Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease




{kind=link}
Abstract
1. Introduction
2. Iron Dyshomeostasis is Associated with AD
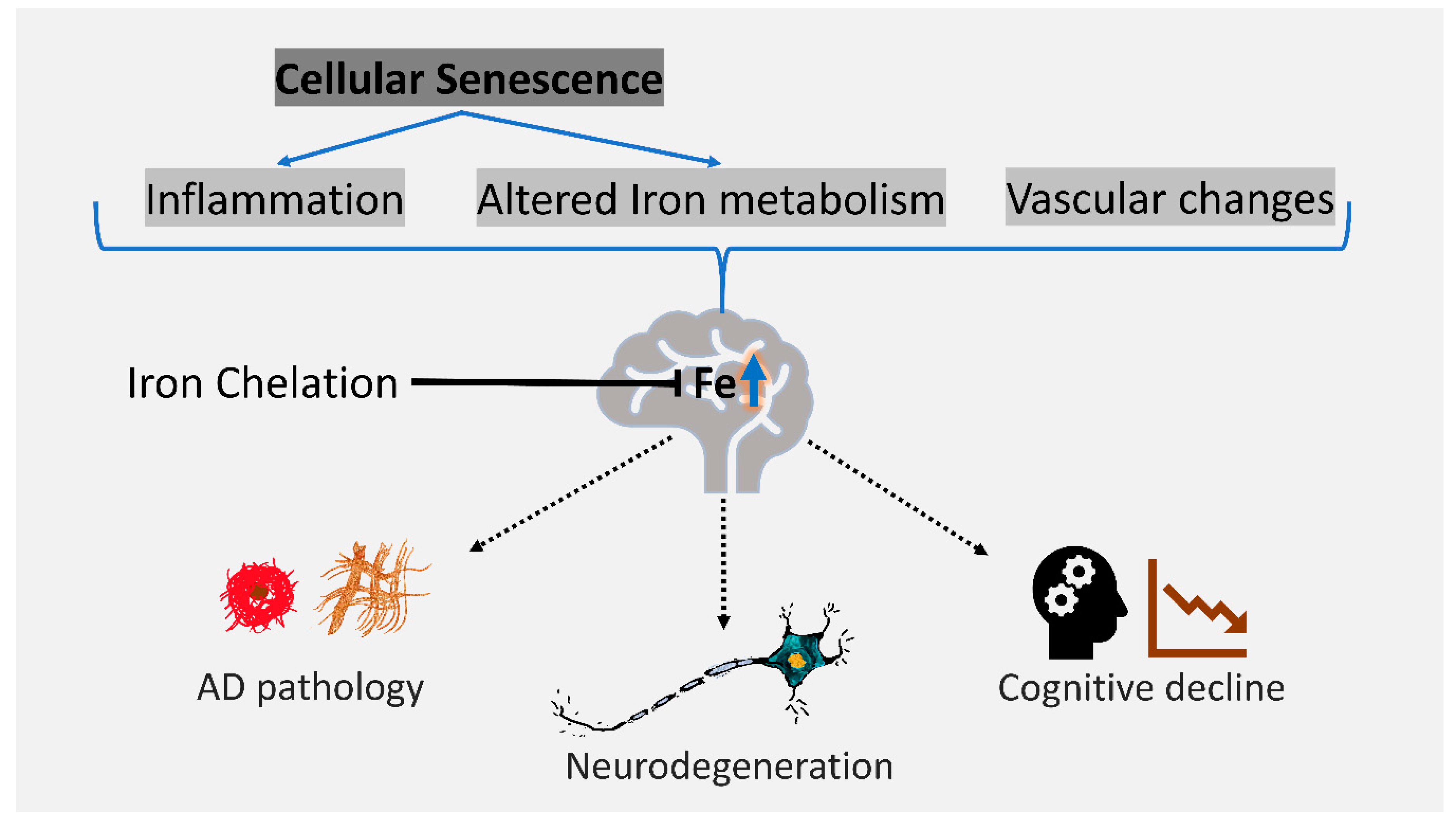
3. Cellular Senescence is Associated with AD and Iron Dyshomeostasis
4. Iron as a Therapeutic Target in AD
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nikseresht, S.; Bush, A.I.; Ayton, S. Treating Alzheimer’s disease by targeting iron. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Alborzinia, H.; Ignashkova, T.I.; Dejure, F.R.; Gendarme, M.; Theobald, J.; Wölfl, S.; Lindemann, R.K.; Reiling, J.H. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun. Biol. 2018, 1, 210. [Google Scholar] [CrossRef] [PubMed]
- Biogen and Eisai to Discontinue Phase 3 Engage and Emerge Trials of Aducanumab in Alzheimer’s Disease. Available online: http://investors.Biogen.Com/news-releases/news-release-details/biogen-and-eisai-discontinue-phase-3-engage-and-emerge-trials (accessed on 24 April 2019).
