Modern Electrochemical Methods for Monitoring of Chemical Carcinogens

Abstract
:Introduction
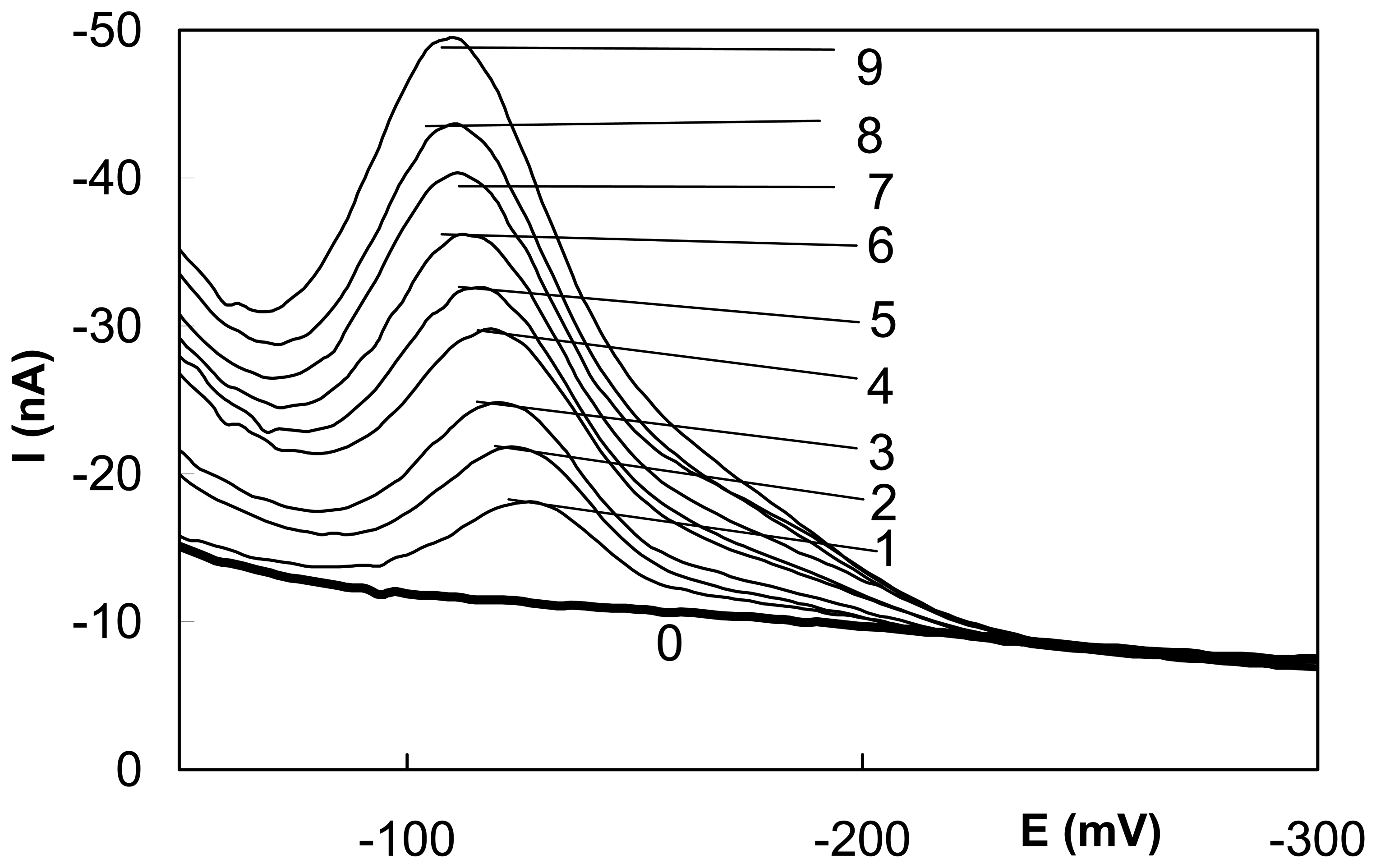
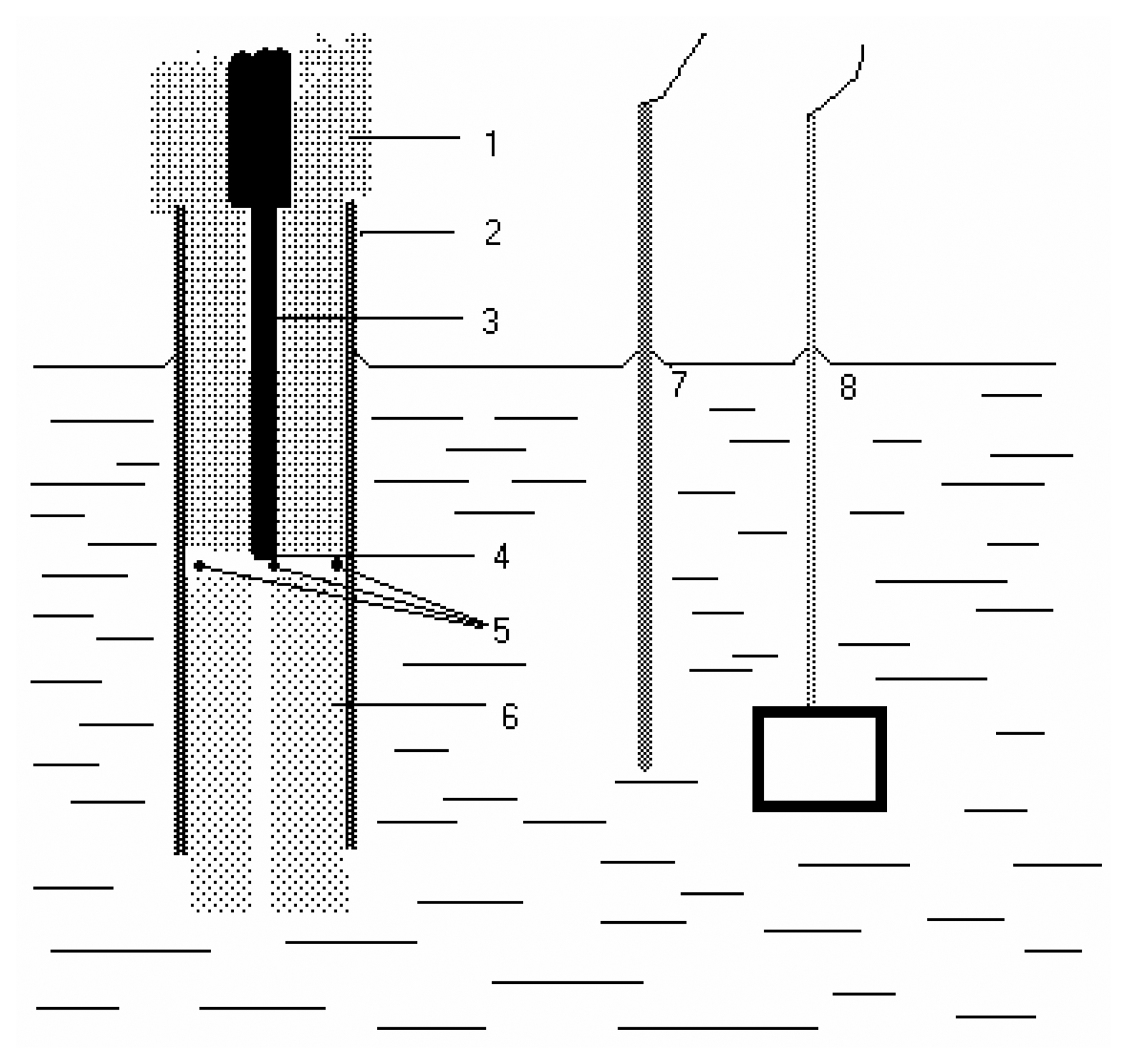
Polarography and voltammetry at mercury electrodes
- Easy renewal of their surface minimizing problems with the passivation,
- extremely broad potential window in cathodic region,
- high sensitivity,
- extremely broad concentration range from 10-3 to 10-10 M,
- broad spectrum of analytes (organic, inorganic, organometallic, macromolecular substances, etc.),
- low investment and running costs,
- high speed,
- molecule of an analyte is a direct source of signal,
- they present an independent alternative to spectrometric and separation methods (this is important from legal point of view because the “proof beyond reasonable doubt” requires several independent methods).
