A Flexible Platform of Electrochemically Functionalized Carbon Nanotubes for NADH Sensors

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Instrumentation
2.3. Fabrication of SWCNT Flexible Electrodes
2.4. Modification of SWCNT Electrodes
2.5. Amperometric Detection of NADH
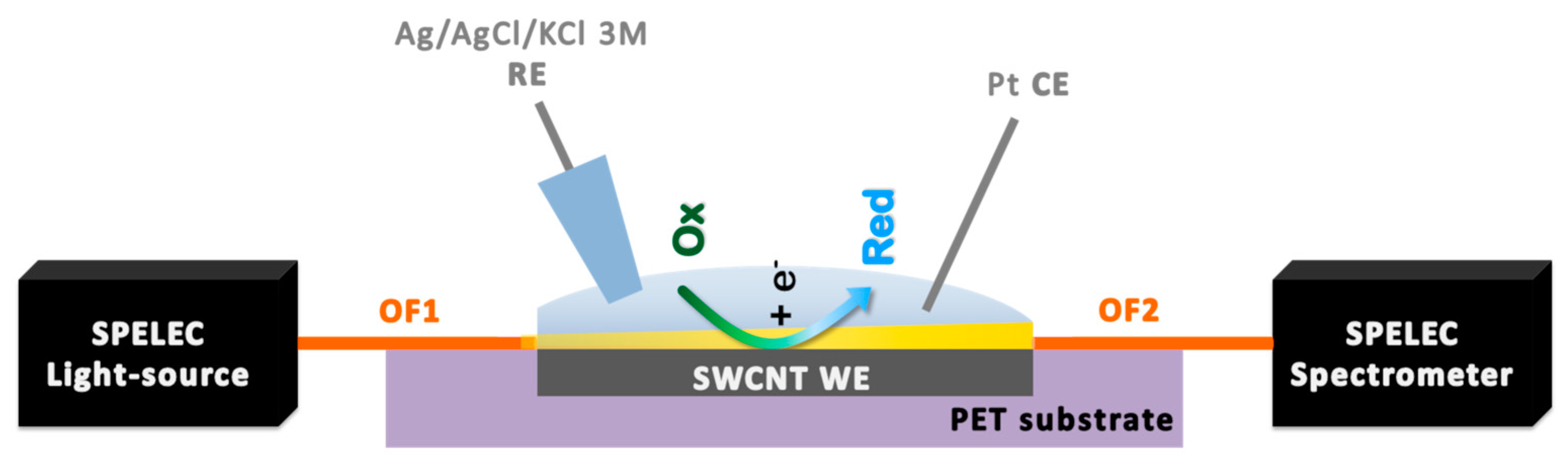
2.6. UV/Vis Absorption Spectroelectrochemical Set-up and Measurement Procedures
3. Results and Discussions
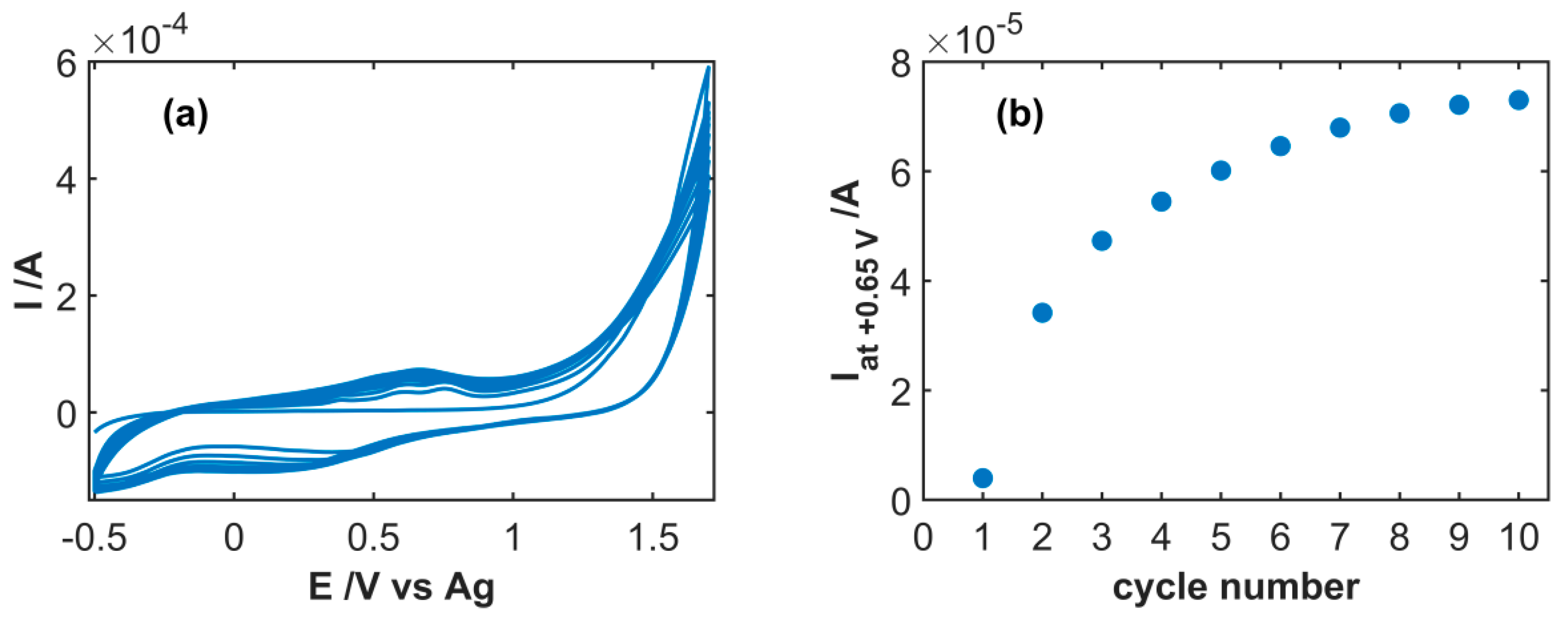
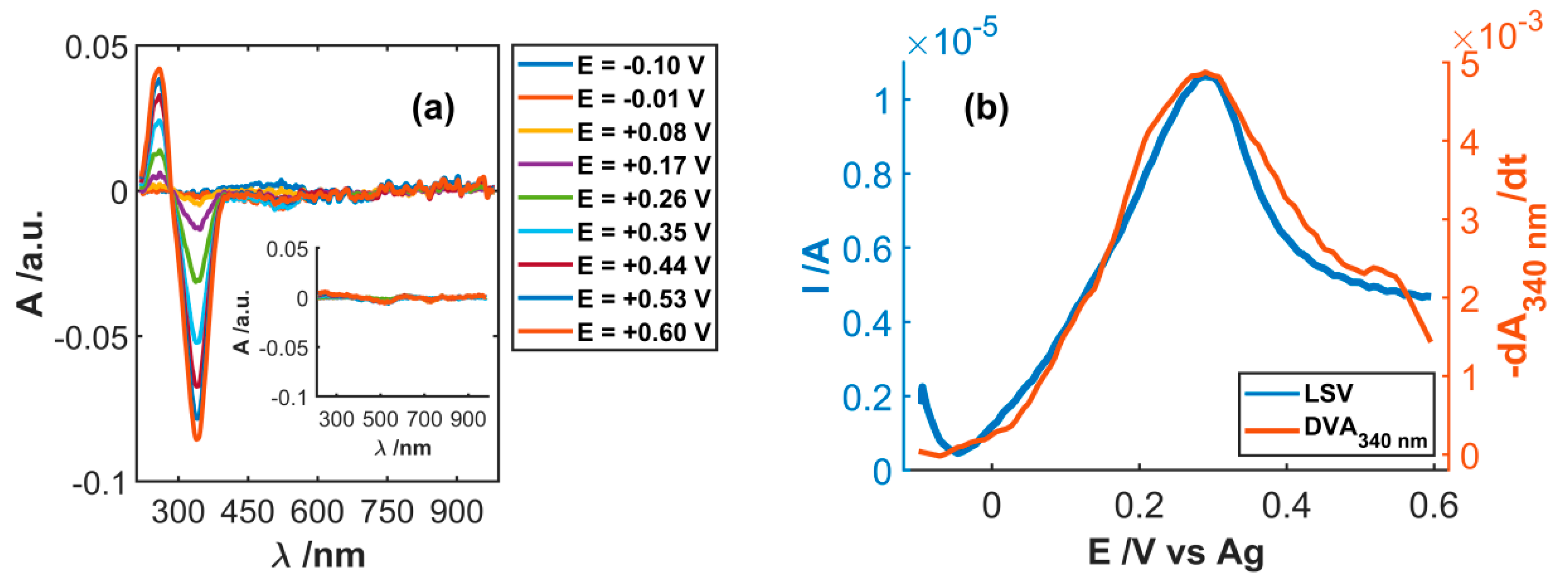
3.1. Spectroelectrochemical Study of Electrochemical Functionalization of SWCNT
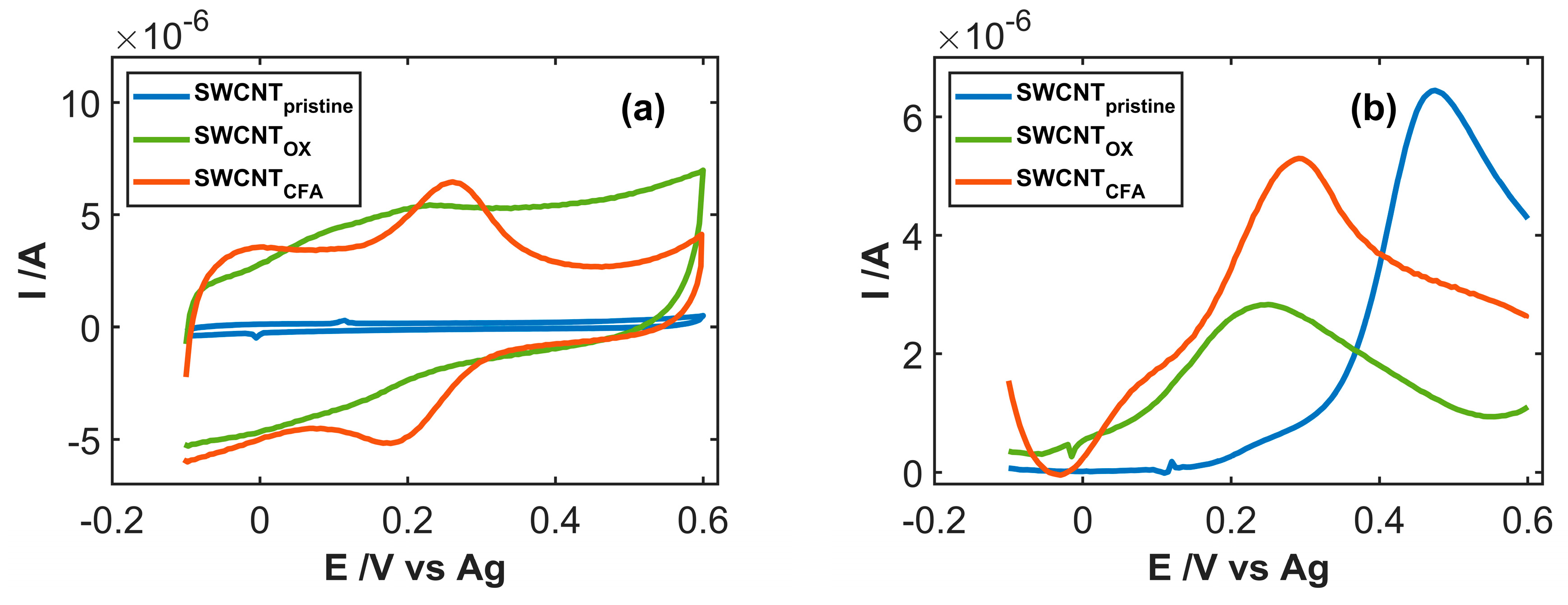
3.2. Study of Electrocatalytic Efficiency of Functionalized SWCNT for NADH Oxidation
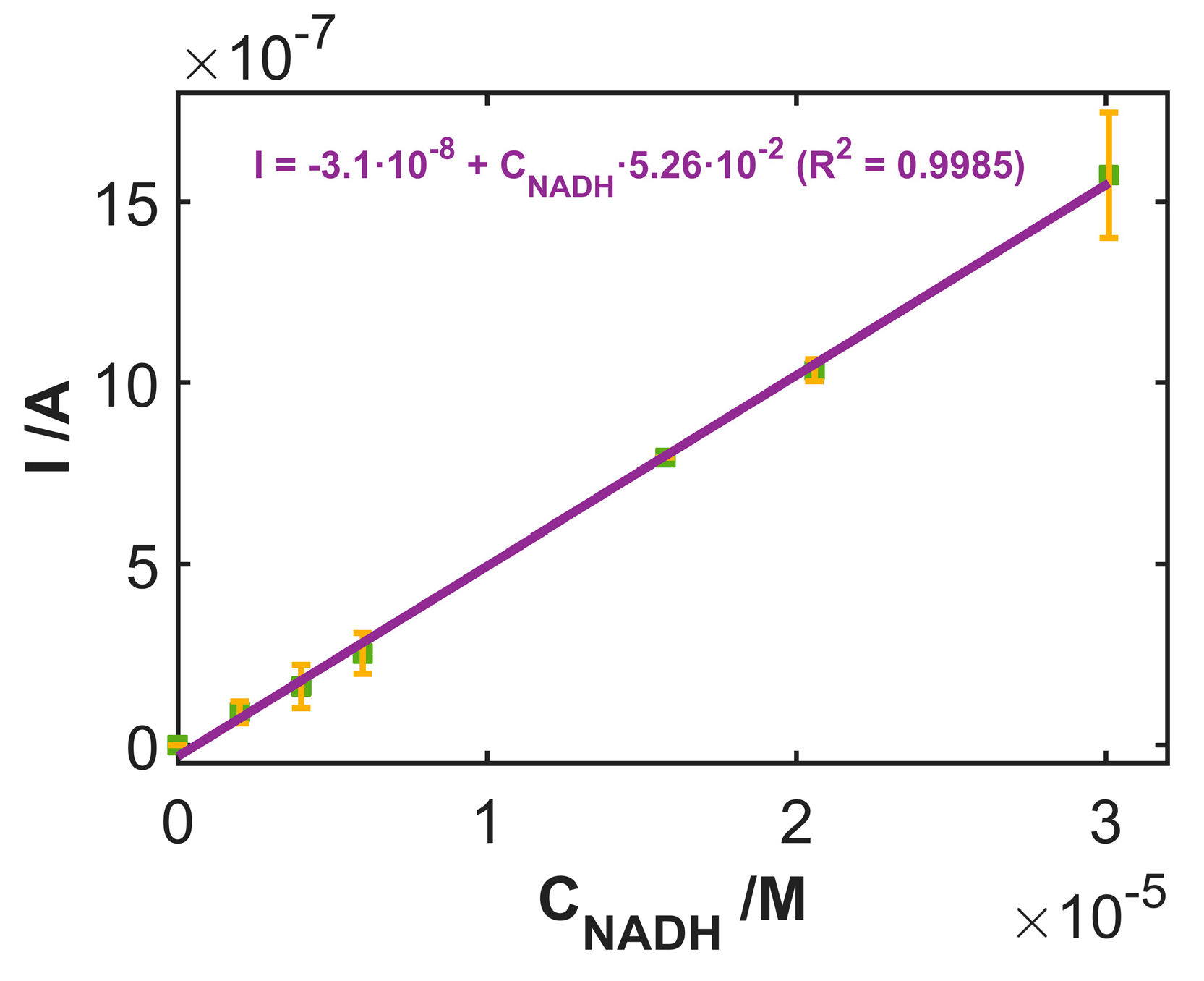
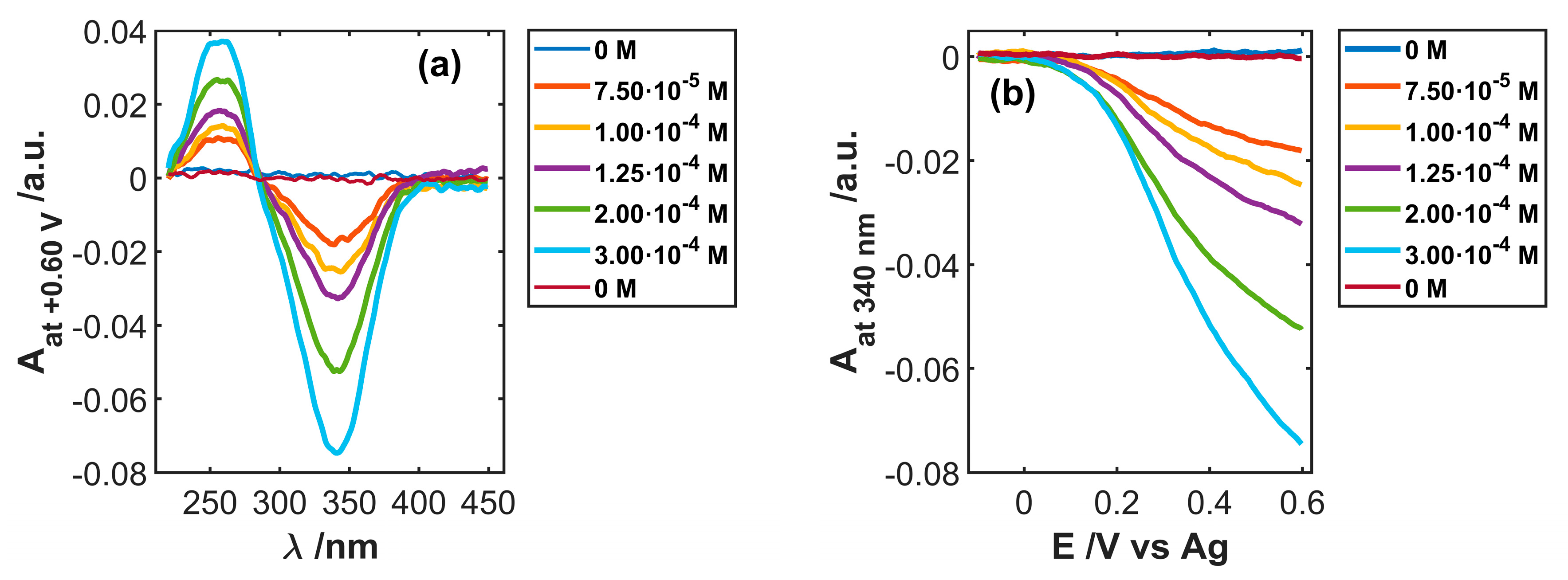
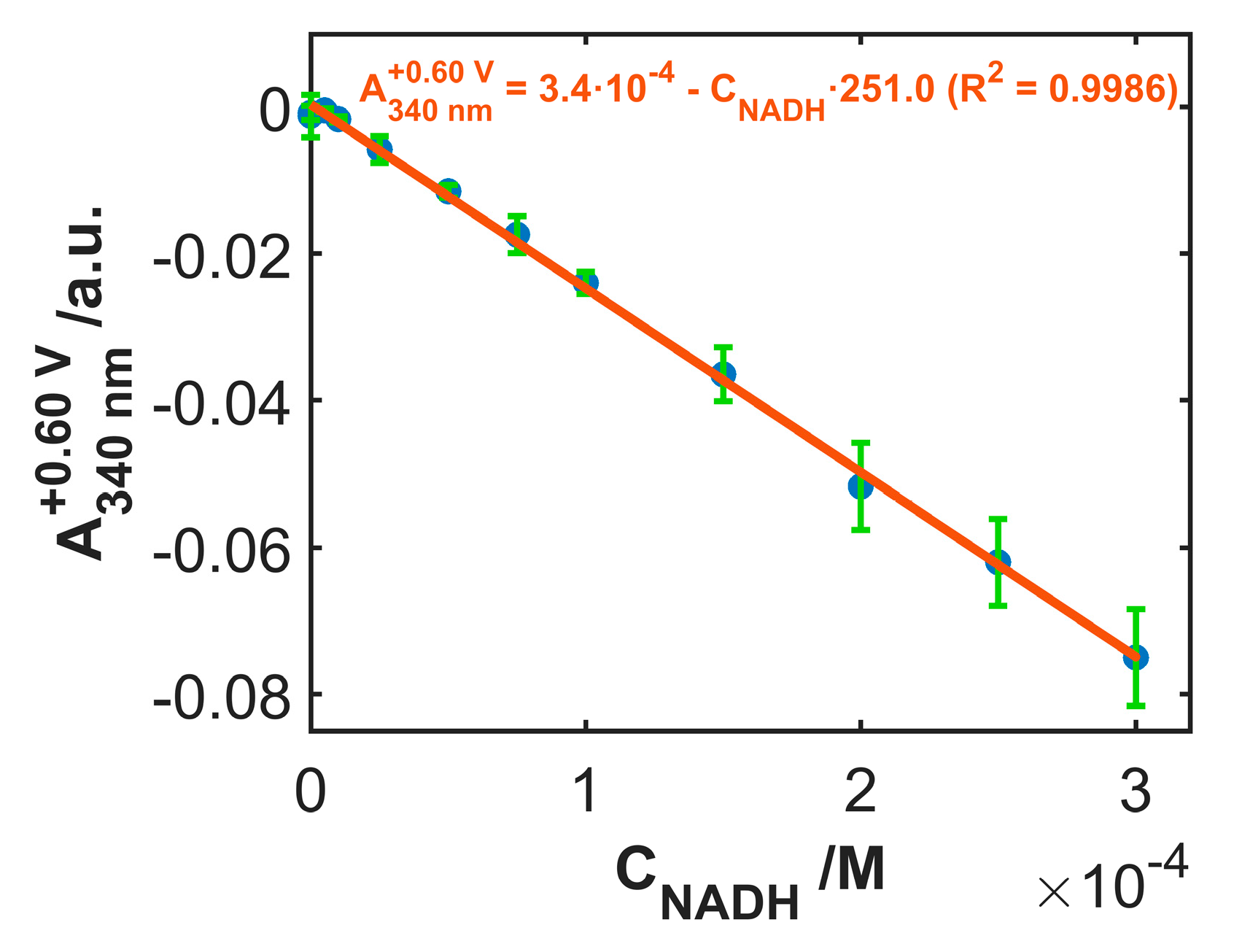
3.3. Use of SWCNTCFA as an Amperometric Sensor
3.4. Use of SWCNTCFA as a Voltabsorptometric Sensor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bartlett, P.N.; Simon, E.; Toh, C.S. Modified electrodes for NADH oxidation and dehydrogenase-based biosensors. Bioelectrochem. 2002, 56, 117–122. [Google Scholar] [CrossRef]
