Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects
by
Olivier Loréal
*, 
Thibault Cavey
,
François Robin
,
Moussa Kenawi
,
Pascal Guggenbuhl
and
Pierre Brissot
INSERM, Univ Rennes, INRA, CHU Rennes, Institut NUMECAN (Nutrition Metabolisms and Cancer), F-35033 Rennes, France
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2018, 11(4), 131; https://doi.org/10.3390/ph11040131
Submission received: 28 August 2018
/
Revised: 16 November 2018
/
Accepted: 19 November 2018
/
Published: 26 November 2018
(This article belongs to the Special Issue Iron as Therapeutic Targets in Human Diseases)
Abstract
:Genetic hemochromatosis is an iron overload disease that is mainly related to the C282Y mutation in the HFE gene. This gene controls the expression of hepcidin, a peptide secreted in plasma by the liver and regulates systemic iron distribution. Homozygous C282Y mutation induces hepcidin deficiency, leading to increased circulating transferrin saturation, and ultimately, iron accumulation in organs such as the liver, pancreas, heart, and bone. Iron in excess may induce or favor the development of complications such as cirrhosis, liver cancer, diabetes, heart failure, hypogonadism, but also complaints such as asthenia and disabling arthritis. Iron depletive treatment mainly consists of venesections that permit the removal of iron contained in red blood cells and the subsequent mobilization of stored iron in order to synthesize hemoglobin for new erythrocytes. It is highly efficient in removing excess iron and preventing most of the complications associated with excess iron in the body. However, this treatment does not target the biological mechanisms involved in the iron metabolism disturbance. New treatments based on the increase of hepcidin levels, by using hepcidin mimetics or inducers, or inhibitors of the iron export activity of ferroportin protein that is the target of hepcidin, if devoid of significant secondary effects, should be useful to better control iron parameters and symptoms, such as arthritis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Iron metabolism must be tightly controlled in order to avoid deleterious situations, including iron overload diseases, especially HFE-related hemochromatosis. In such conditions an iron depletive treatment is engaged in order to avoid the development of complications. However, iron depletive therapy targets only iron excess and alternative, possibly complementary, novel approaches are needed.
1. Normal Iron Metabolism
1.1. The Ferroportin/Hepcidin Duo Controls Systemic Iron Metabolism
Systemic iron metabolism is characterized by a continuous distribution of iron from plasma toward cells which require its presence for participating in biological functions, including oxygen transport and enzymatic processes [1]. Plasma iron must be continuously renewed. Plasma iron originates predominantly from macrophages that recycle iron from senescent erythrocytes during the erythrophagocytosis process. The erythropoietic cells contains 70% of total body iron. The macrophages provide schematically 19 mg out of the 20 mg of iron required from the plasma by cells every day. The second source of iron is digestive absorption. Iron is absorbed in two steps from the nutrients. The first step takes place at the apical pole of the enterocytes, non-heme and heme iron being transferred from nutrients to the enterocyte cytoplasm. The transfer of non-heme iron implicates DMT1 (divalent metal transporter 1) that takes up ferrous iron [2] after reduction by the ferrireductase dCytb [3]. Heme iron is taken up through a receptor that could be HCP1 (heme carrier protein1) (controversial) [4,5,6]. The second step of iron absorption is the transfer of iron from the cytoplasm toward the plasma through the basal membrane of the enterocyte. This process limits the iron entry into the body, and only 1–2 mg are transferred each day toward the plasma, whereas the remaining iron is transiently stored within the cytoplasm and will be lost during enterocyte desquamation. This selective process is the classical mucosal block [7]. Ferroportin is the iron export protein that allows iron egress from enterocytes (concerning both the non-heme and heme iron absorbed at the apical side) and macrophages toward the plasma [8,9,10]. Once transferred into the plasma, and after its oxidation by ferroxidase enzymes—ceruloplasmin [11] and/or hephaestin [12]—ferric iron is associated to transferrin, the protein that can link up to two iron atoms and delivers iron to cells. Cellular uptake of transferrin iron is mediated by the transferrin receptor 1 (TFR1) [13]. Thereafter, endocytosis of the complex permits the transport of iron into endocytic vesicles and subsequently, its transfer to the cytosol by DMT1, that is also expressed in endocytic vesicles, after oxidation of iron by STEAP3 (Six-Transmembrane Epithelial Antigen of the Prostate 3) [14]. Another transferrin receptor, transferrin receptor 2 (TFR2) is expressed by cells, especially hepatocytes, but its affinity coefficient for transferrin is very low [15] so that its function is related to iron sensing rather than iron transport [16].
Hepcidin is a small cysteine rich peptide [17,18,19], mainly expressed by hepatocytes and secreted in the plasma, that interacts with ferroportin [20], the only known protein involved in cellular iron export. An increase of plasma hepcidin level leads to a decrease of both digestive iron absorption and iron leakage by macrophages [21]. Hepcidin decreases plasma iron concentration and the saturation of transferrin by iron. Conversely, a decrease of hepcidin expression favors cell membrane expression of ferroportin, and, in turn, the iron release from cells into the plasma, thus, increasing the transferrin saturation by iron. The regulation of hepcidin expression plays a major role in the maintenance of iron homeostasis.
1.2. Regulation of Hepcidin Expression
Hepcidin expression is regulated by many factors, including iron status [19] and inflammation [19,22] that induce an upregulation of hepcidin expression, and hypoxia/erythropoiesis activity [23,24,25] that decrease hepcidin expression. Mechanisms related to the induction of hepcidin expression by iron status are mainly transcriptional.
The first iron-related mechanism that regulates hepcidin expression is reported to be linked to transferrin saturation level in plasma [26,27]. This pathway involves the HFE gene, located on the chromosome 6, that encodes a HLA like class I protein that is expressed on cell membrane in association with the ß2-microglobulin [28]. It has been reported that HFE protein may interact either with TFR1 [29,30] or TFR2. The mechanism potentially involved in such regulation is a decrease of the physical interaction between TFR1 and HFE proteins when transferrin saturation increases [31]. This could lead to the stimulation of a MAP (mitogen-activated protein kinase) signaling pathway that promotes the hepcidin transcription level [27]. However, while the HFE/TFR2 interaction has been documented in in vitro experiments, the in vivo relevance of these findings is questionable [32]. The increase of hepcidin expression promotes ferroportin degradation, and thus, reduces plasma iron concentration and transferrin saturation by iron (Figure 1).
The second regulatory mechanism implicates the bone morphogenetic protein/hemojuvelin/son of mothers against decapentaplegic homolog (BMP/HJV/SMAD) pathway [33,34,35,36]. When cell iron concentration increases, BMP6 and BMP2 proteins are produced and secreted by hepatocytes and more likely sinusoidal cells [37,38,39]. BMPs interact with BMP receptor proteins that are associated to HJV, acting as BMPs co-receptor. The interaction induces a phosphorylation of SMADs (Son of Mothers Against Decapentaplegic) 1, 5, 8 that are translocated toward the nucleus after association with SMAD4 in the nucleus [40,41]. Then, they interact with BMP-responsive element sequence within the hepcidin gene promoter and promote hepcidin transcription [35,36,42].
