Multispectral Depth-Resolved Fluorescence Lifetime Spectroscopy Using SPAD Array Detectors and Fiber Probes

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Optical Setup
2.2. Reference Flurophores
2.3. Fluorescence Data Analysis
2.4. Calibration
2.5. Agarose Phantoms of Reference Fluorophores
2.6. Rabbit Aorta Specimens
3. Results
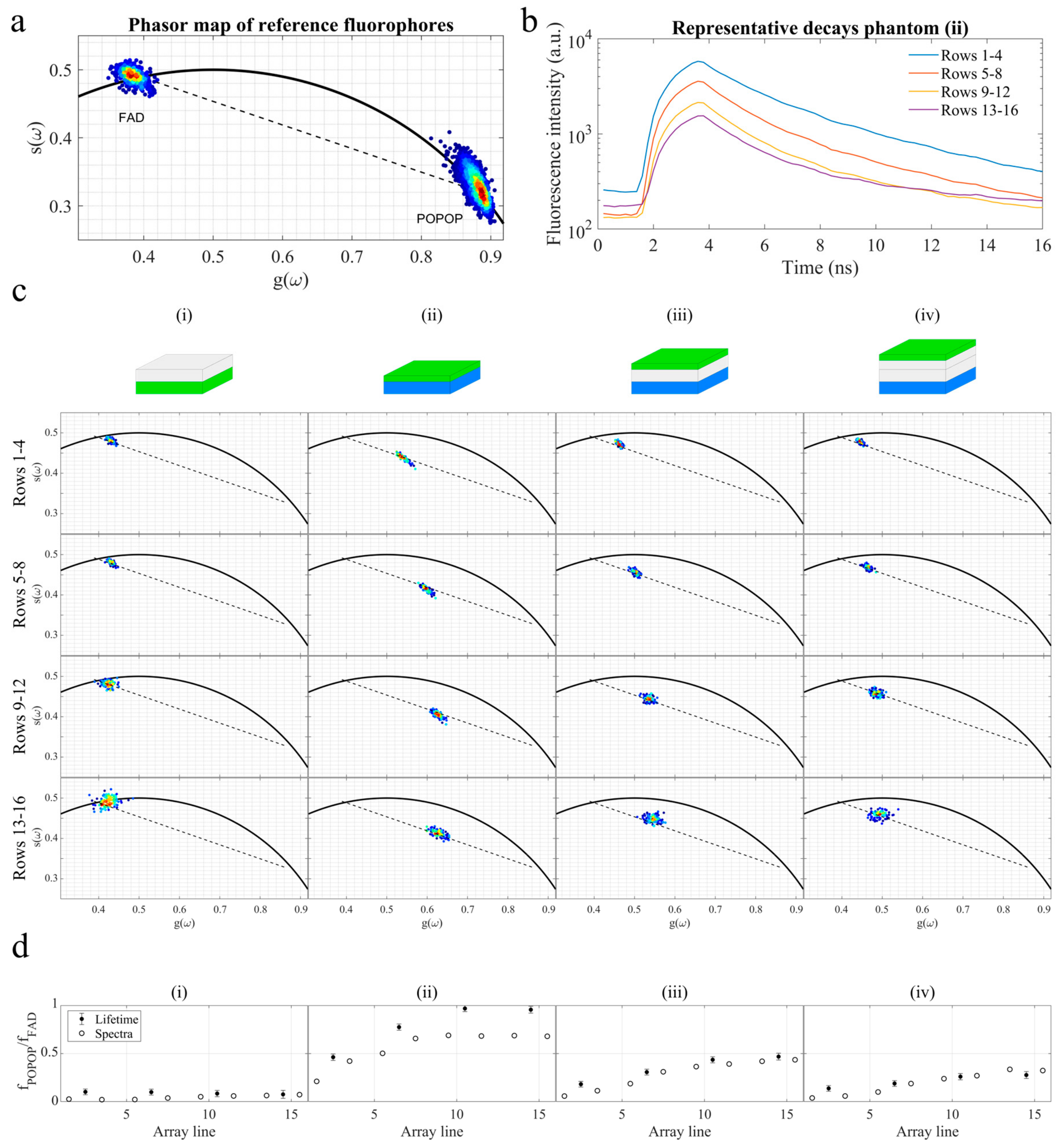
3.1. Fluorescence Measurements of Reference Fluorophores in Agarose
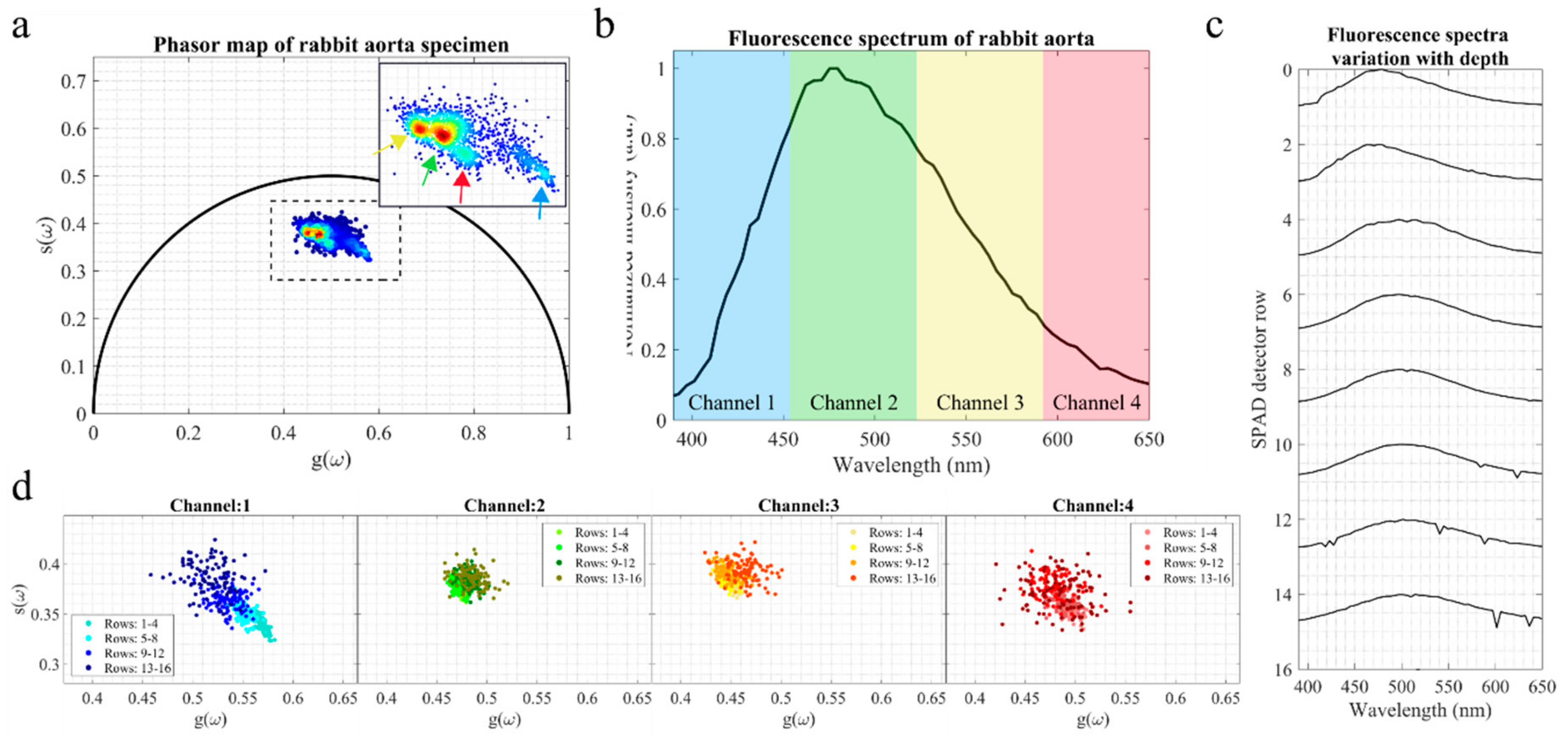
3.2. Fluorescence Measurements of Rabbit Aorta
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
3-D Printing of Molds for Agarose Phantoms
References
- Gillenwater, A.; Jacob, R.; Ganeshappa, R.; Kemp, B.; El-Naggar, A.K.; Palmer, J.L.; Clayman, G.; Mitchell, M.F.; Richards-Kortum, R. Noninvasive diagnosis of oral neoplasia based on fluorescence spectroscopy and native tissue autofluorescence. Arch. Otolaryngol. Head Neck Surg. 1998, 124, 1251–1258. [Google Scholar] [CrossRef]
- Gillenwater, A.; Jacob, R.; Richards-kortum, R. Fluorescence spectroscopy: A technique with potential to improve the early detection of aerodigestive tract neoplasia. Head Neck 1998, 20, 556–562. [Google Scholar] [CrossRef]
- Wu, Y.; Xi, P.; Qu, J.; Cheung, T.-H.; Yu, M.-Y. Depth-resolved fluorescence spectroscopy reveals layered structure of tissue. Opt. Express 2004, 12, 3218–3223. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qu, J.Y. Autofluorescence spectroscopy of epithelial tissues. J. Biomed. Opt. 2006, 11, 054023. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraj, M.; Udayakumar, K.; Jayanth, V.; Prakasa Rao, A.; Bharanidharan, G.; Koteeswaran, D.; Munusamy, B.D.; Murali Krishna, C.; Ganesan, S. Fluorescence spectroscopic characterization of salivary metabolites of oral cancer patients. J. Photochem. Photobiol. B Biol. 2014, 130, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Cicchi, R.; Giordano, F.; Conti, V.; Buccoliero, A.M.; Guerrini, R.; Pavone, F.S. Multimodal fiber-probe spectroscopy allows detecting epileptogenic focal cortical dysplasia in children. J. Biophotonics 2017, 10, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Nazeer, S.S.; Saraswathy, A.; Shenoy, S.J.; Jayasree, R.S. Fluorescence spectroscopy as an efficient tool for staging the degree of liver fibrosis: An in vivo comparison with MRI. Sci. Rep. 2018, 8, 10967. [Google Scholar] [CrossRef]
- Butte, P.V.; Pikul, B.K.; Hever, A.; Yong, W.H.; Black, K.L.; Marcu, L. Diagnosis of meningioma by time-resolved fluorescence spectroscopy. J. Biomed. Opt. 2010, 10, 064026. [Google Scholar] [CrossRef]