- Katakis, I.; Dominguez, E. Catalytic electrooxidation of NADH for dehydrogenase amperometric biosensors. Mikrochim Acta. 1997, 126, 11–32. [Google Scholar] [CrossRef]
- Uppal, A.; Gupta, P.K. Measurement of NADH concentration in normal and malignant human tissues from breast and oral cavity. Biotechnol. Appl. Biochem. 2003, 37, 45–50. [Google Scholar] [CrossRef]
- Schmakel, C.O.; Santhanam, K.S.V.; Elving, P.J. Nicotinamide adenine dinucleotide (NAD+) and related compounds. Electrochemical redox pattern and allied chemical behavior. J. Am. Chem. Soc. 1975, 97, 5083–5092. [Google Scholar] [CrossRef] [PubMed]
- Blaedel, W.J.; Jenkins, R.A. Study of the electrochemical oxidation of reduced nicotinamide adenine dinucleotide. Anal. Chem. 1975, 47, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Moiroux, J.; Elving, P.J. Mechanistic aspects of the electrochemical oxidation of dihydronicotinamide adenine dinucleotide (NADH). J. Am. Chem. Soc. 1980, 102, 6533–6538. [Google Scholar] [CrossRef]
- Maddar, F.M.; Lazenby, R.A.; Patel, A.N.; Unwin, P.R. Electrochemical oxidation of dihydronicotinamide adenine dinucleotide (NADH): Comparison of highly oriented pyrolytic graphite (HOPG) and polycrystalline boron-doped diamond (pBDD) electrodes. Phys Chem Chem Phys. 2016, 18, 26404–26411. [Google Scholar] [CrossRef]
- Gorton, L.; Domínguez, E. Electrocatalytic oxidation of NAD(P)H at mediator-modified electrodes. Rev. Mol. Biotechnol. 2002, 82, 371–392. [Google Scholar] [CrossRef]
- Prieto-Simón, B.; Fàbregas, E. Comparative study of electron mediators used in the electrochemical oxidation of NADH. Biosens. Bioelectron. 2004, 19, 1131–1138. [Google Scholar] [CrossRef]
- Radoi, A.; Compagnone, D. Recent advances in NADH electrochemical sensing design. Bioelectrochem. 2009, 76, 126–134. [Google Scholar] [CrossRef]
- Wang, J. Carbon-nanotube based electrochemical biosensors: a review. Electroanal. 2005, 17, 7–14. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Burghard, M. Biosensors based on carbon nanotubes. Anal. Bioanal. Chem. 2006, 385, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Y. Functionalized carbon nanotubes and nanofibers for biosensing applications. TrAC Trends Anal. Chem. 2008, 27, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Sedeño, P.; Pingarrón, J.M.; Riu, J.; Rius, F.X. Electrochemical sensing based on carbon nanotubes. TrAC Trends Anal. Chem. 2010, 29, 939–953. [Google Scholar] [CrossRef]
