Determination of Parathion and Carbaryl Pesticides in Water and Food Samples Using a Self Assembled Monolayer /Acetylcholinesterase Electrochemical Biosensor

Abstract
:1. Introduction
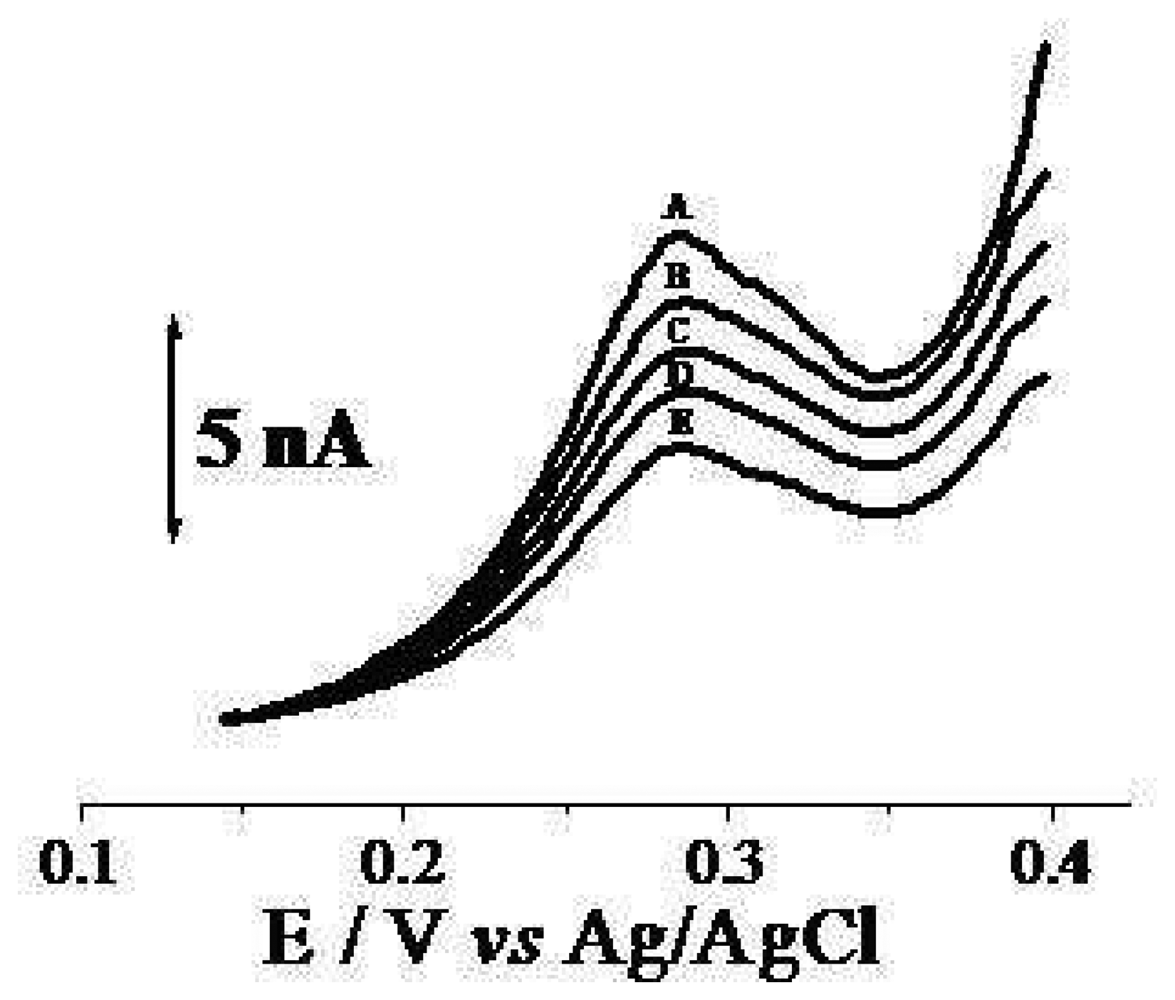
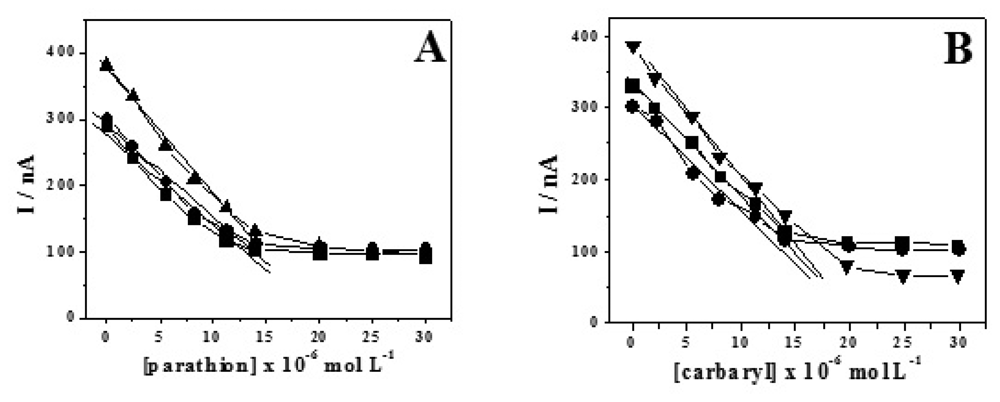
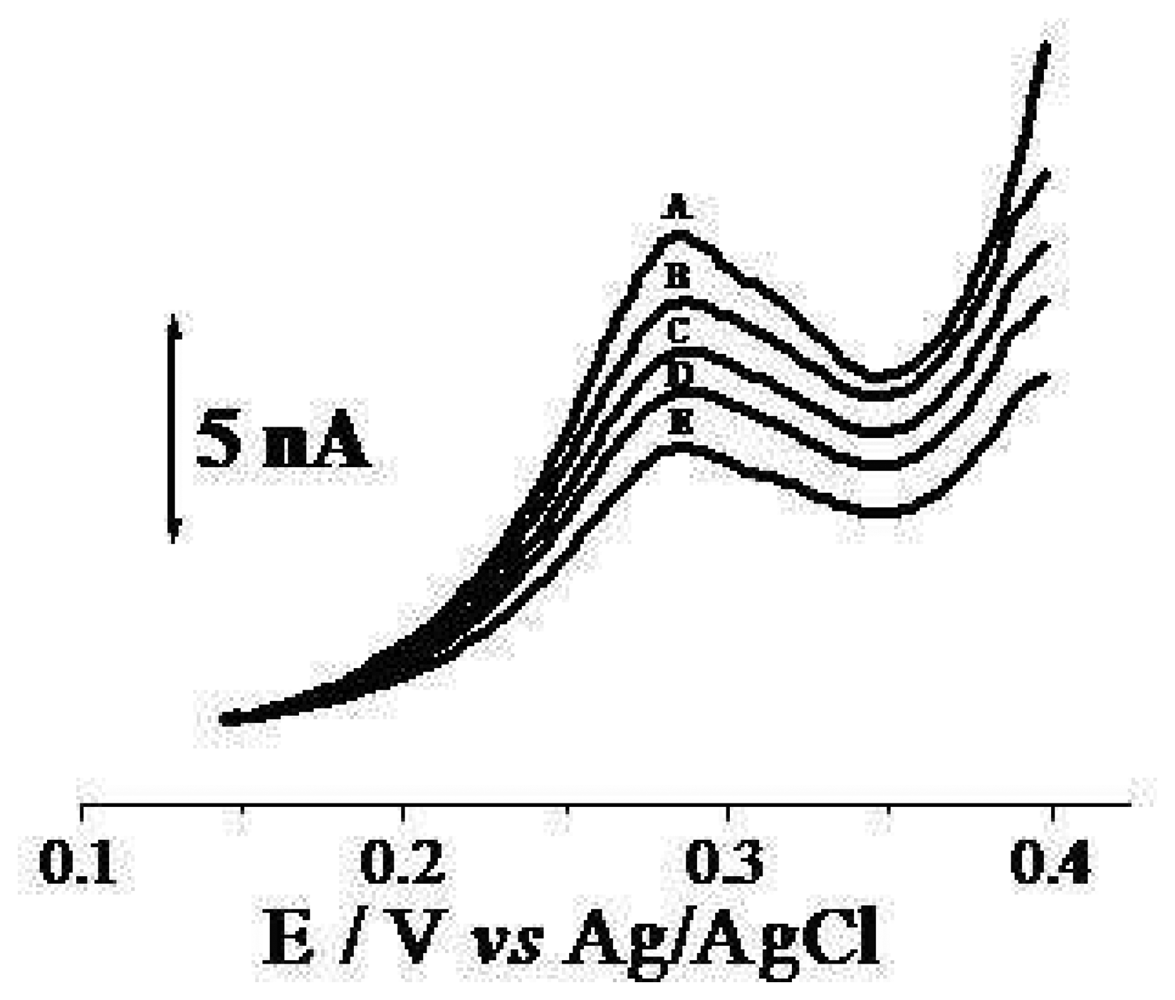
2. Results and Discussion
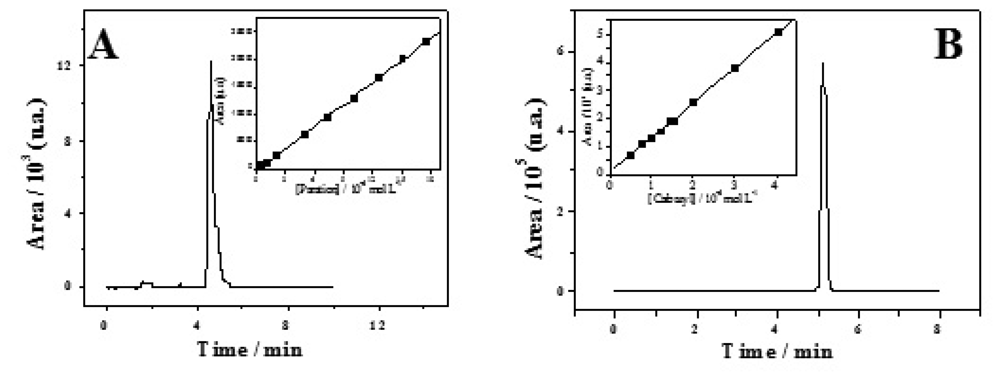
2.1. Water Analysis
2.2. Fruit sample testing
3. Conclusion
4. Experimental Section
4.1 Materials
4.2 Apparatus
4.3 Water Sample collection
4.4. Extraction step in fruits
Sample Treatment for Electrochemical Measurements in Fruits
Extraction for Matrix Solid-Phase Dispersion (MSPD) analysis
Acknowledgments
References and Notes
- Hand, L.H.; Kuet, S.F.; Lane, M.C.G.; Maund, S.J.; Warinton, J.S.; Hill, I.R. Influences of aquatic plants on the fate of the pyrethroid insecticide lambda-cyhalothrin in aquatic environments. Environ. Toxicology Chem. 2001, 20, 1740–1745. [Google Scholar]
- Dyson, J.S.; Beulke, S.; Brown, C.D.; Lane, M.C.G. Adsorption and degradation of the weak acid mesotrione in soil and environmental fate implications. J. Environ. Quality 2002, 31, 613–618. [Google Scholar]
- Mora, A.; Comejo, J.; Revilla, E.; Hermosin, M.C. Persistence and degradation of carbofuran in Spanish soil suspensions. Chemosphere 1996, 32, 1585–1598. [Google Scholar]