Voltammetry at carbon paste electrodes
HPLC with electrochemical detection (ED)
- Application of platinum tubular detector developed in our laboratory [31] for the determination of trace amounts of carcinogenic aromatic amines and its metabolites. This very simple device was used for HPLC ED determination of submicromolar concentrations of 1- and 2-aminonaphthalene [32], 1-,2-, 3- and 4-hydroxyphenanthrene [33] 3-, 5-, 6-, and 8-aminoquinoline [34], and 2- and 4-aminobiphenyl and 2- and 4-hydroxybiphenyl [35].
- Application of a carbon paste electrode based on glassy carbon micro beads for HPLC determination of genotoxic pyrene derivatives, namely 1-aminopyrene and 1-hydroxypyrene [27]. This type of working electrode in a wall-jet arrangement is compatible with a relatively high content of an organic solvent (methanol, acetonitrile, etc.) in a mobile phase.
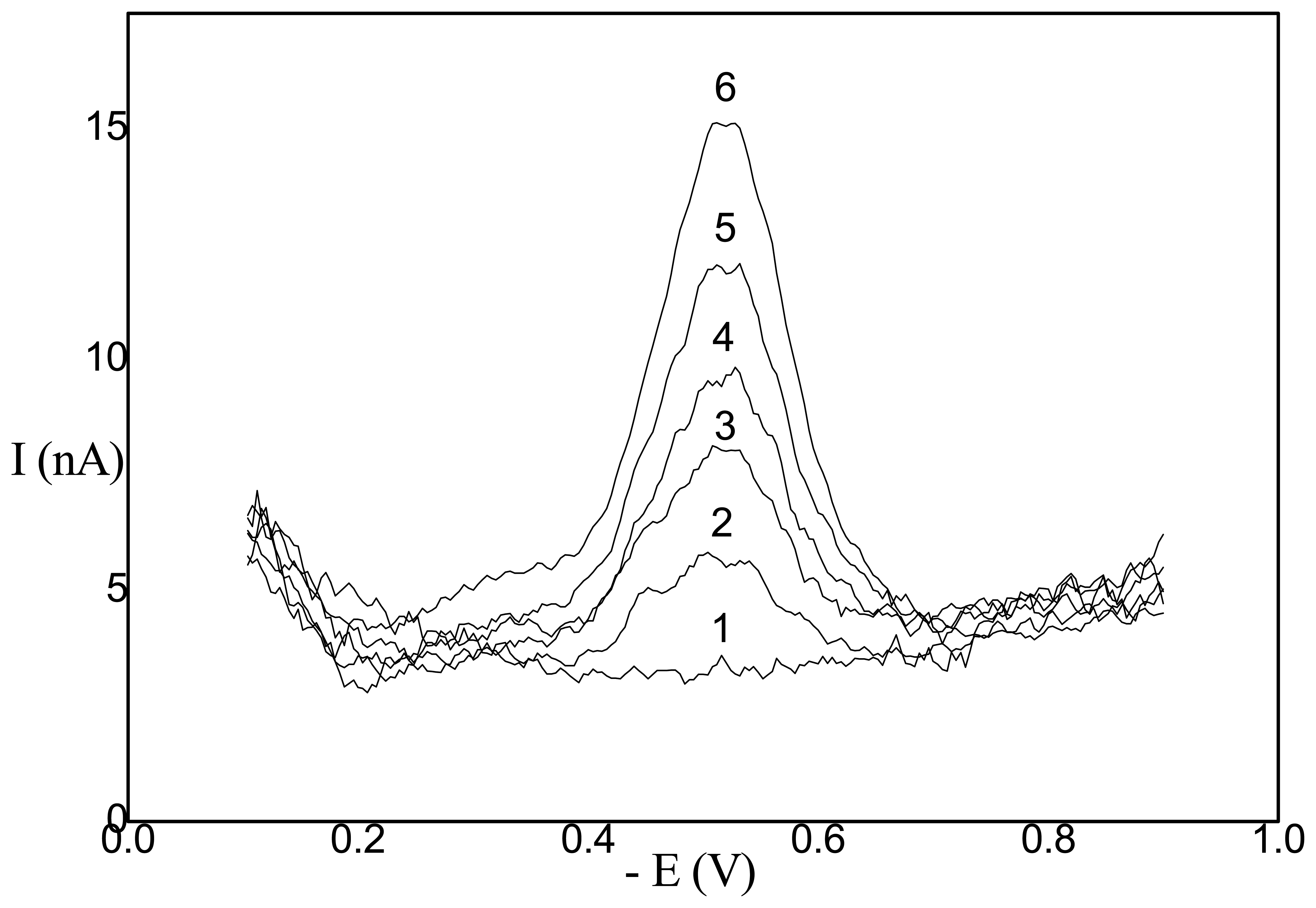
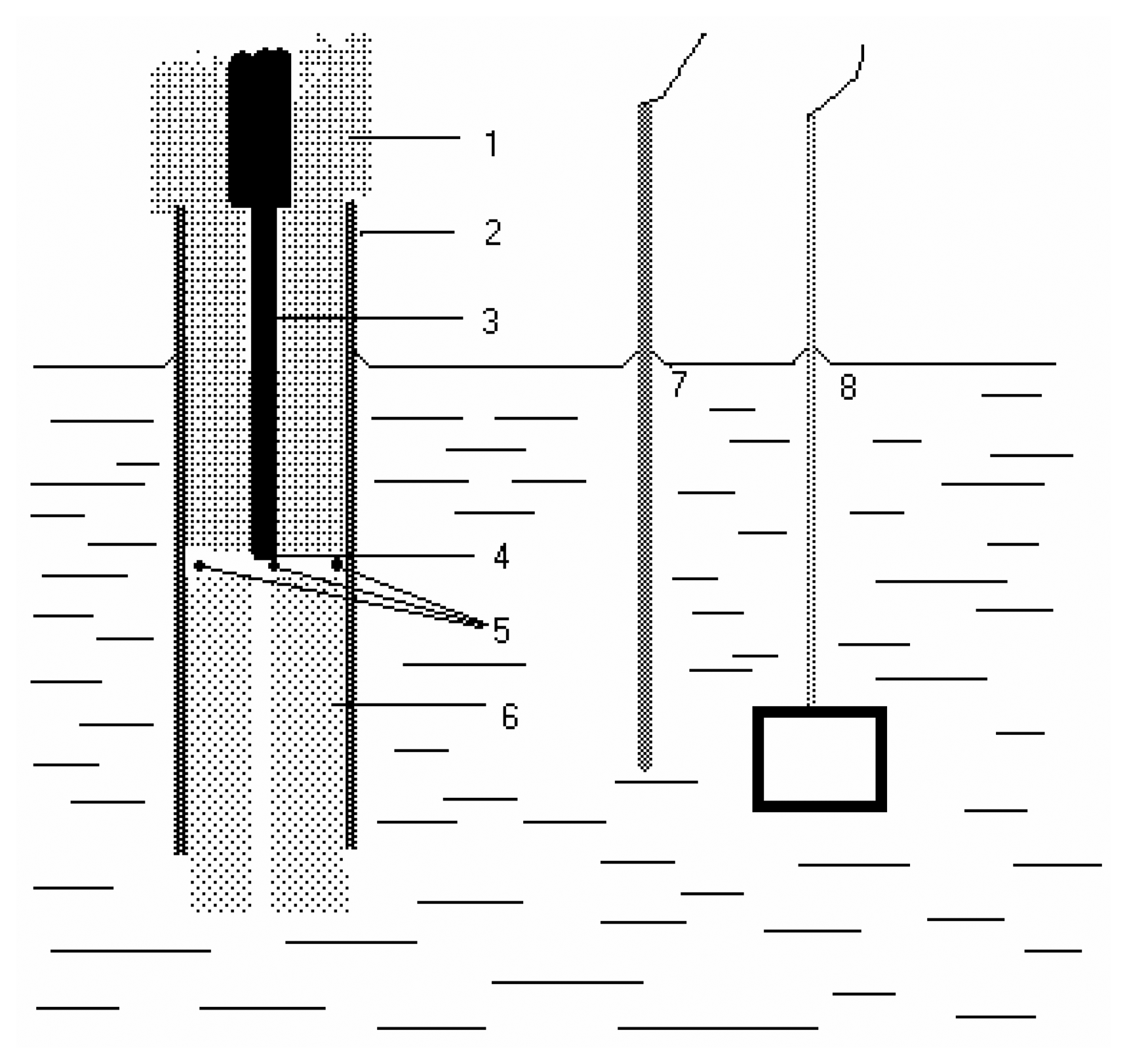
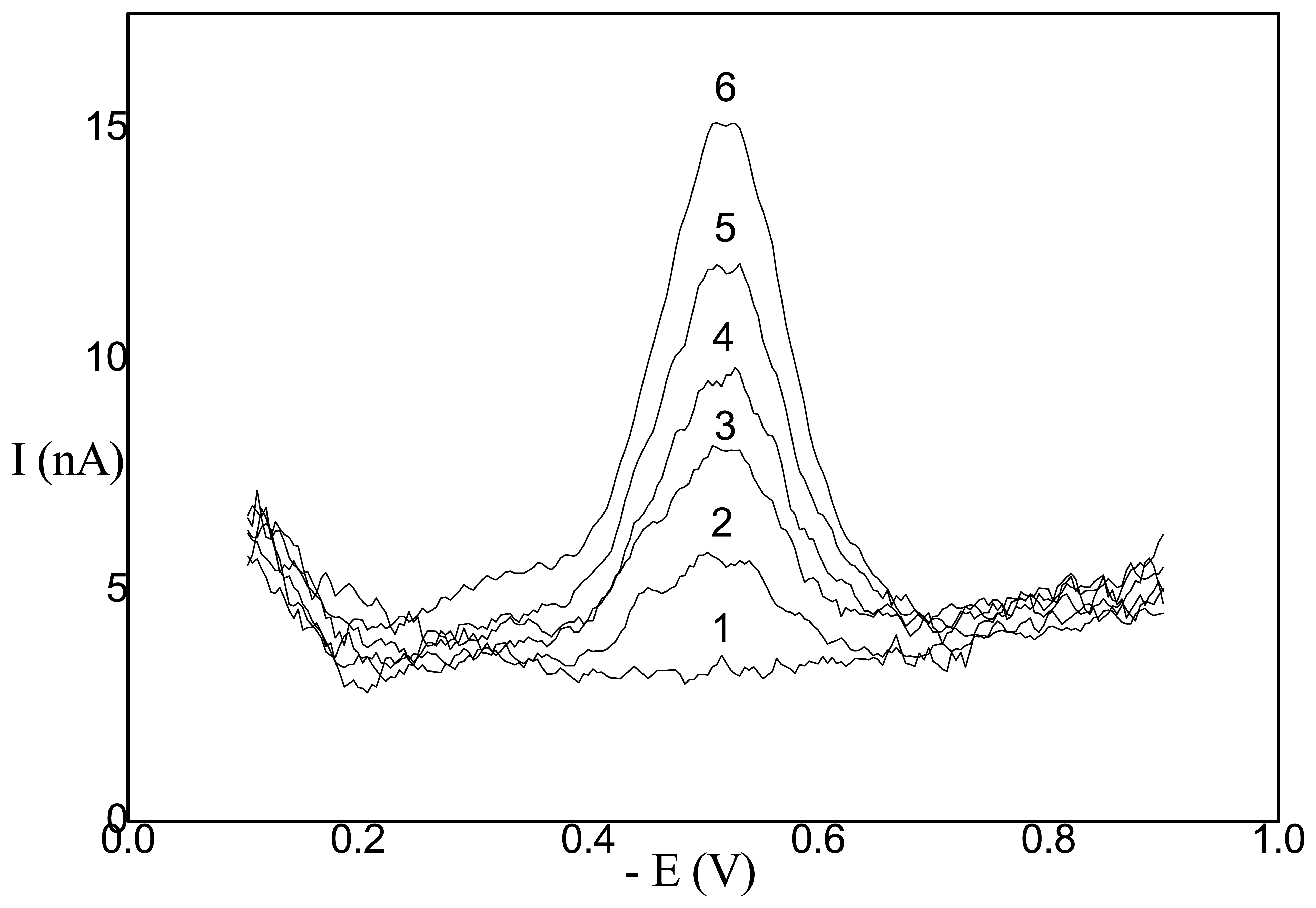
- Application of AgSAE for rather selective HPLC ED determination of polarographically active organic substances [36]. In this case, AgSAE serves as a working electrode in a wall-jet arrangement (see Fig. 3). LOD is around 10-6 M, i.e. comparable with HMDE working electrode. However, AgSAE is mechanically much more stable then HMDE.