- Chen, D.; Feng, H.; Li, J. Graphene oxide: preparation, functionalization, and electrochemical applications. Chem. Rev. 2012, 112, 6027–6053. [Google Scholar] [CrossRef] [PubMed]
- Maccaferri, G.; Zanardi, C.; Xia, Z.Y.; Kovtun, A.; Liscio, A.; Terzi, F.; Palermo, V.; Seeber, R. Systematic study of the correlation between surface chemistry, conductivity and electrocatalytic properties of graphene oxide nanosheets. Carbon 2017, 120, 165–175. [Google Scholar] [CrossRef]
- Arduini, F.; Amine, A.; Majorani, C.; Di Giorgio, F.; De Felicis, D.; Cataldo, F.; Moscone, D.; Palleschi, G. High performance electrochemical sensor based on modified screen-printed electrodes with cost-effective dispersion of nanostructured carbon black. Electrochem. Commun. 2010, 12, 346–350. [Google Scholar] [CrossRef]
- Lei, J.; Ju, H. Signal amplification using functional nanomaterials for biosensing. Chem Soc Rev. 2012, 41, 2122–2134. [Google Scholar] [CrossRef]
- Seeber, R.; Pigani, L.; Terzi, F.; Zanardi, C. Amperometric sensing. A melting pot for material, electrochemical, and analytical sciences. Electrochim. Acta. 2015, 179, 350–363. [Google Scholar] [CrossRef]
- Zanardi, C.; Ferrari, E.; Pigani, L.; Arduini, F.; Seeber, R. Development of an electrochemical sensor for nadh determination based on a caffeic acid redox mediator supported on carbon black. Chemosensors 2015, 3, 118–128. [Google Scholar] [CrossRef]
- Singh, P.; Campidelli, S.; Giordani, S.; Bonifazi, D.; Bianco, A.; Prato, M. Organic functionalisation and characterisation of single-walled carbon nanotubes. Chem. Soc. Rev. 2009, 38, 2214–2230. [Google Scholar] [CrossRef]
- Banerjee, S.; Hemraj-Benny, T.; Wong, S.S. Covalent surface chemistry of single-walled carbon nanotubes. Adv. Mater. 2005, 17, 17–29. [Google Scholar] [CrossRef]
- Garoz-Ruiz, J.; Izquierdo, D.; Colina, A.; Palmero, S.; Heras, A. Optical fiber spectroelectrochemical device for detection of catechol at press-transferred single-walled carbon nanotubes electrodes. Anal. Bioanal. Chem. 2013, 405, 3593–3602. [Google Scholar] [CrossRef]