- Hassal, K.A. The Chemistry of Pesticides: Their Methabolism, Mode of Action and Uses in Crop Protection; MacMillan: New York, 1983; pp. 264–285. [Google Scholar]
- Simonian, A.L.; Rainina, E.I.; Wild, J.R. A new approach for discriminative detection of organophosphate neurotoxins in the presence of other cholinesterase inhibitors. Anal. Lett. 1997, 30, 2453–2468. [Google Scholar]
- Petropoulou, S.S.E.; Tsarbopoulos, A.; Siskos, P.A. Determination of carbofuran, carbaryl and their main metabolites in plasma samples of agricultural populations using gas chromatographytandem mass spectrometry. Anal. Bioanal. Chem. 2006, 385, 1444–1451. [Google Scholar]
- Brito, N.M.; Navickiene, S.; Polese, L.; Jardim, E.F.G.; Abakerli, R.B.; Ribeiro, M.L. Determination of pesticide residues in coconut water by liquid-liquid extraction and gas chromatography with electron-capture plus thermionic specific detection and solid-phase extraction and high-performance liquid chromatography with ultraviolet detection. J. Chromatography A 2002, 957, 201–209. [Google Scholar]
- Prado, A.G.S.; Airoldi, C. The effect of the herbicide diuron on soil microbial activity. Pest Management Science 2001, 57, 640–644. [Google Scholar]
- Pedrosa, V.A.; Machado, S.A.S.; Avaca, L.A. Application of a Deconvolutive Procedure to Analyze Several Chlorophenol Species in Natural Waters by Square-Wave Voltammetry on the Boron-Doped Diamond Electrode. Anal. Lett. 2006, 39, 1955–1965. [Google Scholar]