Acknowledgments
References and notes
- Barek, J.; Cvačka, J.; Muck, A.; Quaiserová, V.; Zima, J. Electrochemical methods for monitoring of environmental carcinogens. Fresenius J. Anal. Chem. 2001, 369, 556. [Google Scholar]
- Barek, J.; Cvačka, J.; Muck, A.; Quaiserová, V.; Zima, J. Polarographic and voltammetric determination of carcinogenic nitro and amino derivatives of polycyclic aromatic hydrocarbons. Electroanalysis 2001, 13, 799. [Google Scholar]
- Kalvoda, R. Instrumentation in analytical chemistry; Zýka, J., Ed.; Horwood: London, 1994; p. p. 54. [Google Scholar]
- Kalvoda, R. Adsorptive accumulation in stripping voltammetry. Anal. Chim. Acta 1982, 138, 11. [Google Scholar]
- Kalvoda, R. Adsorptive stripping voltammetry of electroactive organic compounds. Anal. Chim. Acta 1984, 162, 197. [Google Scholar]
- Štulík, K.; Pacáková, V. Instrumentation in analytical chemistry; Vol. 1, Zýka, J., Ed.; Ellis Horwood: Chichester, 1991; p. p. 84. [Google Scholar]
- Barek, J.; Fogg, A.G.; Muck, A.; Zima, J. Polarography and voltammetry at mercury electrodes. Crit. Rev. Anal. Chem. 2001, 31, 291. [Google Scholar]
- Yosypchuk, B.; Novotný, L. Nontoxic electrodes of solid amalgams. Crit. Rev. Anal. Chem. 2002, 32, 141. [Google Scholar]
- Švancara, I.; Vytřas, K.; Barek, J.; Zima, J. Carbon paste electrodes in modern electroanalysis. Crit. Rev. Anal. Chem. 2001, 31, 311. [Google Scholar]
- Barek, J.; Muck, A.; Wang, J.; Zima, J. Study of voltammetric determination of carcinogenic 1-nitropyrene and 1-aminopyrene using a glassy carbon paste electrode. Sensors 2004, 4, 47. [Google Scholar]
- Economou, A.; Fielden, P.R. Applications, potentialities and limitations of adsorptive stripping analysis on mercury film electrodes. Trends Anal. Chem. 1997, 16, 286. [Google Scholar]
- Emons, H.; Baade, A.; Schöning, M.J. Voltammetric determination of heavy metals in microvolumes of rain water. Electroanalysis 2000, 12, 1171. [Google Scholar]
- Novotný, L. Compression of the interfacial boundary electrode-solution as the basis of new compression accumulation (stripping) techniques. Electroanalysis 1996, 8, 135. [Google Scholar]