- Garoz-Ruiz, J.; Heras, A.; Palmero, S.; Colina, A. Development of a novel bidimensional spectroelectrochemistry cell using transfer single-walled carbon nanotubes films as optically transparent electrodes. Anal. Chem. 2015, 87, 6233–6239. [Google Scholar] [CrossRef]
- Garoz-Ruiz, J.; Heras, A.; Colina, A. Direct determination of ascorbic acid in a grapefruit: paving the way for in vivo spectroelectrochemistry. Anal. Chem. 2017, 89, 1815–1822. [Google Scholar] [CrossRef]
- Garoz-Ruiz, J.; Guillen-Posteguillo, C.; Heras, A.; Colina, A. Simplifying the assessment of parameters of electron-transfer reactions by using easy-to-use thin-layer spectroelectrochemistry devices. Electrochem. Commun. 2018, 86, 12–16. [Google Scholar] [CrossRef]
- Garoz-Ruiz, J.; Guillen-Posteguillo, C.; Colina, A.; Heras, A. Application of spectroelectroanalysis for the quantitative determination of mixtures of compounds with highly overlapping signals. Talanta 2019, 195, 815–821. [Google Scholar] [CrossRef]
- Palacios-Santander, J.M.; Terzi, F.; Zanardi, C.; Pigani, L.; Cubillana-Aguilera, L.M.; Naranjo-Rodriguez, I.; Seeber, R. Electrocatalytic and antifouling properties of CeO2-glassy carbon electrodes. J. Solid State Electrochem. 2016, 20, 3125–3131. [Google Scholar] [CrossRef]