2. Pathophysiology of HFE Hemochromatosis
2.1. HFE Hemochromatosis
HFE hemochromatosis is a disease mainly related to homozygosity of the C282Y (p.Cys282Tyr) mutation in the HFE gene [45]. The p.Cys282Tyr mutation alters the structure of the HFE protein due to the substitution of a cysteine that is engaged in intra-molecular disulfide bounds, that play a role in the protein shape, by a threonine. Thus, the expression of HFE protein on cell membrane, as well as its interaction with the Beta2 microglobulin are altered [29]. Some exceptional private mutations in the HFE gene can also lead to hemochromatosis, when present either at the homozygous state or in association with the C282Y mutation [46]. Homozygosity for H63D (p.His63Asp) that is present at homozygous state in approximately 2% of the Caucasian population cannot by itself generate clinically significant iron excess, so that, when associated to iron overload, one must search for associated genetic or acquired associated factors that promote iron excess [47]. It must be pointed out that the penetrance of HFE-related hemochromatosis is very incomplete. In terms of clinical penetrance, some studies have estimated a prevalence of 25–60%, while a single study reported a prevalence of 28% in males and 1% in females [48]. However, in the same study the biochemical penetrance, assessed by increased ferritinemia, was much higher (82% and 55%, respectively) [48].
2.2. Pathophysiology of Iron Overload during HFE Hemochromatosis
In hemochromatosis, iron overload is a two-hit phenomenon.
The first hit (Figure 1), which is related to the structural change of the protein is a deficiency in hepcidin expression and secretion—meaning hepcidin deficiency—compared to the iron stores [22,26]. In other words, hepcidin expression is lower than expected when considering plasma transferrin saturation and body iron stores [49]. This hepcidin deficiency results from the mutation in the HFE gene that alters the efficacy of the transduction pathway regulating hepcidin expression. Consecutively, despite iron excess, ferroportin expression on cell membranes of enterocytes and macrophages remains elevated and favors an increase of both plasma iron concentration and transferrin iron saturation [20]. As previously mentioned, transferrin iron ingress into cells is modulated by the expression level of its receptor TFR1. In physiological situation, TFR1 expression on cell membranes is downregulated when cellular iron is in excess [50], in order to avoid cellular iron accumulation with subsequent toxicity, especially through the production of reactive oxygen species (ROS) [51,52]. The iron responsive element/iron regulatory protein system (IRE/IRP) regulates TFR1 and ferritin expression, adapting iron entry into the cell (TFR1) and the capacity of iron storage in cells (ferritin), to the variations of cellular iron content [50,53].
The second hit (Figure 1) involved in the development of iron overload in hemochromatosis results from the appearance of the non-transferrin-bound form of iron (NTBI) [54]. Indeed, transferrin saturation increase favors the presence of NTBI in plasma [55]. The NTBI is constituted of low molecular forms of iron linked to citrate or acetate [56]. The NTBI, in contrast to transferrin iron, constantly enters the cells, especially through the Zip14 transporter [57], even when they are already overloaded [58,59], whereas transferrin iron ingress is physiologically reduced due to the decrease of TFR1 on cell membrane [60,61]. The transporters involved in the uptake of NTBI are mainly expressed in the liver, the pancreas [57,62], and the heart, explaining that these organs are the primary targets of iron excess.
It is important to note that rare or very rare non-HFE mutations may also favor hepcidin deficiency. Homozygous and compound heterozygous mutations in the HAMP [63] or HJV [64] genes induce an early and severe iron overload disease (juvenile hemochromatosis) that is related to severe hepcidin deficiency with major complications that quickly impact well-being and life expectancy. In addition, mutations in the TFR2 gene induce an hepcidin deficiency that provokes a clinical iron overload phenotype which is in between juvenile hemochromatosis and the classical HFE-related hemochromatosis form [65].
2.3. Pathophysiology of Organ Damage in Hemochromatosis
Complications of HFE-related genetic hemochromatosis, and more globally of hemochromatosis related to hepcidin deficiency, include hepatic damage with the development of liver fibrosis, with the risks of cirrhosis and hepatocellular carcinoma, diabetes, and at a lesser degree, heart dysfunction, which are sources of morbidity and mortality [66,67]. The risk of hepatic fibrosis increases with the severity of iron overload [68], and it is recommended to perform a liver biopsy in patients exhibiting very high levels of ferritinemia [69]. This risk is associated with the presence of sideronecrotic lesions of hepatocytes [70], that likely corresponds to the recently identified ferroptotic cell death process [71,72], a new cell death pathway in cells containing high iron content. Whereas hepatocyte iron loading is the pathophysiological basic feature of iron overload in hemochromatosis, it is noteworthy that Kupffer cell iron load finally occurs in advanced iron hemochromatosis and also represents a risk for the development of hepatic damage [70]. The role of an iron-related induction of TGF (transforming growth factor)-beta in the development of fibrosis has been reported [73,74]. It should be underlined that hepatocellular carcinoma, that mostly develops in patients with a cirrhotic liver, can also be rarely found in non-cirrhotic patients which suggests the role of iron itself [75] and/or additional cofactors in the development of hepatocellular carcinoma (HCC) (see below). Finally, patients exhibiting cirrhosis before iron depletive treatment still present a risk for developing HCC even despite completion of iron depletive treatment [67].
Other complications may include hypogonadism—mostly in juvenile hemochromatosis—and much more frequently osteoporosis and arthritis that have a strong impact on the quality of life. Arthritis is characterized by absence of systemic inflammation and sometimes presence of calcium pyrophosphate crystals in the synovial fluids and visible on X-rays (review in [76]).
The ability of excessive iron to generate oxygen reactive species (ROS) (Figure 2) through the Haber–Weiss and Fenton reactions is strongly involved in the development of tissue lesions [77]. Indeed, ROS induces peroxidation that alters lipids, proteins, and DNA, generating dysfunctions of organelles, including mitochondria and cells, thus leading to tissue and organ damage [78,79,80]. One of the abnormal forms of NTBI, called Labile Plasma iron-LPI (or reactive plasma iron), is found in plasma when transferrin saturation reaches 80% [81]. This iron species is highly reactive and participates strongly in oxidative stress. Labile plasma iron is considered as a major determinant in the development of organ damage during hemochromatosis [82].
In addition to the typical disease causing mutations associated with hemochromatosis, additional genetic modifiers have recently been described to elucidate in part, the large disparity in disease manifestations in patients. In this context, the importance of gene polymorphisms, e.g., GNPAT (glyceronephosphate O-acyltransferase) remains controversial and awaits additional confirmatory studies [83,84,85,86,87,88]. Alcohol consumption [68,89], non-alcoholic fatty liver disease [90], and viral hepatitis [91,92] may also be involved in disease penetrance, as well as a polymorphism in the PCSK7 [93] or PNPLA3 [94] genes.