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2018, 133, 221–233. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. 1), 179–197. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Arab, N.T.T.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta 2017, 1864, 399–430. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Sultzer, D.; Mintz, J.; Holt, L.E.; Marx, P.; Phelan, C.K.; Marder, S.R. In vivo evaluation of brain iron in Alzheimer’s disease and normal subjects using MRI. Biol. Psychiatry 1994, 35, 480–487. [Google Scholar] [CrossRef]
- Bartzokis, G.; Tishler, T.A. MRI evaluation of basal ganglia ferritin iron and neurotoxicity in Alzheimer’s and Huntingon’s disease. Cell. Mol. Biol. 2000, 46, 821–833. [Google Scholar]
- Ding, B.; Chen, K.M.; Ling, H.W.; Sun, F.; Li, X.; Wan, T.; Chai, W.M.; Zhang, H.; Zhan, Y.; Guan, Y.J. Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J. Magn. Reson. Imaging 2009, 29, 793–798. [Google Scholar] [CrossRef]
- Pfefferbaum, A.; Adalsteinsson, E.; Rohlfing, T.; Sullivan, E.V. MRI estimates of brain iron concentration in normal aging: Comparison of field-dependent (FDRI) and phase (SWI) methods. NeuroImage 2009, 47, 493–500. [Google Scholar] [CrossRef]
- Bilgic, B.; Pfefferbaum, A.; Rohlfing, T.; Sullivan, E.V.; Adalsteinsson, E. MRI estimates of brain iron concentration in normal aging using quantitative susceptibility mapping. NeuroImage 2012, 59, 2625–2635. [Google Scholar] [CrossRef]
- Luo, Z.; Zhuang, X.; Kumar, D.; Wu, X.; Yue, C.; Han, C.; Lv, J. The correlation of hippocampal T2-mapping with neuropsychology test in patients with Alzheimer’s disease. PLoS ONE 2013, 8, e76203. [Google Scholar] [CrossRef] [PubMed]
- Langkammer, C.; Ropele, S.; Pirpamer, L.; Fazekas, F.; Schmidt, R. MRI for iron mapping in Alzheimer’s disease. Neuro Degener. Dis. 2014, 13, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, Y.; Rogers, J.T.; Wang, F. Perturbed iron distribution in Alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 42, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Ghadery, C.; Pirpamer, L.; Hofer, E.; Langkammer, C.; Petrovic, K.; Loitfelder, M.; Schwingenschuh, P.; Seiler, S.; Duering, M.; Jouvent, E.; et al. R2* mapping for brain iron: Associations with cognition in normal aging. Neurobiol. Aging 2015, 36, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Bulk, M.; Abdelmoula, W.M.; Nabuurs, R.J.A.; van der Graaf, L.M.; Mulders, C.W.H.; Mulder, A.A.; Jost, C.R.; Koster, A.J.; van Buchem, M.A.; Natte, R.; et al. Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early- and late-onset Alzheimer’s disease. Neurobiol. Aging 2018, 62, 231–242. [Google Scholar] [CrossRef]
- Bulk, M.; Kenkhuis, B.; van der Graaf, L.M.; Goeman, J.J.; Natte, R.; van der Weerd, L. Postmortem t2*- weighted MRI imaging of cortical iron reflects severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 65, 1125–1137. [Google Scholar] [CrossRef]
- Van Duijn, S.; Bulk, M.; van Duinen, S.G.; Nabuurs, R.J.A.; van Buchem, M.A.; van der Weerd, L.; Natte, R. Cortical iron reflects severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 60, 1533–1545. [Google Scholar] [CrossRef]
- Bulk, M.; van der Weerd, L.; Breimer, W.; Lebedev, N.; Webb, A.; Goeman, J.J.; Ward, R.J.; Huber, M.; Oosterkamp, T.H.; Bossoni, L. Quantitative comparison of different iron forms in the temporal cortex of Alzheimer patients and control subjects. Sci. Rep. 2018, 8, 6898. [Google Scholar] [CrossRef]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2019, 24. [Google Scholar] [CrossRef]
- Goodman, L. Alzheimer’s disease: A clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. J. Nerv. Ment. Dis. 1953, 118, 97–130. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Chebrolu, H.; Wekstein, D.R.; Schmitt, F.A.; Jicha, G.A.; Cooper, G.; Markesbery, W.R. Brain structural alterations before mild cognitive impairment. Neurology 2007, 68, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Meadowcroft, M.D.; Peters, D.G.; Dewal, R.P.; Connor, J.R.; Yang, Q.X. The effect of iron in MRI and transverse relaxation of amyloid-beta plaques in Alzheimer’s disease. NMR Biomed. 2015, 28, 297–305. [Google Scholar] [CrossRef]
- Everett, J.; Collingwood, J.F.; Tjendana-Tjhin, V.; Brooks, J.; Lermyte, F.; Plascencia-Villa, G.; Hands-Portman, I.; Dobson, J.; Perry, G.; Telling, N.D. Nanoscale synchrotron x-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale 2018, 10, 11782–11796. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Ponce, A.; Collingwood, J.F.; Arellano-Jimenez, M.J.; Zhu, X.; Rogers, J.T.; Betancourt, I.; Jose-Yacaman, M.; Perry, G. High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci. Rep. 2016, 6, 24873. [Google Scholar] [CrossRef]