- McCreery, R.L. Carbon electrodes: structural effects on electron transfer kinetics. In Electroanalytical chemistry; Bard, A.J., Ed.; Marcel Dekker: New York, NY, USA, 1991; Volume 17, pp. 221–374. [Google Scholar]
- Pariente, F.; Tobalina, F.; Darder, M.; Lorenzo, E.; Abruña, H.D. Electrodeposition of redox-active films of dihydroxybenzaldehydes and related analogs and their electrocatalytic activity toward NADH oxidation. Anal. Chem. 1996, 68, 3135–3142. [Google Scholar] [CrossRef]
- Zare, H.R.; Golabi, S.M. Electrocatalytic oxidation of reduced nicotinamide adenine dinucleotide (NADH) at a chlorogenic acid modified glassy carbon electrode. J. Electroanal. Chem. 1999, 464, 14–23. [Google Scholar] [CrossRef]
- Zare, H.R.; Golabi, S.M. Caffeic acid modified glassy carbon electrode for electrocatalytic oxidation of reduced nicotinamide adenine dinucleotide (NADH). J. Solid State Electrochem. 2000, 4, 87–94. [Google Scholar]
- Huang, X.; El-Sayed, I.H.; Yi, X.; El-Sayed, M.A. Gold nanoparticles: Catalyst for the oxidation of NADH to NAD+. J. Photochem. Photobio. B: Biology 2005, 81, 76–83. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SWCNTCFA-1 | SWCNTCFA-2 | SWCNTCFA-3 | |