3. Iron Is Presently the Main Therapeutic Target in HFE-Related Hemochromatosis
It is well known that iron depletion is a treatment of choice for hemochromatosis patients [95]. Indeed, when performed in patients prior to severe complications, the iron depletive treatment restores normal life expectancy, meaning that hepatic, pancreatic, and cardiac dysfunctions can be fully prevented [67]. In addition, partial regression of liver fibrosis after completion of iron removal has been reported [96].
3.1. Venesections Are the Mainstay Treatment for Iron Removal
The principle of repeated venesections [47,95] (Figure 3) is to remove red blood cells that are known to be very rich in iron, as part of hemoglobin. Erythropoiesis is stimulated to compensate hematocrit loss, and iron is taken up from the plasma by erythroblasts to produce new erythrocytes. In order to maintain enough plasma iron for the stimulated erythropoiesis, iron is released from macrophages and enterocytes but also from iron storage cells, including parenchymal cells such as hepatocytes. Removing iron at a faster rate than that excessively reaccumulated by intestinal absorption is critical to negative iron balance, but determining this rate prior to starting depletive treatment is difficult due to individual differences between hemochromatosis patients. It should also be recalled that hepcidin synthesis can be further suppressed by increasing erythropoiesis as a consequence of erythroferrone production. In practice, the depletive treatment includes two successive phases. The first one, called induction phase, aims at totally removing the iron excess present at the time of diagnosis. It usually consists of weekly venesections (≤7.5 mL/kg body weight per venesection). Once excess iron has been removed, the second phase, called maintenance therapy, aims to avoid recurrent iron overload using lifelong venesections, performed every 1–4 months.
3.2. Use of Chelation Therapy
Another way to remove iron is the use of iron chelators that promote iron mobilization and excretion. However, in contrast to venesections, these drugs, including mainly today new oral iron-chelators, may have potential side effects [97]. Therefore during genetic hemochromatosis, oral chelation is only used, and as an off-label drug, in rare situations including cardiac failure, recent cerebral ischemic stroke, venous access problems limiting the possibility of venesections, or psychological intolerance to venesections.
3.3. Biochemical Follow-Up of Venesection Therapy
Three main parameters are classically followed: plasma ferritin and transferrin saturation levels that reflect the efficacy of iron depletive treatment, whereas hemoglobin levels reflect the tolerance of venesections. During the induction phase ferritinemia—that reliably reflects the amount of iron excess in hemochromatosis (provided other frequent acquired causes of hyperferritinemia have been excluded, such as inflammation, alcoholism, cytolysis or metabolic syndrome; genetic hyperferritinemia, as seen in the ferritin-cataract syndrome, is much rarer)—is the first parameter that decreases. This decrease is only slowly progressive, especially in patients exhibiting a severe iron load phenotype. Conversely, the decrease of transferrin saturation levels, that reflect plasma iron bioavailability, is a very late event. Normalization of transferrin saturation occurs only when the induction treatment is in its final phase. It is noteworthy that during hemochromatosis, despite the impressive frequency of venesections, hemoglobin levels remain stable due to the high capacity of cellular iron release by ferroportin hyperactivity, related to hepcidin deficiency. However, transient hypoxia and relative iron deficiency occurring after venesection, together with increased erythropoiesis, tend to decrease hepcidin plasma levels, thus contributing to further enhance iron entry into the plasma [98,99]. When considering NTBI during the induction therapy, its plasma concentration decreases parallel to transferrin saturation levels [55]. Altogether, these data suggest that induction therapy must be fully completed to remove iron excess, but also to prevent the appearance of toxic forms of iron, including plasma NTBI.
4. Iron Removal Is not the Unique Therapeutic Target in HFE-Related Hemochromatosis
4.1. Other Preventive Actions to Avoid Iron Overload Complications
Besides iron removal, it is important to prevent associated causes of organ damage, as mentioned previously. This holds especially true for hepatic lesions knowing the impact of excessive alcohol intake and viral hepatitis as cofactors of liver fibrosis, cirrhosis, and hepatocellular carcinoma. Moreover, NAFLD (Non Alcoholic Fatty Liver Disease) with the risk of NASH (Non-Alcoholic Steato-Hepatitis) represents an additional risk for hemochromatosis patients. The practitioner must complete the iron removal approach by nutritional recommendations on alcohol and glucido-lipidic regimens [47,95]. Moreover, avoiding the occurrence of viral B hepatitis through vaccination is critical. In addition, tea [100] has been demonstrated to limit iron absorption, and therefore, could be used as adjuvant to avoid recurrence of excessive iron stores after completion of iron removal. In the same way, the use of therapeutic oral calcium channel blockers has also been proposed due to its positive impact on urinary iron excretion related to DMT1 activity increase in the kidney [101]. The use of proton-pump inhibitors could also represent an interesting adjunct [102].
4.2. The Development of a Pathophysiological Treatment Is a Major Goal
Iron removal is a treatment that only cures iron excess, but clearly does not correct the hepcidin deficiency that generates iron metabolism disturbances, and some clinical observations do suggest that the removal of iron excess is not sufficient to treat hemochromatosis. Considering arthropathy, it is noteworthy that, despite a well-conducted iron depletive treatment, the symptomatology may persist and even become more severe [76]. This suggests that: (i) synovial iron deposition [103] may represent an inaccessible compartment for iron depletion by venesections because synovial fluid is not contiguous with serum; (ii) synovitis, that has been documented in hemochromatosis [104,105] is irreversible despite effective iron depletion; and/or (iii) hepcidin deficiency leading to iron excess could be involved directly in symptomatic arthropathy and/or (iv) the mutations of the HFE gene, that encodes an HLA-like class I protein, could be involved directly in disease expression [106].
Considering the biochemical follow-up of patients, it is noteworthy that transferrin saturation levels may be found to be frequently increased during the maintenance therapeutic period, despite the absence of increased body iron stores. This could contribute to the appearance of clinical manifestations such as fatigue and arthropathy. It should be emphasized that, during the maintenance period, serum hepcidin levels are even lower than in iron overloaded patients [98,107]. Such hypohepcidinemia is expected to favor transferrin saturation increase and the appearance of NTBI that could participate in the arthritis of hemochromatosis patients.
Targeted treatment in the setting of hepcidin deficiency itself could represent an efficient way, as suggested by clinical observations obtained in hemochromatosis patients that have been transplanted for hepatocellular carcinoma [108]. The development of substitutive hepcidin treatment and/or of drugs stimulating hepcidin expression is under development and could be useful to complete the therapeutic arsenal, as suggested by results obtained with mini-hepcidins [109] and BMP known to induce hepcidin expression [110]. Clinical trials with hepcidin supplementation are ongoing (Clinical.trial.gov). Using antisense oligonucleotides for increasing hepcidin synthesis [111] or ferroportin antagonists [112] represents also further interesting innovative approaches. These different treatments could be especially indicated as an adjunct to venesections during the induction phase, and possibly as replacing venesections during maintenance therapy. Considering the overall safety and efficacy of the venesections, every putative treatment of hemochromatosis targeting iron metabolism must be without side-effects and easy to follow, suggesting especially that oral treatments providing exogenous hepcidin or inducing endogenous hepcidin synthesis might be preferable.
