Piperazine- and Piperidine-Containing Thiazolo[5,4-d]pyrimidine Derivatives as New Potent and Selective Adenosine A2A Receptor Inverse Agonists

 ,
,
Abstract
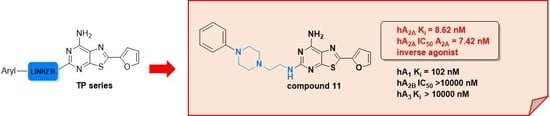
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Pharmacological Assays
3. Discussion
3.1. Structure-Activity Relationships
3.2. In Silico ADME Prediction
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. General Procedure for the Synthesis of 1–8, 10–21
4.1.3. General Procedure for the Synthesis of 31–32
4.1.4. General Procedure for the Synthesis of 33–36
4.1.5. Ethyl 4-(4-(2-aminoethyl)piperazin-1-yl)benzoate (37)
4.1.6. General Procedure for the Synthesis of 39–42
4.1.7. General Procedure for the Synthesis of 45–50
4.1.8. Ethyl 4-(4-(2-(1,3-dioxoisoindolin-2-yl)ethyl)piperazin-1-yl)benzoate (51)
4.1.9. General Procedure for the Synthesis of 58–61
4.2. Pharmacological Assays
4.2.1. Cell Culture and Membrane Preparation
4.2.2. Competition Binding Experiments
4.2.3. Cyclic AMP Assays
4.2.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: The bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [Green Version]
- Al-Attraqchi, O.H.A.; Attimarad, M.; Venugopala, K.N.; Nair, A.; Al-Attraqchi, N.H.A. Adenosine A2A receptor as a potential drug target—Current status and future perspectives. Curr. Pharm. Des. 2019, 25, 2716–2740. [Google Scholar] [CrossRef]
- Domenici, M.R.; Ferrante, A.; Martire, A.; Chiodi, V.; Pepponi, R.; Tebano, M.T.; Popoli, P. Adenosine A(2A) receptor as potential therapeutic target in neuropsychiatric disorders. Pharmacol Res. 2019, 147, 104338. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, X.; Zhen, X. Development of adenosine A(2A) receptor antagonists for the treatment of Parkinson’s Disease: A recent update and challenge. ACS Chem. Neurosci. 2019, 20, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Flor, A.M.; Moreau, J.L.; Poli, S.M.; Riemer, C.; Steward, L. 4-Hydroxy-4-methyl-piperidine-1-carboxylic acid-(4-methoxy-7-morpholin-4-yl-benzothiazol-2-yl) Amide. US Patent US20050261289, 24 November 2005. [Google Scholar]
- Minetti, P.; Tinti, M.O.; Carminati, P.; Castorina, M.; Di Cesare, M.A.; Di Serio, S.; Gallo, G.; Ghirardi, O.; Giorgi, F.; Giorgi, L.; et al. 2-n-Butyl-9-methyl-8-[1,2,3]-triazol-2-yl-9H-purin-6-ylamine and analogues as A2A adenosine receptor antagonists. Design, synthesis, and pharmacological characterization. J. Med. Chem. 2005, 48, 6887–6896. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.J.; Bamford, S.J.; Botting, R.; Comer, M.; Denny, S.; Gaur, S.; Griffin, M.; Jordan, A.M.; Knight, A.R.; Lerpiniere, J.; et al. Antogonists of the human A(2A) adenosine receptor. Design, synthesis, and preclinical evaluation of 7-aryltriazolo[4,5-d]pyrimidines. J. Med. Chem. 2009, 52, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, R.A.; Bedard, P.J.; Varty, G.B.; Kazdoba, T.M.; Di Paolo, T.; Grzelak, M.E.; Pond, A.J.; Hadjtahar, A.; Belanger, N.; Gregoire, L.; et al. Preladenant, a selective A2A receptor antagonist, is active in primate models of movement disorders. Exp. Neurol. 2010, 225, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A. Adenosine A2A receptor antagonists in Parkinson’s disease: Progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.G.; Souza, D.O.; Cunha, R.A.; Lara, D.L. Caffeine and adenosine A2a receptor antagonists β-amyloid (25–35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Brown, G.A.; Borodovsky, A.; Lamb, M.L. Targeting adenosine A2A receptor antagonism for treatment of cancer. Expert Opin. Drug Discov. 2018, 13, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Merighi, S.; Battistello, E.; Giacomelli, L.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. Targeting A3 and A2A adenosine receptors in the fight against cancer. Exp. Opin. Ther. Targets 2019, 23, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Merck Sharp and Dohme Corp. A phase Ib/II Study to Evaluate the Safety and Tolerability of Preladenant as a Single Agent and in Combination with Pembrolizumab in Subjects with Advanced Malignancies. ClinicalTrials.gov NLM Identifier: NCT03099161. Available online: https://clinicaltrials.gov/ct2/show/NCT03099161 (accessed on 4 April 2017).