- Butte, P.V.; Mamelak, A.N.; Nuno, M.; Bannykh, S.I.; Black, K.L.; Marcu, L. Fluorescence lifetime spectroscopy for guided therapy of brain tumors. Neuroimage 2011, 54 (Suppl. 1), S125–S135. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.J.; Coda, S.; Sørensen, M.B.; Kennedy, G.; Patalay, R.; Waitong-Brämming, U.; De Beule, P.A.A.; Neil, M.A.A.; Andersson-Engels, S.; Bendsøe, N.; et al. In vivo measurements of diffuse reflectance and time-resolved autofluorescence emission spectra of basal cell carcinomas. J. Biophotonics 2012, 5, 240–254. [Google Scholar] [CrossRef]
- Fereidouni, F.; Bader, A.N.; Colonna, A.; Gerritsen, H.C. Phasor analysis of multiphoton spectral images distinguishes autofluorescence components of in vivo human skin. J. Biophotonics 2014, 7, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Coda, S.; Thompson, A.J.; Kennedy, G.T.; Roche, K.L.; Ayaru, L.; Bansi, D.S.; Stamp, G.W.; Thillainayagam, A.V.; French, P.M.W.; Dunsby, C. Fluorescence lifetime spectroscopy of tissue autofluorescence in normal and diseased colon measured ex vivo using a fiber-optic probe. Biomed. Opt. Express 2014, 5, 515–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagarto, J.; Dyer, B.T.; Talbot, C.; Sikkel, M.B.; Peters, N.S.; French, P.M.W.; Lyon, A.R.; Dunsby, C. Application of time-resolved autofluorescence to label-free in vivo optical mapping of changes in tissue matrix and metabolism associated with myocardial infarction and heart failure. Biomed. Opt. Express 2015, 6, 324–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bec, J.; Phipps, J.E.; Gorpas, D.; Ma, D.; Fatakdawala, H.; Margulies, K.B.; Southard, J.A.; Marcu, L. In vivo label-free structural and biochemical imaging of coronary arteries using an integrated ultrasound and multispectral fluorescence lifetime catheter system. Sci. Rep. 2017, 7, 8960–8969. [Google Scholar] [CrossRef] [PubMed]
- De Beule, P.A.A.; Dunsby, C.; Galletly, N.P.; Stamp, G.W.; Chu, A.C.; Anand, U.; Anand, P.; Benham, C.D.; Naylor, A.; French, P. A hyperspectral fluorescence lifetime probe for skin cancer diagnosis. Rev. Sci. Instrum. 2007, 78, 123101. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.H. Distinction of brain tissue, low grade and high grade glioma with time-resolved fluorescence spectroscopy. Front. Biosci. 2006, 11, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Yankelevich, D.R.; Ma, D.; Liu, J.; Sun, Y.; Sun, Y.; Bec, J.; Elson, D.S.; Marcu, L. Design and evaluation of a device for fast multispectral time-resolved fluorescence spectroscopy and imaging. Rev. Sci. Instrum. 