5. Conclusions
Iron remains the major therapeutic target during HFE-related hemochromatosis. However, targeting directly the iron stores by iron depletive treatment does not correct the pathophysiological defect of the disease. The development of treatments aimed at restoring iron homeostasis, especially by correcting hepcidin deficiency, could be useful to optimize the treatment, especially during the maintenance phase by controlling transferrin saturation with hopefully a favorable impact on the arthritis of hemochromatosis patients.
Funding
This research received no external funding.
Acknowledgments
The authors thank the Associations Fer-Métaux Essentiels-Recherche-Santé (AFeMERS), Fédération Française des Associations de Malades de l’Hémochromatose (FFAMH) and Association Hémochromatose Ouest for their supports.
Conflicts of Interest
The authors have no conflict of interest regarding this work.
References
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, G.; Di Sabatino, A.; Pasini, A.; Ubezio, C.; Costanzo, F.; Grataroli, D.; Masotti, M.; Alvisi, C.; Corazza, G.R. Intestinal expression of genes implicated in iron absorption and their regulation by hepcidin. Clin. Nutr. Edinb. Scotl. 2017, 36, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, T.H.; Pirzio-Biroli, G.; Finch, C.A. Iron absorption. I. Factors influencing absorption. J. Lab. Clin. Med. 1958, 51, 24–36. [Google Scholar] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [PubMed]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar] [PubMed]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Roetto, A.; Cali, A.; De Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 2000, 25, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Cox, T.M. Co-localization of the mammalian hemochromatosis gene product (HFE) and a newly identified transferrin receptor (TFR2) in intestinal tissue and cells. J. Histochem. Cytochem. 2003, 51, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Neitz, S.; Magert, H.J.; Schulz, A.; Forssmann, W.G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loreal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, A New Iron Regulatory Peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrke, S.G.; Kulaksiz, H.; Herrmann, T.; Riedel, H.D.; Bents, K.; Veltkamp, C.; Stremmel, W. Expression of hepcidin in hereditary hemochromatosis: Evidence for a regulation in response to serum transferrin saturation and non-transferrin-bound iron. Blood 2003, 102, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Ramey, G.; Deschemin, J.C.; Vaulont, S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009, 94, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feder, J.N.; Tsuchihashi, Z.; Irrinki, A.; Lee, V.K.; Mapa, F.A.; Morikang, E.; Prass, C.E.; Starnes, S.M.; Wolff, R.K.; Parkkila, S.; et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J. Biol. Chem. 1997, 272, 14025–14028. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Penny, D.M.; Irrinki, A.; Lee, V.K.; Lebron, J.A.; Watson, N.; Tsuchihashi, Z.; Sigal, E.; Bjorkman, P.J.; Schatzman, R.C. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc. Natl. Acad. Sci. USA 1998, 95, 1472–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates HFE-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.; Andrews, N.C. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 2006, 281, 28494–28498. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Fleming, M.D. Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am. J. Hematol. 2012, 87, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 2009, 87, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Island, M.L.; Jouanolle, A.M.; Mosser, A.; Deugnier, Y.; David, V.; Brissot, P.; Loreal, O. A new mutation in the hepcidin promoter impairs its BMP response and contributes to a severe phenotype in HFE related hemochromatosis. Haematologica 2009, 94, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.-Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Rausa, M.; Pagani, A.; Nai, A.; Campanella, A.; Gilberti, M.E.; Apostoli, P.; Camaschella, C.; Silvestri, L. Bmp6 expression in murine liver non parenchymal cells: A mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS ONE 2015, 10, e0122696. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.-S.; Olsavszky, V.; Ulbrich, F.; Sticht, C.; Demory, A.; Leibing, T.; Henzler, T.; Meyer, M.; Zierow, J.; Schneider, S.; et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017, 129, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Meynard, D.; Monnier, A.; Darnaud, V.; Bouvet, R.; Wang, R.H.; Deng, C.; Vaulont, S.; Mosser, J.; Coppin, H.; et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 2008, 112, 1503–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verga Falzacappa, M.V.; Casanovas, G.; Hentze, M.W.; Muckenthaler, M.U. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. 2008, 86, 531–540. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins HFE, TFR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Iron regulation of hepcidin through HFE and HJV: Common or distinct pathways? Hepatology 2015, 62, 1922–1923. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Loreal, O. Iron metabolism and related genetic diseases: A cleared land, keeping mysteries. J. Hepatol 2016, 64, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdi-Roze, H.; Beaumont-Epinette, M.P.; Ben Ali, Z.; Le Lan, C.; Loustaud-Ratti, V.; Causse, X.; Loreal, O.; Deugnier, Y.; Brissot, P.; Jouanolle, A.M.; et al. Rare HFE variants are the most frequent cause of hemochromatosis in non-c282y homozygous patients with hemochromatosis. Am. J. Hematol. 2016, 91, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primer 2018, 4, 18016. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.J.; Gurrin, L.C.; Constantine, C.C.; Osborne, N.J.; Delatycki, M.B.; Nicoll, A.J.; McLaren, C.E.; Bahlo, M.; Nisselle, A.E.; Vulpe, C.D.; et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N. Engl. J. Med. 2008, 358, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, B.A.C.; Laarakkers, C.M.M.; Klaver, S.M.; Jacobs, E.M.G.; van Tits, L.J.H.; Janssen, M.C.H.; Swinkels, D.W. Serum hepcidin levels are innately low in HFE-related haemochromatosis but differ between C282Y-homozygotes with elevated and normal ferritin levels. Br. J. Haematol. 2008, 142, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Cellular iron metabolism. Kidney Int. Suppl. 1999, 69, S2–S11. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Lok, C.N. The transferrin receptor: Role in health and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1111–1137. [Google Scholar] [CrossRef]
- Arosio, P.; Carmona, F.; Gozzelino, R.; Maccarinelli, F.; Poli, M. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem. J. 2015, 472, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, C.; Peto, T.E. Non-transferrin plasma iron. Br. J. Haematol 1987, 66, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Loreal, O.; Gosriwatana, I.; Guyader, D.; Porter, J.; Brissot, P.; Hider, R.C. Determination of non-transferrin-bound iron in genetic hemochromatosis using a new HPLC-based method. J. Hepatol. 2000, 32, 727–733. [Google Scholar] [CrossRef]
- Grootveld, M.; Bell, J.D.; Halliwell, B.; Aruoma, O.I.; Bomford, A.; Sadler, P.J. Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy. J. Biol. Chem. 1989, 264, 4417–4422. [Google Scholar] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissot, P.; Wright, T.L.; Ma, W.L.; Weisiger, R.A. Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J. Clin. Investig. 1985, 76, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Bolder, U.; Schteingart, C.D.; Arnaud, J.; Hofmann, A.F. Intestinal absorption and enterohepatic cycling of biliary iron originating from plasma non-transferrin-bound iron in rats. Hepatology 1997, 25, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, N.; Lescoat, G.; Sciot, R.; Moirand, R.; Jego, P.; Leroyer, P.; Brissot, P. Regulation of ferritin and transferrin receptor expression by iron in human hepatocyte cultures. J. Hepatol. 1993, 18, 301–312. [Google Scholar] [CrossRef]
- Sciot, R.; Verhoeven, G.; Van Eyken, P.; Cailleau, J.; Desmet, V.J. Transferrin receptor expression in rat liver: Immunohistochemical and biochemical analysis of the effect of age and iron storage. Hepatology 1990, 11, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Jenkitkasemwong, S.; Wang, C.Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003, 33, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Samuels, M.E.; Ludwig, E.H.; MacDonald, M.L.; Franchini, P.L.; Dube, M.P.; Andres, L.; MacFarlane, J.; Sakellaropoulos, N.; Politou, M.; et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 2004, 36, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Cunat, S.; Beaumont-Epinette, M.-P.; Kannengiesser, C.; Causse, X.; Sauvion, S.; Pouliquen, B.; Deugnier, Y.; David, V.; Loréal, O.; et al. Variable age of onset and clinical severity in transferrin receptor 2 related haemochromatosis: Novel observations. Br. J. Haematol. 2013, 162, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Sonnenberg, A.; Stremmel, W.; Trampisch, H.J.; Strohmeyer, G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med. 1985, 313, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Purschel, A.; Stremmel, W.; Haussinger, D.; Strohmeyer, G. Long-term survival in patients with hereditary hemochromatosis [see comments]. Gastroenterology 1996, 110, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Loreal, O.; Deugnier, Y.; Moirand, R.; Lauvin, L.; Guyader, D.; Jouanolle, H.; Turlin, B.; Lescoat, G.; Brissot, P. Liver fibrosis in genetic hemochromatosis. Respective roles of iron and non-iron-related factors in 127 homozygous patients. J. Hepatol. 1992, 16, 122–127. [Google Scholar] [PubMed]
- Guyader, D.; Jacquelinet, C.; Moirand, R.; Turlin, B.; Mendler, M.H.; Chaperon, J.; David, V.; Brissot, P.; Adams, P.; Deugnier, Y. Noninvasive prediction of fibrosis in C282Y homozygous hemochromatosis. Gastroenterology 1998, 115, 929–936. [Google Scholar] [CrossRef]
- Deugnier, Y.M.; Loreal, O.; Turlin, B.; Guyader, D.; Jouanolle, H.; Moirand, R.; Jacquelinet, C.; Brissot, P. Liver pathology in genetic hemochromatosis: A review of 135 homozygous cases and their bioclinical correlations. Gastroenterology 1992, 102, 2050–2059. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Houglum, K.; Bedossa, P.; Chojkier, M. TGF-beta and collagen-alpha 1 (I) gene expression are increased in hepatic acinar zone 1 of rats with iron overload. Am. J. Physiol. 1994, 267, G908–G913. [Google Scholar] [CrossRef] [PubMed]
- Gualdi, R.; Casalgrandi, G.; Montosi, G.; Ventura, E.; Pietrangelo, A. Excess iron into hepatocytes is required for activation of collagen type I gene during experimental siderosis. Gastroenterology 1994, 107, 1118–1124. [Google Scholar] [CrossRef]