- Mediavilla-Verela, M.; Castro, J.; Chiappori, A.; Noyes, D.; Hernandez, D.C.; Allard, B.; Stagg, J.; Antonia, S.J. A novel antagonist of the immune checkpoint protein adenosine A2A receptor restores tumor infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia 2017, 19, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Corvus Pharmaceuticals, Inc. A phase 1/1b, Open Label, Multicenter, Repeat-Dose, Dose-Selection Study of CPI-444 as a Single Agent and in Combination with Atezolizumab in Patients with Selected Incurable Cancers. ClinicalTrials.gov, NML Identifier NCT02655822. Available online: https://clinicaltrials.gov/ct2/show/NCT02655822 (accessed on 14 January 2016).
- Congreve, M.; Andrews, S.P.; Dorè, A.S.; Hollestenin, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Betti, M.; Falsini, M.; Ravani, A.; Borea, P.A.; Colotta, V.; Varani, K. Design, synthesis and pharmacological characterization of 2-(2-furanyl)thiazolo[5,4-d]pyrimidine-5,7-diamine derivatives: New potent A2A adenosine receptor inverse agonists with antinociceptive activity. J. Med. Chem. 2016, 59, 10564–10576. [Google Scholar] [CrossRef]
- Poli, D.; Falsini, M.; Varano, F.; Betti, M.; Varani, K.; Vincenzi, F.; Pugliese, A.M.; Pedata, F.; Dal Ben, D.; Thomas, A.; et al. Imidazo[1,2-a]pyrazin-8-amine core for the design of new adenosine receptor antagonists: Structural exploration to target the A3 and A2A subtypes. Eur. J. Med. Chem. 2017, 125, 611–628. [Google Scholar] [CrossRef]
- Squarcialupi, L.; Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Ravani, A.; Pasquini, S.; Vincenzi, F.; Salmaso, V.; Sturlese, M.; et al. The role of 5-arylalkylamino- and 5-piperazino moieties on the 7-aminopyrazolo[4,3-d]pyrimidine core in affecting adenosine A1 A2A receptor affinity and selectivity profiles. J. Enzym. Inhib. Med. Chem. 2017, 32, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Falsini, M.; Squarcialupi, L.; Catarzi, D.; Varano, F.; Betti, M.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; De Vita, T.; et al. The 1,2,4-triazolo[4,3-a]pyrazin-3-one as a versatile scaffold for the design of potent adenosine human receptor antagonists. Structural investigations to target the A2A receptor subtype. J. Med. Chem. 2017, 60, 5772–5790. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Falsini, M.; Vincenzi, F.; Pasquini, S.; Varani, K.; Colotta, V. Identification of novel thiazolo[5,4-d]pyrimidine derivatives as human A1 and A2A adenosine receptor antagonists/inverse agonists. Bioorg. Med. Chem. 2018, 26, 3688–3695. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Falsini, M.; Pasquini, S.; Borea, P.A.; Colotta, V.; Varani, K. Structure-activity relationship studies and pharmacological ch.; aracterization of N5-heteroarylalkyl-substituted-2-(2-furanyl)-thiazolo[5,4-d]pyrimidine-5,7-diamine-based derivatives as inverse agonists at human A2A adenosine receptor. Eur. J. Med. Chem. 2018, 155, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Falsini, M.; Dal Ben, D.; Buccioni, M.; Marucci, G.; Volpini, R.; Colotta, V. Novel human adenosine receptor antagonists based on the 7-amino-thiazolo[5,4-d]pyrimidine scaffold. Structural investigations at the 2-, 5- and 7- positions to enhance affinity and tune selectivity. Bioorg. Med. Chem. Lett. 2019, 29, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Falsini, M.; Catarzi, D.; Varano, F.; Ceni, C.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; Di Cesare Mannelli, L.; Lucarini, E.; et al. Antioxidant-conjugated 1,2,4-triazolo[4,3-a]pyrazin-3-one derivatives: Highly potent and selective human A2A adenosine receptor antagonists possessing protective efficacy in neuropathic pain. J. Med. Chem. 2019, 62, 8511–8531. [Google Scholar] [CrossRef] [PubMed]