2014, 85, 034303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, L.; Nogueira, M.S.; Pratavieira, S.; Moriyama, L.T.; Kurachi, C. Time-resolved fluorescence lifetime for cutaneous melanoma detection. Biomed. Opt. Express 2014, 5, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagnieres, G.A.; Star, W.M.; Wilson, B.C. In vivo fluorescence spectroscopy and imaging for oncological applications. Photochem. Photobiol. 1998, 68, 603–632. [Google Scholar] [CrossRef]
- Gillies, R.; Zonios, G.; Anderson, R.R.; Kollias, N. Fluorescence excitation spectroscopy provides information about human skin in vivo. J. Investig. Dermatol. 2000, 115, 704–707. [Google Scholar] [CrossRef]
- Anderson, R.R.; Parrish, J.A. The optics of human skin. J. Investig. Dermatol. 1981, 77, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kholodtsova, M.N.; Grachev, P.V.; Savelieva, T.A.; Kalyagina, N.A.; Blondel, W.; Loschenov, V.B. Scattered and fluorescent photon track reconstruction in a biological tissue. Int. J. Photoenergy 2014, 2014, 517510. [Google Scholar] [CrossRef]
- Jacques, S.L. Optical properties of biological tissues: A review. Phys. Med. Biol. 2013, 58, R37–R61. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, N.; Al-Ebraheem, A.; Le, D.; Cornacchi, S.; Fang, Q.; Farrell, T.; Lovrics, P.; Gohla, G.; Reid, S.; Hodgson, N.; et al. Time-resolved fluorescence (TRF) and diffuse reflectance spectroscopy (DRS) for margin analysis in breast cancer. Lasers Surg. Med. 2018, 50, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Lagarto, J.L.; Dyer, B.T.; Talbot, C.B.; Peters, N.S.; French, P.M.W.; Lyon, A.R.; Dunsby, C. Characterization of NAD(P)H and FAD autofluorescence signatures in a Langendorff isolated-perfused rat heart model. Biomed. Opt. Express 2018, 9, 4961–4978. [Google Scholar] [CrossRef] [PubMed]
- Lukina, M.; Orlova, A.; Shirmanova, M.; Shirokov, D.; Pavlikov, A.; Neubauer, A.; Studier, H.; Becker, W.; Zagaynova, E.; Yoshihara, T.; et al. Interrogation of metabolic and oxygen states of tumors with fiber-based luminescence lifetime spectroscopy. Opt. Lett. 2017, 42, 731–734. [Google Scholar] [CrossRef]
- Sanchez, W.Y.; Prow, T.W.; Sanchez, W.H.; Grice, J.E.; Roberts, M.S. Analysis of the metabolic deterioration of ex vivo skin from ischemic necrosis through the imaging of intracellular NAD(P)H by multiphoton tomography and fluorescence lifetime imaging microscopy. J. Biomed. Opt. 2010, 15, 046008. [Google Scholar] [CrossRef] [PubMed]