- Deugnier, Y.M.; Guyader, D.; Crantock, L.; Lopez, J.M.; Turlin, B.; Yaouanq, J.; Jouanolle, H.; Campion, J.P.; Launois, B.; Halliday, J.W.; et al. Primary liver cancer in genetic hemochromatosis: A clinical, pathological, and pathogenetic study of 54 cases. Gastroenterology 1993, 104, 228–234. [Google Scholar] [CrossRef]
- Guggenbuhl, P.; Brissot, P.; Loreal, O. Miscellaneous non-inflammatory musculoskeletal conditions. Haemochromatosis: The bone and the joint. Best Pract. Res. Clin. Rheumatol 2011, 25, 6649–6664. [Google Scholar] [CrossRef]
- Wardman, P.; Candeias, L.P. Fenton chemistry: An introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Britton, R.S. Hepatic injury in chronic iron overload. Role of lipid peroxidation. Chem. Biol. Interact. 1989, 70, 183–226. [Google Scholar] [CrossRef]
- Bacon, B.R.; Tavill, A.S.; Brittenham, G.M.; Park, C.H.; Recknagel, R.O. Hepatic lipid peroxidation in vivo in rats with chronic iron overload. J. Clin. Investig. 1983, 71, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; O’Neill, R.; Britton, R.S. Hepatic mitochondrial energy production in rats with chronic iron overload. Gastroenterology 1993, 105, 1134–1140. [Google Scholar] [CrossRef]
- Le Lan, C.; Loreal, O.; Cohen, T.; Ropert, M.; Glickstein, H.; Laine, F.; Pouchard, M.; Deugnier, Y.; Le Treut, A.; Breuer, W.; et al. Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood 2005, 105, 4527–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabantchik, Z.I.; Breuer, W.; Zanninelli, G.; Cianciulli, P. LPI-labile plasma iron in iron overload. Best Pr. Res. Clin. Haematol 2005, 18, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Milet, J.; Dehais, V.; Bourgain, C.; Jouanolle, A.M.; Mosser, A.; Perrin, M.; Morcet, J.; Brissot, P.; David, V.; Deugnier, Y.; et al. Common variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance. Am. J. Hum. Genet. 2007, 81, 799–807. [Google Scholar] [CrossRef] [PubMed]
- De Tayrac, M.; Roth, M.-P.; Jouanolle, A.-M.; Coppin, H.; le Gac, G.; Piperno, A.; Férec, C.; Pelucchi, S.; Scotet, V.; Bardou-Jacquet, E.; et al. Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis. J. Hepatol. 2015, 62, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, C.E.; Emond, M.J.; Subramaniam, V.N.; Phatak, P.D.; Barton, J.C.; Adams, P.C.; Goh, J.B.; McDonald, C.J.; Powell, L.W.; Gurrin, L.C.; et al. Exome sequencing in HFE C282Y homozygous men with extreme phenotypes identifies a GNPAT variant associated with severe iron overload. Hepatology 2015, 62, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Tchernitchko, D.; Scotet, V.; Lefebvre, T.; L’Hostis, C.; Gourlaouen, I.; Merour, M.-C.; Rebah, K.; Peoc’h, K.; Assari, S.; Ferec, C.; et al. GNPAT polymorphism rs11558492 is not associated with increased severity in a large cohort of HFE p.Cys282Tyr homozygous patients. Hepatology 2017, 65, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Levstik, A.; Stuart, A.; Adams, P.C. GNPAT variant (D519G) is not associated with an elevated serum ferritin or iron removed by phlebotomy in patients referred for C282Y-linked hemochromatosis. Ann. Hepatol. 2016, 15, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Greni, F.; Valenti, L.; Mariani, R.; Pelloni, I.; Rametta, R.; Busti, F.; Ravasi, G.; Girelli, D.; Fargion, S.; Galimberti, S.; et al. GNPAT rs11558492 is not a Major Modifier of Iron Status: Study of Italian Hemochromatosis Patients and Blood Donors. Ann. Hepatol. 2017, 16, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, L.M.; Dixon, J.L.; Purdie, D.M.; Powell, L.W.; Crawford, D.H.G. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 2002, 122, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Bhattacharya, R.; Lindor, K.D.; Chalasani, N.; Raaka, S.; Heathcote, E.J.; Miskovsky, E.; Shaffer, E.; Rulyak, S.J.; Kowdley, K.V. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology 2007, 46, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Vegetti, A.; Saitta, C.; Ferrara, F.; Corradini, E.; Raffa, G.; Pietrangelo, A.; Raimondo, G. Hepatitis B virus DNA integration in tumour tissue of a non-cirrhotic HFE-haemochromatosis patient with hepatocellular carcinoma. J. Hepatol. 2013, 58, 190–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwakaran, H.H.; Befeler, A.S.; Britton, R.S.; Brunt, E.M.; Bacon, B.R. Accelerated hepatic fibrosis in patients with combined hereditary hemochromatosis and chronic hepatitis C infection. J. Hepatol. 2002, 36, 687–691. [Google Scholar] [CrossRef]
- Stickel, F.; Buch, S.; Zoller, H.; Hultcrantz, R.; Gallati, S.; Österreicher, C.; Finkenstedt, A.; Stadlmayr, A.; Aigner, E.; Sahinbegovic, E.; et al. Evaluation of genome-wide loci of iron metabolism in hereditary hemochromatosis identifies PCSK7 as a host risk factor of liver cirrhosis. Hum. Mol. Genet. 2014, 23, 3883–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, L.; Maggioni, P.; Piperno, A.; Rametta, R.; Pelucchi, S.; Mariani, R.; Dongiovanni, P.; Fracanzani, A.L.; Fargion, S. Patatin-like phospholipase domain containing-3 gene I148M polymorphism, steatosis, and liver damage in hereditary hemochromatosis. World J. Gastroenterol. 2012, 18, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.; Altes, A.; Brissot, P.; Butzeck, B.; Cabantchik, I.; Cançado, R.; Distante, S.; Evans, P.; Evans, R.; Ganz, T.; et al. Contributors and Hemochromatosis International Taskforce Therapeutic recommendations in HFE hemochromatosis for p.Cys282Tyr (C282Y/C282Y) homozygous genotype. Hepatol. Int. 2018, 12, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Falize, L.; Guillygomarc’h, A.; Perrin, M.; Laine, F.; Guyader, D.; Brissot, P.; Turlin, B.; Deugnier, Y. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: A study of 36 cases. Hepatology 2006, 44, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1779. [Google Scholar] [CrossRef] [PubMed]