- Falsini, M.; Catarzi, D.; Varano, F.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; Di Cesare Mannelli, L.; Ghelardini, C.; Colotta, V. Novel 8-amino-1,2,4-triazolo[4,3-a]pyrazin-3-one derivatives as potent human adenosine A1 and A2Areceptorantagonists. Evaluation of their protective effect against β-amyloid induced neurotoxicity in SHSY5Y cells. Bioorg. Chem. 2019, 87, 380–394. [Google Scholar] [CrossRef]
- Falsini, M.; Ceni, C.; Catarzi, D.; Varano, F.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Navia, A.M.; Volpini, R.; Colotta, V. New 8-amino-1,2,4-triazolo[4,3-a]pyrazin-3-one derivatives. Evaluation of different moieties on the 6-aryl ring to obtain potent and selective human A2A adenosine receptor antagonists. Bioorg. Med. Chem. Lett. 2020, 30, 127126. [Google Scholar] [CrossRef]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Pasquini, S.; Pelletier, J.; Lopes Rangel Fietto, J.; Espindola Gelsleichter, N.; Sarlandie, M.; Guilbaud, A.; Sevigny, J.; et al. Structural investigation on thiazolo[5,4-d]pyrimidines to obtain dual-acting blockers of CD73 and adenosine A2A receptor as potential antitumor agents. Bioorg. Med. Chem. Lett. 2020, 30, 127067. [Google Scholar] [CrossRef]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef]
- Long, J.Z.; Jin, X.; Adibekian, A.; Li, W.; Cravatt, B.F. Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J. Med. Chem. 2010, 53, 1830–1842. [Google Scholar] [CrossRef] [Green Version]
- Silverman, L.S.; Caldwell, J.P.; Greenlee, W.J.; Kiselgof, E.; Matasi, J.J.; Tulshian, D.B.; Arik, L.; Foster, C.; Bertorelli, R.; Monopoli, A.; et al. 3H-[1,2,4]-Triazolo[5,1-i]purin-5-amine derivatives as adenosine A2A antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 1659–1662. [Google Scholar] [CrossRef] [PubMed]
- Chun, C.; Schmitzer, A.R. A pseudorotaxane umbrella thread with chloride transmembrane transport properties. Med. Chem. Commun. 2011, 2, 987–990. [Google Scholar] [CrossRef]
- Mejuch, T.; Garivet, G.; Hofer, W.; Kaiser, N.; Fansa, E.K.; Ehrt, C.; Koch, O.; Baumann, M.; Ziegler, S.; Wittinghofer, A.; et al. Small-molecule inhibition of the UNC119-cargo interaction. Angew. Chem. Int. Ed. 2017, 56, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Tomas, D.; Hiremathad, A.; Capriati, V.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Donezepil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enzym. Inhib. Med. Chem. 2018, 33, 1212–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, D.; Ishikawa, M.; Yamamoto, M.; Murakami, S.; Hachisu, M.; Katano, K.; Ajito, K. Tricyclic pharmacophore-based molecules as novel integrin αvβ3 antagonists. Part 1: Design and synthesis of a lead compound exhibiting αvβ3/αIIbβ3 dual antagonistic activity. Bioorg. Med. Chem. 2006, 2006, 2089–2108. [Google Scholar] [CrossRef]
- Diouf, O.; Depreux, P.; Chavatte, P.; Paupaert, J.H. Synthesis and preliminary pharmacological results on new naphthalene derivatives as 5-HT4 receptor ligands. Eur. J. Med. Chem. 2000, 35, 699–706. [Google Scholar] [CrossRef]
- Furlotti, G.; Alisi, M.A.; Cazzola, N.; Ceccacci, F.; Garrone, B.; Gasperi, T.; La Bella, A.; Leonelli, F.; Loreto, M.A.; Magarò, G.; et al. Targeting serotonin 2A and adrenergic α1 receptors for ocular antihypertensive agents: Discovery of 3,4-dihydropyrazino[1,2-b]indazol-1(2H)-one derivatives. ChemMedChem 2018, 13, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Caulkett, P.W.R.; Jones, G.; Collis, M.G.; Poucher, S.M. Preparation of (amino)heteroaryl[1,2,4]triazolo[1,5-a]triazines and Related Compounds as Adenosine A2 Receptor Antagonists. Eur Patent Appl EP 459702, 23 May 1991. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a potent A1 adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Massara, A.; Vincenzi, F.; Tosi, A.; Padovan, M.; Trotta, F.; Borea, P.A. Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum. 2009, 60, 2880–2891. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Merighi, S.; Gessi, S.; Klotz, K.N.; Leung, E.; Baraldi, P.G.; Cacciari, B.; Romagnoli, R.; Spalluto, G.; Borea, P.A. [3H]MRE 3008F20: A novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol. Pharmacol. 2000, 57, 968–975. [Google Scholar] [PubMed]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and function of A2A and A3 adenosine receptors in patients with ankylosing spondylitis, psoriatic arthritis and rheumatoid arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| R5 | hA1AR a Ki (nM) (I%) e | hA2AAR b Ki (nM) | hA2BAR c IC50 (nM) (I%) e | hA3AR d Ki (nM) (I%) e | |
|---|---|---|---|---|---|
| 1 |  | 586 ± 37 | 594 ± 48 | >10,000 (21%) | >10,000 (8%) |
| 2 |  | 296 ± 22 | 64 ± 5 | >10,000 (15%) | >10,000 (18%) |
| 3 |  | 180 ± 15 | 58 ± 5 | 10,000 (26%) | 868 ± 82 |
| 4 |  | 1213 ± 114 | 237 ± 17 | >10,000 (11%) | 197 ± 16 |
| 5 |  | 2355 ± 213 | 326 ± 27 | >10,000 (7%) | 127 ± 15 |
| 6 |  | 2766 ± 249 | 137 ± 11 | >10,000 (10%) | 816 ± 74 |
| 7 |  | 345 ± 28 | 29 ± 3 | >10,000 (23%) | >10,000 (34%) |
| 8 |  | 638 ± 56 | 15.1 ± 1.3 | >10,000 (19%) | >10,000 (25%) |
| 9f |  | 4536 ± 312 | 279 ± 23 | >10,000 (38%) | 2679 ± 221 |
| 10 |  | 725 ± 67 | 187 ± 16 | >10,000 (22%) | >10,000 (22%) |
| 11 |  | 102 ± 9 | 8.62 ± 0.74 | >10,000 (13%) | >10,000 (15%) |
| 12 |  | 798 ± 72 | 92 ± 8 | >10,000 (26%) | >10,000 (17%) |
| 13 |  | 522 ± 47 | 37 ± 3 | >10,000 (29%) | >10,000 (33%) |
| 14 |  | 452 ± 38 | 18.3 ± 1.9 | >10,000 (17%) | 1492 ± 126 |
| 15 |  | 436 ± 36 | 10.8 ± 1.0 | >10,000 (16%) | >10,000 (23%) |
| 16 |  | >10,000 (31%) | 264 ± 24 | >10,000 (26%) | >10,000 (34%) |
| 17 |  | >10,000 (29%) | >10,000 (33%) | >10,000 (17%) | >10,000 (28%) |
| 18 |  | 524 ± 41 | 802 ± 73 | >10,000 (11%) | >10,000 (28%) |
| 19 |  | 365 ± 29 | 15.2 ± 1.7 | >10,000 (18%) | >10,000 (35%) |
| 20 |  | 152 ± 11 | 88 ± 9 | >10,000 (24%) | >10,000 (29%) |
| 21 |  | 247 ± 18 | 483 ± 34 | >10,000 (27%) | >10,000 (21%) |
| ZM 241385 | 188 ± 16 | 0.94 ± 0.07 | 51 ± 4 | 672 ± 51 | |
| Compounds | Potency IC50, nM | Intrinsic Activity | Pharmacological Behavior |
|---|---|---|---|
| 8 | 13.8 ± 1.2 | −44 ± 3 | Inverse agonist |
| 11 | 7.42 ± 0.68 | −52 ± 4 | Inverse agonist |
| 14 | 15.2 ± 1.3 | −51 ± 5 | Inverse agonist |
| 15 | 9.42 ± 0.87 | −67 ± 5 | Inverse agonist |
| 19 | 14.8 ± 1.4 | −64 ± 4 | Inverse agonist |
| ZM 241385 | 1.42 ± 0.11 | −48 ± 4 | Inverse agonist |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varano, F.; Catarzi, D.; Vigiani, E.; Vincenzi, F.; Pasquini, S.; Varani, K.; Colotta, V. Piperazine- and Piperidine-Containing Thiazolo[5,4-d]pyrimidine Derivatives as New Potent and Selective Adenosine A2A Receptor Inverse Agonists. Pharmaceuticals 2020, 13, 161. https://doi.org/10.3390/ph13080161
Varano F, Catarzi D, Vigiani E, Vincenzi F, Pasquini S, Varani K, Colotta V. Piperazine- and Piperidine-Containing Thiazolo[5,4-d]pyrimidine Derivatives as New Potent and Selective Adenosine A2A Receptor Inverse Agonists. Pharmaceuticals. 2020; 13(8):161. https://doi.org/10.3390/ph13080161
Chicago/Turabian StyleVarano, Flavia, Daniela Catarzi, Erica Vigiani, Fabrizio Vincenzi, Silvia Pasquini, Katia Varani, and Vittoria Colotta. 2020. "Piperazine- and Piperidine-Containing Thiazolo[5,4-d]pyrimidine Derivatives as New Potent and Selective Adenosine A2A Receptor Inverse Agonists" Pharmaceuticals 13, no. 8: 161. https://doi.org/10.3390/ph13080161