- Pedrosa, V.A.; Codognoto, L.; Avaca, L.A. Electroanalytical determination of 4-nitrophenol by square wave voltammetry on diamond electrodes. J Braz. Chem. Soc. 2003, 14, 530–535. [Google Scholar]
- Prieto-Simon, B.; Campas, M.; Andreescu, S.; Marty, J.L. Trends in flow-based biosensing systems for pesticide assessment. Sensors 2006, 6, 1161–1186. [Google Scholar]
- Li, X.H.; Xie, Z.H.; Min, H.; Xian, Y.Z.; Jin, L.T. Amperometric biosensor based on immobilization acetyleholinesterase on manganese porphyrin nanoparticles for detection of trichlorfon with flow-injection analysis system. Electroanalysis 2007, 19, 2551–2557. [Google Scholar]
- Suwansa-Ard, S.; Kanatharana, P.; Asawatreratanakul, P.; Limsakul, C.; Wongkittisuksa, B.; Thavarungkul, P. Semi disposable reactor biosensors for detecting carbamate pesticides in water. Biosens. Bioelectron. 2005, 21, 445–454. [Google Scholar]
- Shahgaldian, P.; Pieles, U. Cyclodextrin derivatives as chiral supramolecular receptors for enantioselective sensing. Sensors 2006, 6, 593–615. [Google Scholar]
- Arya, S.K.; Solanki, P.R.; Singh, S.P.; Kaneto, K.; Pandey, M.K.; Datta, M.; Malhotra, B.D. Poly-(3-hexylthiophene) self-assembled monolayer based cholesterol biosensor using surface plasmon resonance technique. Biosen. Bioelectron. 2007, 22, 2516–2524. [Google Scholar]