- Novotný, L. Voltammetry of biologically active species and surfactants on new miniaturized and contractible (compressible) mercury electrodes. Fresenius J. Anal. Chem. 1999, 363, 55. [Google Scholar]
- Barek, J.; Zima, J. Electrochemistry of environmentally important organic substances. In Electrochemistry for environmental protection; Štulík, K., Kalvoda, R., Eds.; UNESCO Technical Report No. 25; UNESCO Regional Office for Science and Technology for Europe (ROSTE): Venice, 1996; p. 137. [Google Scholar]
- Barek, J.; Pumera, M.; Muck, A.; Kadeřábková, M.; Zima, J. Polarographic and voltammetric determination of selected nitrated polycyclic aromatic hydrocarbons. Anal. Chim. Acta 1999, 393, 141. [Google Scholar]
- Yosypchuk, B.; Novotný, L. Solid amalgam electrodes. In US-CZ Workshop on Electrochemical Sensors; Barek, J., Drašar, P., Eds.; Czech Chemical Society: Prague, 2001; p. 26. [Google Scholar]
- Pecková, K. Diploma Thesis, Charles University, Faculty of Science, Prague, 2001.
- Štěpán, R. Diploma Thesis, Charles University, Faculty of Science, Prague, 2001.
- Kolářová, J. Diploma Thesis, Charles University, Faculty of Science, Prague, 2002.
- Fantová, N. Diploma Thesis, Charles University, Faculty of Science, Prague, 2001.