- Seidenari, S.; Arginelli, F.; Bassoli, S.; Cautela, J.; French, P.M.W.; Guanti, M.; Guardoli, D.; König, K.; Talbot, C.; Dunsby, C. Multiphoton laser microscopy and fluorescence lifetime imaging for the evaluation of the skin. Dermatol. Res. Pract. 2012, 2012, 810749. [Google Scholar] [CrossRef]
- Dancik, Y.; Favre, A.; Loy, C.J.; Zvyagin, A.V.; Roberts, M.S. Use of multiphoton tomography and fluorescence lifetime imaging to investigate skin pigmentation in vivo. J. Biomed. Opt. 2013, 18, 026022. [Google Scholar] [CrossRef]
- Knorr, F.; Yankelevich, D.R.; Liu, J.; Wachsmann-Hogiu, S.; Marcu, L. Two-photon excited fluorescence lifetime measurements through a double-clad photonic crystal fiber for tissue micro-endoscopy. J. Biophotonics 2012, 5, 14–19. [Google Scholar] [CrossRef]
- Hage, C.H.; Leclerc, P.; Brevier, J.; Fabert, M.; Le Nezet, C.; Kudlinski, A.; Heliot, L.; Louradour, F. Towards two-photon excited endogenous fluorescence lifetime imaging microendoscopy. Biomed. Opt. Express 2018, 9, 142–156. [Google Scholar] [CrossRef]
- Krstajić, N.; Levitt, J.; Poland, S.; Ameer-beg, S.; Henderson, R. 256 × 2 SPAD line sensor for time resolved fluorescence spectroscopy. Opt. Express 2015, 23, 5653–5669. [Google Scholar] [CrossRef] [PubMed]
- Krstajić, N.; Poland, S.; Levitt, J.; Walker, R.; Erdogan, A.; Ameer-Beg, S.; Henderson, R.K. 0.5 billion events per second time correlated single photon counting using CMOS SPAD arrays. Opt. Lett. 2015, 40, 4305–4308. [Google Scholar] [CrossRef] [PubMed]
- Popleteeva, M.; Haas, K.T.; Stoppa, D.; Pancheri, L.; Gasparini, L.; Kaminski, C.F.; Cassidy, L.D.; Venkitaraman, A.R.; Esposito, A. Fast and simple spectral FLIM for biochemical and medical imaging. Opt. Express 2015, 23, 23511–23525. [Google Scholar] [CrossRef] [PubMed]
- Bronzi, D.; Villa, F.; Tisa, S.; Tosi, A.; Zappa, F. SPAD figures of merit for photon-counting, photon-timing, and imaging applications: A review. IEEE Sens. J. 2016, 16, 3–12. [Google Scholar] [CrossRef]
- Castello, M.; Tortarolo, G.; Buttafava, M.; Deguchi, T.; Villa, F.; Koho, S.; Pesce, L.; Oneto, M.; Pelicci, S.; Lanzanó, L.; et al. A robust and versatile platform for image scanning microscopy enabling super-resolution FLIM. Nat. Methods 2019, 16, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Scully, A.D.; MacRobert, A.J.; Botchway, S.; O’Neill, P.; Parker, A.W.; Ostler, R.B.; Phillips, D. Development of a laser-based fluorescence microscope with subnanosecond time resolution. J. Fluoresc. 1996, 6, 119–125. [Google Scholar] [CrossRef]