- Rombout-Sestrienkova, E.; Koek, G.H.; Neslo, R.; van Kraaij, M.; Menheere, P.P.; Masclee, A.; Swinkels, D.W. Course of iron parameters in HFE-hemochromatosis patients during initial treatment with erythrocytapheresis compared to phlebotomy. J. Clin. Apheresis 2016, 31, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E.; Schlumpf, K.S.; Wright, D.J.; Johnson, B.; Glynn, S.A.; Busch, M.P.; Olbina, G.; Westerman, M.; Nemeth, E.; Ganz, T. NHLBI Retrovirus Epidemiology Donor Study-II (REDS-II) Hepcidin level predicts hemoglobin concentration in individuals undergoing repeated phlebotomy. Haematologica 2013, 98, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Kaltwasser, J.P.; Werner, E.; Schalk, K.; Hansen, C.; Gottschalk, R.; Seidl, C. Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis. Gut 1998, 43, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwiczek, S.; Theurl, I.; Muckenthaler, M.U.; Jakab, M.; Mair, S.M.; Theurl, M.; Kiss, J.; Paulmichl, M.; Hentze, M.W.; Ritter, M.; et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat. Med. 2007, 13, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Vanclooster, A.; van Deursen, C.; Jaspers, R.; Cassiman, D.; Koek, G. Proton Pump Inhibitors Decrease Phlebotomy Need in HFE Hemochromatosis: Double-Blind Randomized Placebo-Controlled Trial. Gastroenterology 2017, 153, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, H.R. Articular cartilage in the degenerative arthropathy of hemochromatosis. Arthritis Rheum. 1982, 25, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiland, G.R.; Aigner, E.; Dallos, T.; Sahinbegovic, E.; Krenn, V.; Thaler, C.; Weiss, G.; Distler, J.H.; Datz, C.; Schett, G.; et al. Synovial immunopathology in haemochromatosis arthropathy. Ann. Rheum. Dis. 2010, 69, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Carroll, G.J.; Sharma, G.; Upadhyay, A.; Jazayeri, J.A. Ferritin concentrations in synovial fluid are higher in osteoarthritis patients with HFE gene mutations (C282Y or H63D). Scand. J. Rheumatol. 2010, 39, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Chung, J.W.; Lapointe, R.; Santos, M.M. The hemochromatosis protein HFE 20 years later: An emerging role in antigen presentation and in the immune system. Immun. Inflamm. Dis. 2017, 5, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Lainé, F.; Guggenbuhl, P.; Morcet, J.; Jézéquel, C.; Guyader, D.; Moirand, R.; Deugnier, Y. Worse Outcomes of Patients with HFE Hemochromatosis With Persistent Increases in Transferrin Saturation During Maintenance Therapy. Clin. Gastroenterol. Hepatol. 2017, 15, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Philip, J.; Lorho, R.; Ropert, M.; Latournerie, M.; Houssel-Debry, P.; Guyader, D.; Loreal, O.; Boudjema, K.; Brissot, P. Liver transplantation normalizes serum hepcidin level and cures iron metabolism alterations in HFE hemochromatosis. Hepatology 2014, 59, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corradini, E.; Schmidt, P.J.; Meynard, D.; Garuti, C.; Montosi, G.; Chen, S.; Vukicevic, S.; Pietrangelo, A.; Lin, H.Y.; Babitt, J.L. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in HFE knockout mice. Gastroenterology 2010, 139, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.L.; Biswas, K.; Rottman, J.; Allen, J.R.; Long, J.; Miranda, L.P.; Winters, A.; Arvedson, T.L. Identification of Antibody and Small Molecule Antagonists of Ferroportin-Hepcidin Interaction. Front. Pharmacol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed]
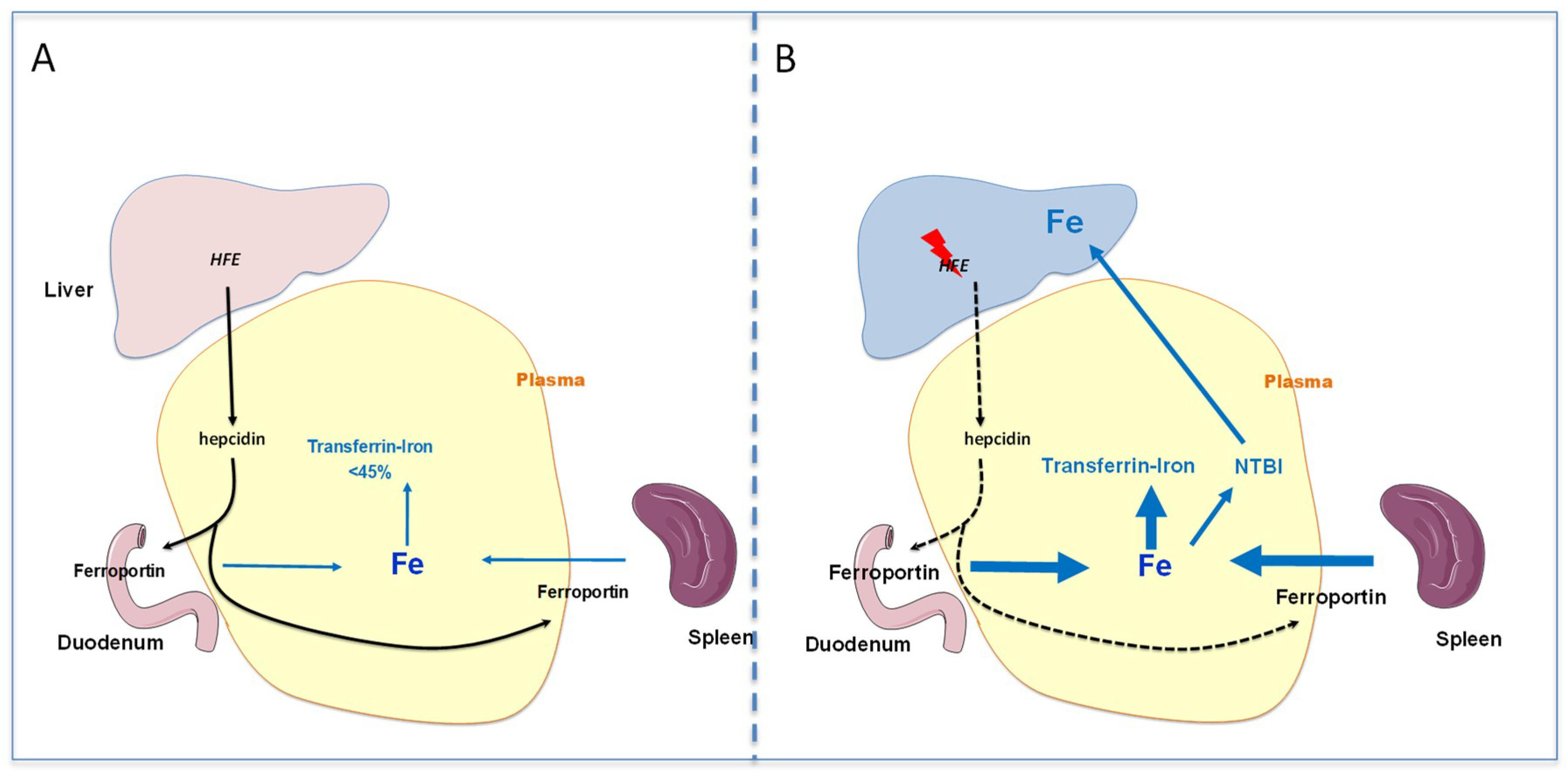
Figure 1.
Schematic representation of the pathophysiological mechanisms leading to the development of iron overload during HFE-related hemochromatosis. (A): Normal situation with adequate HFE signaling allowing control of transferrin saturation level (<45%). (B): Genetic hemochromatosis with low activity of HFE-related signaling that limits hepcidin expression and in turn favors iron release in plasma, despite the presence of a sufficient amount of iron. Thus, the transferrin saturation increases and non-transferrin bound iron appears and targets organs such as liver, leading to abnormal iron accumulation.
Figure 1.
Schematic representation of the pathophysiological mechanisms leading to the development of iron overload during HFE-related hemochromatosis. (A): Normal situation with adequate HFE signaling allowing control of transferrin saturation level (<45%). (B): Genetic hemochromatosis with low activity of HFE-related signaling that limits hepcidin expression and in turn favors iron release in plasma, despite the presence of a sufficient amount of iron. Thus, the transferrin saturation increases and non-transferrin bound iron appears and targets organs such as liver, leading to abnormal iron accumulation.

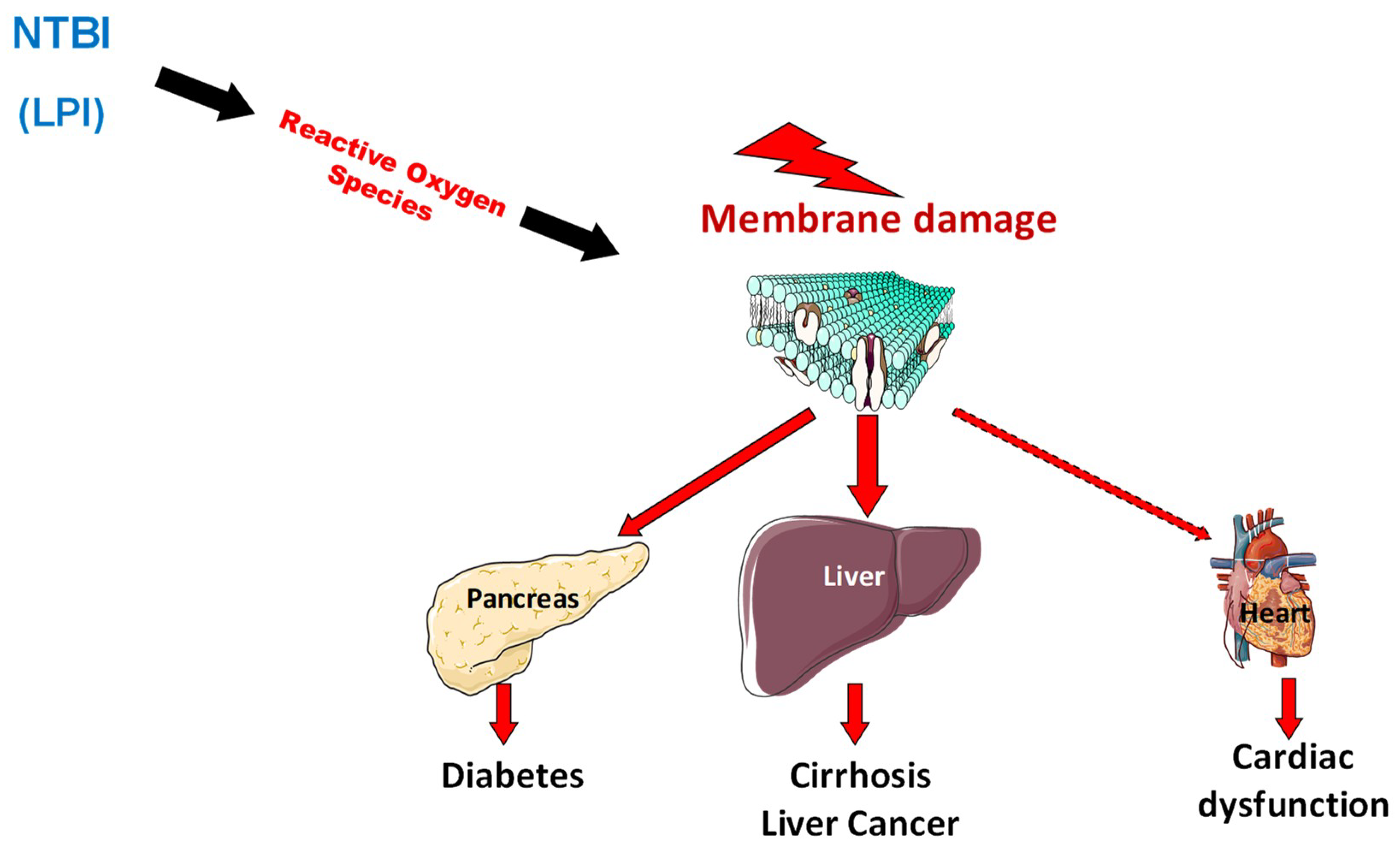
Figure 2.