- Telling, N.D.; Everett, J.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Gallagher, J.J.; Wang, J.; Hitchcock, A.P. Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer’s disease. Cell Chem. Biol. 2017, 24, 1205–1215. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Ghilardi, J.R.; Rogers, S.; DeMaster, E.; Allen, C.J.; Stimson, E.R.; Maggio, J.E. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J. Neurochem. 1993, 61, 1171–1174. [Google Scholar] [CrossRef]
- Schubert, D.; Chevion, M. The role of iron in beta amyloid toxicity. Biochem. Biophys. Res. Commun. 1995, 216, 702–707. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J. Biol. Inorg. Chem. 2004, 9, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef]
- Kwiatek-Majkusiak, J.; Dickson, D.W.; Tacik, P.; Aoki, N.; Tomasiuk, R.; Koziorowski, D.; Friedman, A. Relationships between typical histopathological hallmarks and the ferritin in the hippocampus from patients with Alzheimer’s disease. Acta Neurobiol. Exp. 2015, 75, 391–398. [Google Scholar]
- Gong, N.J.; Dibb, R.; Bulk, M.; van der Weerd, L.; Liu, C. Imaging beta amyloid aggregation and iron accumulation in Alzheimer’s disease using quantitative susceptibility mapping MRI. NeuroImage 2019, 191, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Tahirbegi, I.B.; Pardo, W.A.; Alvira, M.; Mir, M.; Samitier, J. Amyloid Abeta 42, a promoter of magnetite nanoparticle formation in Alzheimer’s disease. Nanotechnology 2016, 27, 465102. [Google Scholar] [CrossRef]
- Huang, X.; Cuajungco, M.P.; Atwood, C.S.; Hartshorn, M.A.; Tyndall, J.D.; Hanson, G.R.; Stokes, K.C.; Leopold, M.; Multhaup, G.; Goldstein, L.E.; et al. Cu(ii) potentiation of Alzheimer Abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 1999, 274, 37111–37116. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Rogers, J.T.; Leiter, L.M.; McPhee, J.; Cahill, C.M.; Zhan, S.S.; Potter, H.; Nilsson, L.N. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 1999, 274, 6421–6431. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type ii in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.H.; Klevanski, M.; Saar, M.; Muller, U.C. Roles of the amyloid precursor protein family in the peripheral nervous system. Mech. Dev. 2013, 130, 433–446. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. Apoe2, apoe3, and apoe4 differentially stimulate app transcription and abeta secretion. Cell 2017, 168, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Bodovitz, S.; Falduto, M.T.; Frail, D.E.; Klein, W.L. Iron levels modulate alpha-secretase cleavage of amyloid precursor protein. J. Neurochem. 1995, 64, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Camaschella, C. A potential pathogenetic role of iron in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 1548–1550. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. The Lancet. Neurology 2014, 13, 1045–1060. [Google Scholar] [PubMed]
- Yamamoto, A.; Shin, R.W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (iii) induces aggregation of hyperphosphorylated tau and its reduction to iron (ii) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xiong, S.; Xie, C.; Davies, P.; Markesbery, W.R. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimer’s Dis. 2004, 6, 659–671, discussion 673–681. [Google Scholar] [CrossRef]
- Chan, A.; Shea, T.B. Dietary and genetically-induced oxidative stress alter tau phosphorylation: Influence of folate and apolipoprotein e deficiency. J. Alzheimer’s Dis. 2006, 9, 399–405. [Google Scholar] [CrossRef]
- Amit, T.; Avramovich-Tirosh, Y.; Youdim, M.B.; Mandel, S. Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 2008, 22, 1296–1305. [Google Scholar] [CrossRef]
- Wang, D.; Hui, Y.; Peng, Y.; Tang, L.; Jin, J.; He, R.; Li, Y.; Zhang, S.; Li, L.; Zhou, Y.; et al. Overexpression of heme oxygenase 1 causes cognitive decline and affects pathways for tauopathy in mice. J. Alzheimer’s Dis. 2015, 43, 519–534. [Google Scholar] [CrossRef]
- Perry, G.; Nunomura, A.; Hirai, K.; Zhu, X.; Perez, M.; Avila, J.; Castellani, R.J.; Atwood, C.S.; Aliev, G.; Sayre, L.M.; et al. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free Radic. Biol. Med. 2002, 33, 1475–1479. [Google Scholar] [CrossRef]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Faux, N.G.; Bush, A.I.; Weiner, M.W.; Aisen, P.; Petersen, R.; Jack Jr, C.R.; Jagust, W.; Trojanowki, J.Q.; Toga, A.W. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by apoe. Nat. Commun. 2015, 6, 6760. [Google Scholar] [CrossRef] [PubMed]