- Gerritsen, H.C.; Sanders, R.; Draaijer, A.; Ince, C.; Levine, Y.K. Fluorescence lifetime imaging of oxygen in living cells. J. Fluoresc. 1997, 7, 11–15. [Google Scholar] [CrossRef]
- Bronzi, D.; Villa, F.; Tisa, S.; Tosi, A.; Zappa, F.; Durini, D.; Weyers, S.; Brockherde, W. 100000 frames/s 64 × 32 single-photon detector array for 2-D imaging and 3-D ranging. IEEE J. Sel. Top. Quantum Electron. 2014, 20, 354–363. [Google Scholar] [CrossRef]
- Bronzi, D.; Zou, Y.; Villa, F.; Tisa, S.; Tosi, A.; Zappa, F. Automotive three-dimensional vision through a single-photon counting SPAD camera. IEEE Trans. Intell. Transp. Syst. 2016, 17, 782–795. [Google Scholar] [CrossRef]
- Islam, M.S.; Honma, M.; Nakabayashi, T.; Kinjo, M.; Ohta, N. pH dependence of the fluorescence lifetime of FAD in solution and in cells. Int. J. Mol. Sci. 2013, 14, 1952–1963. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.; Auchet, J.C.; Visser, A.J.W.G.; Muller, F. Time resolved fluorescence of flavin adenine dinucleotide. FEBS Lett. 1974, 44, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, T.; Islam, M.S.; Ohta, N. Fluorescence decay dynamics of flavin adenine dinucleotide in a mixture of alcohol and water in the femtosecond and nanosecond time range. J. Phys. Chem. B 2010, 114, 15254–15260. [Google Scholar] [CrossRef] [PubMed]
- Delfino, I.; Esposito, R.; Portaccio, M.; Lepore, M. Dynamical and structural properties of flavin adenine dinucleotide in aqueous solutions. In Proceedings of the Biophotonics: Photonic Solutions for Better Health Care VI, Strasbourg, France, 23–26 April 2018; Popp, J., Tuchin, V.V., Pavone, F.S., Eds.; SPIE: Washington, DC, USA, 2018; p. 169. [Google Scholar]
- König, K.; Berns, M.W.; Tromberg, B.J. Time-resolved and steady-state fluorescence measurements of β-nicotinamide adenine dinucleotide-alcohol dehydrogenase complex during UVA exposure. J. Photochem. Photobiol. B Biol. 1997, 37, 91–95. [Google Scholar] [CrossRef]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 2008, 94, L14–L16. [Google Scholar] [CrossRef] [PubMed]
- Stringari, C.; Cinquin, A.; Cinquin, O.; Digman, M.A.; Donovan, P.J.; Gratton, E. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc. Nalt. Acad. Sci. USA 2011, 108, 13582–13587. [Google Scholar] [CrossRef] [Green Version]
- Ranjit, S.; Malacrida, L.; Jameson, D.M.; Gratton, E. Fit-free analysis of fluorescence lifetime imaging data using the phasor approach. Nat. Protoc. 2018, 13, 1979–2004. [Google Scholar] [CrossRef]