- Lowinsohn, D.; Bertotti, M. Flow injection analysis of blood L-lactate by using a Prussian Blue-based biosensor as amperometric detector. Anal. Biochem. 2007, 365, 260–265. [Google Scholar]
- Freire, R.S.; Pessoa, C.A.; Mello, L.D.; Kubota, L.T. Direct electron transfer: An approach for electrochemical biosensors with higher selectivity and sensitivity. J. Braz. Chem. Soc. 2003, 14, 230–243. [Google Scholar]
- Zhou, N.; Wang, J.; Chen, T.; Yu, Z.G.; Li, G.X. Enlargement of gold nanoparticles on the surface of a self-assembled monolayer modified electrode: A mode in biosensor design. Anal. Chem. 2006, 78, 5227–5230. [Google Scholar]
- Chen, H.X.; Lee, M.; Choi, S.W.; Kim, J.H.; Choi, H.J.; Kim, S.H.; Lee, J.B.; Koh, K.N. Comparative study of protein immobilization properties on calixarene monolayers. Sensors 2007, 7, 1091–1107. [Google Scholar]
- Luo, X.L.; Xu, J.J.; Zhang, Q.; Yang, G.J.; Chen, H.Y. Electrochemically deposited chitosan hydrogel for horseradish peroxidase immobilization through gold nanoparticles self-assembly. Biosen. Bioelectron. 2005, 21, 190–196. [Google Scholar]
- Pedrosa, V.A.; Paixao, T.R.C.; Freire, R.S.; Bertotti, M. Studies on the electrochemical behavior of a cystine self-assembled monolayer modified electrode using ferrocyanide as a probe. J Electroanal. Chem. 2007, 602, 149–155. [Google Scholar]
- Environmental Protection Agency. Fer. Regist. 1979, 44, 233.