|---|---|---|---|
| Slope /M−1 | −267.7 | −259.6 | −274.7 |
| Intercept /a.u. | 0.0015 | 0.0011 | 0.0007 |
| R2 | 0.9998 | 0.9986 | 0.9960 |
| Sy/x | 0.4·10−3 | 1.1·10−3 | 2.0·10−3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heras, A.; Vulcano, F.; Garoz-Ruiz, J.; Porcelli, N.; Terzi, F.; Colina, A.; Seeber, R.; Zanardi, C. A Flexible Platform of Electrochemically Functionalized Carbon Nanotubes for NADH Sensors. Sensors 2019, 19, 518. https://doi.org/10.3390/s19030518
Heras A, Vulcano F, Garoz-Ruiz J, Porcelli N, Terzi F, Colina A, Seeber R, Zanardi C. A Flexible Platform of Electrochemically Functionalized Carbon Nanotubes for NADH Sensors. Sensors. 2019; 19(3):518. https://doi.org/10.3390/s19030518
Chicago/Turabian StyleHeras, Aranzazu, Fabio Vulcano, Jesus Garoz-Ruiz, Nicola Porcelli, Fabio Terzi, Alvaro Colina, Renato Seeber, and Chiara Zanardi. 2019. "A Flexible Platform of Electrochemically Functionalized Carbon Nanotubes for NADH Sensors" Sensors 19, no. 3: 518. https://doi.org/10.3390/s19030518