The appearance of non-transferrin bound forms of iron in plasma favors organ iron deposition but also the occurrence of oxidative stress that alters organelles in cells especially in iron overloaded organs.
Figure 2.
The appearance of non-transferrin bound forms of iron in plasma favors organ iron deposition but also the occurrence of oxidative stress that alters organelles in cells especially in iron overloaded organs.

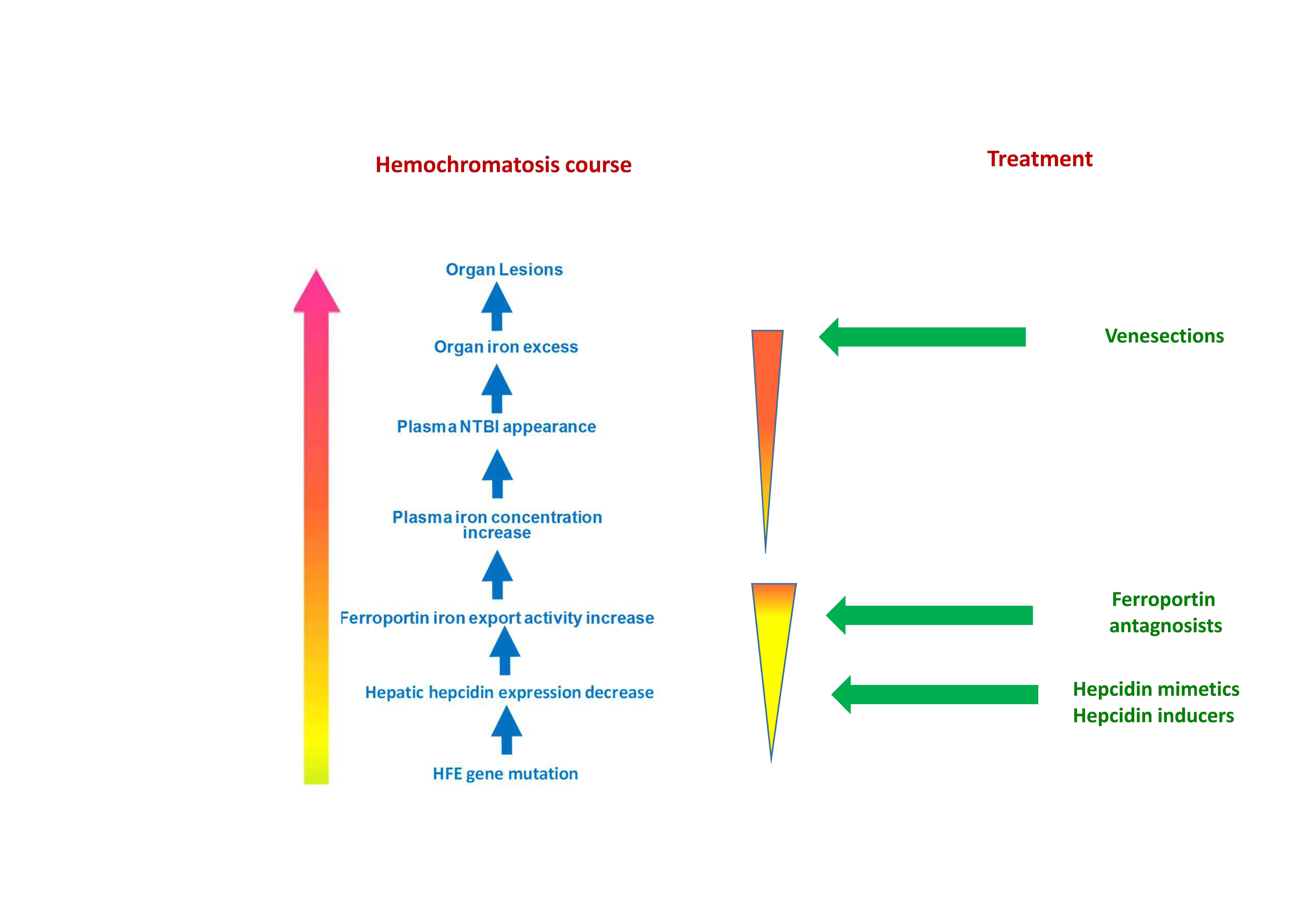
Figure 3.
Schematic representation of currently used iron removal treatments during hemochromatosis and potential pathophysiological actions. (1) Iron depletive treatment is classically operated through repeated venesections that remove red blood cells. In some rare cases, iron chelators can be also used as complementary or suppletive treatment. (2) Hepcidin supplementation by endogenous or exogenous hepcidin. (3) Alternatively (4) the use of ferroportin antagonists could be also a way of treatment.
Figure 3.
Schematic representation of currently used iron removal treatments during hemochromatosis and potential pathophysiological actions. (1) Iron depletive treatment is classically operated through repeated venesections that remove red blood cells. In some rare cases, iron chelators can be also used as complementary or suppletive treatment. (2) Hepcidin supplementation by endogenous or exogenous hepcidin. (3) Alternatively (4) the use of ferroportin antagonists could be also a way of treatment.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Loréal, O.; Cavey, T.; Robin, F.; Kenawi, M.; Guggenbuhl, P.; Brissot, P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals 2018, 11, 131. https://doi.org/10.3390/ph11040131
AMA Style
Loréal O, Cavey T, Robin F, Kenawi M, Guggenbuhl P, Brissot P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals. 2018; 11(4):131. https://doi.org/10.3390/ph11040131
Chicago/Turabian StyleLoréal, Olivier, Thibault Cavey, François Robin, Moussa Kenawi, Pascal Guggenbuhl, and Pierre Brissot. 2018. "Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects" Pharmaceuticals 11, no. 4: 131. https://doi.org/10.3390/ph11040131
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.