- Pedrosa, V.A.; Caetano, J.; Machado, S.A.S.; Freire, R.S.; Bertotti, M. Acetylcholinesterase immobilization on 3-mercaptopropionic acid self assembled monolayer for determination of pesticides. Electroanalysis 2007, 19, 1415–1420. [Google Scholar]
- Pedrosa, V.A.; Codognoto, L.; Machado, S.A.S.; Avaca, L.A. Is the boron-doped diamond electrode a suitable substitute for mercury in pesticide analyses? A comparative study of 4-nitrophenol quantification in pure and natural waters. J. Electroanal. Chem. 2004, 573, 11–18. [Google Scholar]
- Long, L.G.; Winefordner, J.D. Limit of detection. Anal. Chem. 1983, 55, 712. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Pesticide | Sample | r | Sb (μA) | b (A/mol L-1) | DL (μg L-1) | Recovery (%) | |
|---|---|---|---|---|---|---|---|
| SAM-AchE | Paration | Pure | 0.994 | 0.017 | 1.9×10-4 | 9.3 | - |
| Point 1 | 0.989 | 0.014 | 1.8×10-4 | 10.3 | 96 ± 2 | ||
| Point 2 | 0.980 | 0.013 | 1.5×10-4 | 9.7 | 94 ± 1 | ||
| Carbaryl | Pure | 0.970 | 0.015 | 2.6×10-4 | 9.0 | - | |
| Point 1 | 0.992 | 0.014 | 1.6×10-4 | 9.6 | 95 ± 1 | ||
| Point 2 | 0.990 | 0.013 | 1.7×10-4 | 9.8 | 94 ± 2 | ||
| HPLC* | Parathion | Pure | 0.999 | 16.85 | 1.3×1010 | 1.1 | - |
| Point 1 | 0.997 | 14.30 | 0.2×1010 | 5.1 | 95 ± 3 | ||
| Point 2 | 0.994 | 13.90 | 0.1×1010 | 6.4 | 93 ± 2 | ||
| Carbaryl | Pure | 0.999 | 8.80 | 0.2×1010 | 3.1 | - | |
| Point 1 | 0.998 | 7.90 | 8.0×109 | 8.3 | 94 ± 2 | ||
| Point 2 | 0.993 | 7.80 | 8.3×109 | 9.0 | 92 ± 1 | ||
| Sensor | Sample | Pesticide | Concentratio n added | Concentratio n found | Recovery (%) |
|---|---|---|---|---|---|
| Parathion | 6.0×10-6 | 5.7×10-6 | 95 ± 2 | ||
| 5.0×10-5 | 4.6×10-5 | 92 ± 3 | |||
| SAM-AchE | Tomato | Carbaryl | 6.0×10-6 | 5.5×10-6 | 92 ± 3 |
| 5.0×10-5 | 4.7×10-5 | 94 ± 3 | |||
| Parathion | 6.0×10-6 | 5.2×10-6 | 87 ± 2 | ||
| 5.0×10-5 | 4.4×10-5 | 8.8 ± 1 | |||
| HPLC | Tomato | Carbaryl | 6.0×10-6 | 4.7×10-6 | 78 ± 2 |
| 5.0×10-5 | 4.2×10-5 | 84 ± 2 | |||
| Parathion | 6.0×10-6 | 6.1×10-6 | 101 ± 3 | ||
| 5.0×10-5 | 4.9×10-5 | 98 ± 3 | |||
| SAM-AchE | Apple | Carbaryl | 6.0×10-6 | 5.8×10-6 | 97 ± 2 |
| 5.0×10-5 | 4.7×10-5 | 94 ± 2 | |||
| Parathion | 6.0×10-6 | 5.4×10-6 | 90 ± 1 | ||
| 5.0×10-5 | 4.4×10-5 | 90 ± 2 | |||
| HPLC | Apple | Carbaryl | 6.0×10-6 | 4.6×10-6 | 77 ± 2 |
| 5.0×10-5 | 4.3×10-5 | 82 ± 3 | |||
| Parathion | 6.0×10-6 | 5.8×10-6 | 97 ± 3 | ||
| 5.0×10-5 | 4.9×10-5 | 98 ± 2 | |||
| SAM-AchE | Orange | Carbaryl | 6.0×10-6 | 5.6×10-6 | 93 ±2 |
| 5.0×10-5 | 4.8×10-5 | 96 ± 2 | |||
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pedrosa, V.A.; Caetano, J.; Machado, S.A.S.; Bertotti, M. Determination of Parathion and Carbaryl Pesticides in Water and Food Samples Using a Self Assembled Monolayer /Acetylcholinesterase Electrochemical Biosensor. Sensors 2008, 8, 4600-4610. https://doi.org/10.3390/s8084600
Pedrosa VA, Caetano J, Machado SAS, Bertotti M. Determination of Parathion and Carbaryl Pesticides in Water and Food Samples Using a Self Assembled Monolayer /Acetylcholinesterase Electrochemical Biosensor. Sensors. 2008; 8(8):4600-4610. https://doi.org/10.3390/s8084600
Chicago/Turabian StylePedrosa, Valber A., Josiane Caetano, Sergio A. S. Machado, and Mauro Bertotti. 2008. "Determination of Parathion and Carbaryl Pesticides in Water and Food Samples Using a Self Assembled Monolayer /Acetylcholinesterase Electrochemical Biosensor" Sensors 8, no. 8: 4600-4610. https://doi.org/10.3390/s8084600
APA StylePedrosa, V. A., Caetano, J., Machado, S. A. S., & Bertotti, M. (2008). Determination of Parathion and Carbaryl Pesticides in Water and Food Samples Using a Self Assembled Monolayer /Acetylcholinesterase Electrochemical Biosensor. Sensors, 8(8), 4600-4610. https://doi.org/10.3390/s8084600