S.R.; Genchev, G.; Lu, H.; Fernandez, J.M. A Single-Molecule Perspective on the Role of Solvent Hydrogen Bonds in Protein Folding and Chemical Reactions. Chem. Phys. Chem. 2008, 9, 2836–2847. [Google Scholar] [CrossRef]
- Bachovchin, W.W. Contributions of NMR spectroscopy to the study of hydrogen bonds in serine protease active sites. Magn. Reson. Chem. 2001, 39, S199–S213. [Google Scholar] [CrossRef]
- Patel, A.K.; Mishra, S.K.; Krishnamurthy, K.; Suryaprakash, N. Retention of strong intramolecular hydrogen bonds in high polarity solvents in binaphthalene–benzamide derivatives: Extensive NMR studies. RSC Adv. 2019, 9, 32759–32770. [Google Scholar] [CrossRef] [Green Version]










| Compd | Formation Energies | ||
|---|---|---|---|
| Conformation 1 (Intramolecular H-Bond) | Conformation 2 (No Intramolecular H-Bond | Intramolecular H-Bond | |
| E1 (Hartree) | E2 (Hartree) | ΔE = E1 − E2 (Kcal/mol) | |
| 4 | −1478.7221 | −1478.7188 | −2.1 |
| 6 | −1686.585 | −1686.585 | 0.0 |
| 7 | −1383.427 | −1383.4216 | −3.4 |
| 10 | −1457.8725 | −1457.8675 | −3.1 |
| 12 | −1024.6594 | −1024.6409 | −11.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catarzi, D.; Varano, F.; Vigiani, E.; Calenda, S.; Melani, F.; Varani, K.; Vincenzi, F.; Pasquini, S.; Mennini, N.; Nerli, G.; et al. 4-Heteroaryl Substituted Amino-3,5-Dicyanopyridines as New Adenosine Receptor Ligands: Novel Insights on Structure-Activity Relationships and Perspectives. Pharmaceuticals 2022, 15, 478. https://doi.org/10.3390/ph15040478
Catarzi D, Varano F, Vigiani E, Calenda S, Melani F, Varani K, Vincenzi F, Pasquini S, Mennini N, Nerli G, et al. 4-Heteroaryl Substituted Amino-3,5-Dicyanopyridines as New Adenosine Receptor Ligands: Novel Insights on Structure-Activity Relationships and Perspectives. Pharmaceuticals. 2022; 15(4):478. https://doi.org/10.3390/ph15040478
Chicago/Turabian StyleCatarzi, Daniela, Flavia Varano, Erica Vigiani, Sara Calenda, Fabrizio Melani, Katia Varani, Fabrizio Vincenzi, Silvia Pasquini, Natascia Mennini, Giulia Nerli, and et al. 2022. "4-Heteroaryl Substituted Amino-3,5-Dicyanopyridines as New Adenosine Receptor Ligands: Novel Insights on Structure-Activity Relationships and Perspectives" Pharmaceuticals 15, no. 4: 478. https://doi.org/10.3390/ph15040478