- Pecková, K.; Barek, J.; Dřevínek, M.; Navrátil, T.; Novotný, L.; Yosypchuk, B.; Vaingatová, S.; Zima, J. Determination of nitrated polycyclic aromatic hydrocarbons using a solid-silver amalgam electrode. In US-CZ Workshop on Electrochemical Sensors; Barek, J., Drašar, P., Eds.; Czech Chemical Society: Prague, 2001; p. 32. [Google Scholar]
- Barek, J.; Dodova, E.; Navrátil, T.; Yosypchuk, B.; Novotný, L.; Zima, J. Voltammetric determination of N,N-dimethyl-4-aminocarboxyazobenzene at a silver solid amalgam electrode. Electroanalysis 2003, 15, 1778. [Google Scholar]
- Adams, R.N. Carbon paste electrodes. Anal. Chem. 1958, 30, 1576. [Google Scholar]
- Armalis, S.; Novikova, N.; Kubiliene, E.; Zima, J.; Barek, J. Voltammetric determination of 2-aminofluorene and 2,7-diaminofluorene using carbon paste electrode. Anal. Lett. 2002, 32, 1551. [Google Scholar]
- Ferancová, A.; Korgová, E.; Labuda, J.; Zima, J.; Barek, J. Cyclodextrin modified carbon paste based electrodes as sensors for the determination of carcinogenic polycyclic aromatic amines. Electroanalysis 2002, 14, 1668. [Google Scholar]
- Muck, A. PhD Thesis, Charles University, Faculty of Science, Prague, 2002.