- Diouf, I.; Fazlollahi, A.; Bush, A.I.; Ayton, S.; Alzheimer’s disease Neuroimaging, I. Cerebrospinal fluid ferritin levels predict brain hypometabolism in people with underlying beta-amyloid pathology. Neurobiol. Dis. 2019, 124, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Fazlollahi, A.; Bourgeat, P.; Raniga, P.; Ng, A.; Lim, Y.Y.; Diouf, I.; Farquharson, S.; Fripp, J.; Ames, D. Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain 2017, 140, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Choi, J.; Shendrik, I.; Peacocke, M.; Peehl, D.; Buttyan, R.; Ikeguchi, E.F.; Katz, A.E.; Benson, M.C. Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology 2000, 56, 160–166. [Google Scholar] [CrossRef]
- Te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis. Circulation 2002, 105, 1541. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein ccn1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of pdgf-aa. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by cd158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by abt263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Saint-Germain, E.; Mignacca, L.; Vernier, M.; Bobbala, D.; Ilangumaran, S.; Ferbeyre, G. Socs1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging 2017, 9, 2137–2162. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhang, N.; Ellerby, L.M.; Davalos, A.R.; Zeng, X.; Campisi, J.; Desprez, P.Y. Responses of human embryonic stem cells and their differentiated progeny to ionizing radiation. Biochem. Biophys. Res. Commun. 2012, 426, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; Garcia-Matas, S.; Rodriguez-Farre, E.; Sanfeliu, C.; Cristofol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 2019, 29, 1061–1077. [Google Scholar] [CrossRef]
- Tominaga, T.; Shimada, R.; Okada, Y.; Kawamata, T.; Kibayashi, K. Senescence-associated-beta-galactosidase staining following traumatic brain injury in the mouse cerebrum. PLoS ONE 2019, 14, e0213673. [Google Scholar] [CrossRef]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.J. CD36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018, 19, e45274. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Killilea, D.W.; Wong, S.L.; Cahaya, H.S.; Atamna, H.; Ames, B.N. Iron accumulation during cellular senescence. Ann. N. Y. Acad. Sci. 2004, 1019, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Konig, J.; Hohn, A.; Jung, T.; Grune, T. Reduced autophagy leads to an impaired ferritin turnover in senescent fibroblasts. Free Radic. Biol. Med. 2016, 101, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Smith, Z.M.; Francis, P.S.; Denoyer, D.; Meggyesy, P.M.; Fontaine, S.; Cater, M.A. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018, 16, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Altered processing of beta-amyloid in sh-sy5y cells induced by model senescent microglia. ACS Chem. Neurosci. 2018, 9, 3137–3152. [Google Scholar] [CrossRef] [PubMed]
- James, S.A.; Roberts, B.R.; Hare, D.J.; de Jonge, M.D.; Birchall, I.E.; Jenkins, N.L.; Cherny, R.A.; Bush, A.I.; McColl, G. Direct in vivo imaging of ferrous iron dyshomeostasis in ageing Caenorhabditis elegans. Chem. Sci. 2015, 6, 2952–2962. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Ferrous-glutathione coupling mediates ferroptosis and frailty in Caenorhabditis elegans. bioRxiv 2019. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Moreau, C.; Danel, V.; Devedjian, J.C.; Grolez, G.; Timmerman, K.; Laloux, C.; Petrault, M.; Gouel, F.; Jonneaux, A.; Dutheil, M.; et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal. 2018, 29, 742–748. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masaldan, S.; Belaidi, A.A.; Ayton, S.; Bush, A.I. Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals 2019, 12, 93. https://doi.org/10.3390/ph12020093
Masaldan S, Belaidi AA, Ayton S, Bush AI. Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals. 2019; 12(2):93. https://doi.org/10.3390/ph12020093
Chicago/Turabian StyleMasaldan, Shashank, Abdel Ali Belaidi, Scott Ayton, and Ashley I. Bush. 2019. "Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease" Pharmaceuticals 12, no. 2: 93. https://doi.org/10.3390/ph12020093
APA StyleMasaldan, S., Belaidi, A. A., Ayton, S., & Bush, A. I. (2019). Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals, 12(2), 93. https://doi.org/10.3390/ph12020093