- Svancara, I.; Hvizdalova, M.; Vytras, K.; Kalcher, K.; Novotny, R. A microscopic study on carbon paste electrodes. Electroanalysis 1996, 8, 61. [Google Scholar]
- Wang, J.; Kirgoz, U.A.; Mo, J.W.; Lu, J.M.; Kawde, A.N.; Muck, A. Glassy carbon paste electrodes. Electrochem. Commun. 2001, 3, 203. [Google Scholar]
- Štulík, K.; Pacáková, V. Electroanalytical measurements in flowing liquids.; E. Horwood: Chichester, 1987. [Google Scholar]
- Cvačka, J.; Opekar, F.; Barek, J.; Zima, J. An amperometric detector with a platinum tubular electrode for high performance liquid chromatography. Electroanalysis 2000, 12, 39. [Google Scholar]
- Quaiserová, V. Diploma Thesis, Charles University, Faculty of Science, Prague, 2000.
- Smutná, K. Diploma Thesis, Charles University, Faculty of Science, Prague, 2001.
- Hříbal, Z. Diploma Thesis, Charles University, Faculty of Science, Prague, 2001.
- Vaingátová, S. Diploma Thesis, Charles University, Faculty of Science, Prague, 2002.
- Pastor, F.; Barek, J.; Novotný, L.; Yosypchuk, B.; Zima, J.; Navrátil, T. unpublished results.


{kind=link}
{kind=link}
{kind=link}
| Substance | Technique | Electrode | Medium | LOD, M | Ref. |
|---|---|---|---|---|---|
| Nitrated polycyclic aromatic hydrocarbons | |||||
| 1-nitronaphthalene | DPP | DME | 0.01 M NaOH -MeOH (1:1), pH 12.2 | 1.10-7 | 18 |
| DPV | HMDE | 0.01 M NaOH -MeOH (9:1), pH 12.0 | 3.10-8 | ||
| AdSV | HMDE | 0.001 M LiOH | 2.10-9 | ||
| DPV | AgSAE | BR-MeOH (9:1), pH 7 | 3.10-7 | 18 | |
| 3-nitrobiphenyl | DPP | SMDE | BR-MeOH (1:1), pH 12 | 3.10-8 | 19 |
| DPV | HMDE | BR-MeOH (1:1), pH 12 | 2.10-8 | ||
| AdSV | HMDE | 0.01 M NaOH-MeOH (100:1), pH 12 | 2.10-9 | ||
| DPV | AgSAE | 0.2 M NaOH-MeOH (1:1) | 3.10-7 | 22 | |
| Heterocyclic aromatic hydrocarbons | |||||
| 8-nitroquinoline | DPP | DME | BR-MeOH (9:1), pH 5 | 1.10-7 | 20 |
| DPV | HMDE | BR-MeOH (1:1), pH 4 | 2.10-8 | ||
| AdSV | HMDE | 0.002 M LiOH-MeOH (9:1) | 2.10-8 | ||
| 6-methyl-5- nitroquinoline | DPP | DME | BR-MeOH (1:1), pH 5 | 21 | |
| DPV | HMDE | BR-MeOH(1:1), pH 6 | |||
| AdSV | HMDE | 0.01 M NaOH-MeOH (99:1) | 2.10-8 | ||
| 6-methyl-5-nitrouracil | DPP | DME | BR, pH 6 | 2.10-7 | 21 |
| DPV | HMDE | BR, pH 7 | 2.10-7 | ||
© 2005 by MDPI ( http://www.mdpi.org). Reproduction is permitted for non-commercial purposes.
Share and Cite
Barek, J.; Moreira, J.; Zima, J. Modern Electrochemical Methods for Monitoring of Chemical Carcinogens. Sensors 2005, 5, 148-158. https://doi.org/10.3390/S5040148
Barek J, Moreira J, Zima J. Modern Electrochemical Methods for Monitoring of Chemical Carcinogens. Sensors. 2005; 5(4):148-158. https://doi.org/10.3390/S5040148
Chicago/Turabian StyleBarek, J., J. Moreira, and J. Zima. 2005. "Modern Electrochemical Methods for Monitoring of Chemical Carcinogens" Sensors 5, no. 4: 148-158. https://doi.org/10.3390/S5040148