- Mustari, A.; Nishidate, I.; Wares, M.A.; Maeda, T.; Kawauchi, S.; Sato, S.; Sato, M.; Aizu, Y. Agarose-based tissue mimicking optical phantoms for diffuse reflectance spectroscopy. J. Vis. Exp. 2018, 138, e57578. [Google Scholar] [CrossRef]
- Cicchi, R.; Baria, E.; Matthäus, C.; Lange, M.; Lattermann, A.; Brehm, B.R.; Popp, J.; Pavone, F.S. Non-linear imaging and characterization of atherosclerotic arterial tissue using combined SHG and FLIM microscopy. J. Biophotonics 2015, 8, 347–356. [Google Scholar] [CrossRef]
- Leblond, F.; Ovanesyan, Z.; Davis, S.C.; Valdés, P.A.; Kim, A.; Hartov, A.; Wilson, B.C.; Pogue, B.W.; Paulsen, K.D.; Roberts, D.W. Analytic expression of fluorescence ratio detection correlates with depth in multi-spectral sub-surface imaging. Phys. Med. Biol. 2011, 56, 6823–6837. [Google Scholar] [CrossRef] [Green Version]
- Ash, C.; Dubec, M.; Donne, K.; Bashford, T. Effect of wavelength and beam width on penetration in light-tissue interaction using computational methods. Lasers Med. Sci. 2017, 32, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Colyer, R.A.; Lee, C.; Gratton, E. A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microsc. Res. Tech. 2008, 213, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Lagarto, J.; Hares, J.D.; Dunsby, C.; French, P.M.W. Development of low-cost instrumentation for single point autofluorescence lifetime measurements. J. Fluoresc. 2017, 27, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Munro, I.; McGinty, J.; Galletly, N.; Requejo-Isidro, J.; Lanigan, P.M.P.; Elson, D.S.; Dunsby, C.; Neil, M.A.A.; Lever, M.J.; Stamp, G.W.H.; et al. Toward the clinical application of time-domain fluorescence lifetime imaging. J. Biomed. Opt. 2005, 10, 051403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, H.; Görlitz, F.; Kelly, D.J.; Warren, S.C.; Kellett, P.A.; Garcia, E.; Dymoke-Bradshaw, A.K.L.; Hares, J.D.; Neil, M.A.A.; Dunsby, C.; et al. Characterisation of new gated optical image intensifiers for fluorescence lifetime imaging. Rev. Sci. Instrum. 2017, 88, 013707. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, L.M.; Becker, W.; Milnes, J.; Conneely, T.; Smietana, S.; Le Marois, A.; Jagutzki, O.; Suhling, K. Picosecond wide-field time-correlated single photon counting fluorescence microscopy with a delay line anode detector. Appl. Phys. Lett. 2016, 109, 071101. [Google Scholar] [CrossRef] [Green Version]
- Pavia, J.M.; Wolf, M.; Charbon, E. Measurement and modeling of microlenses fabricated on single-photon avalanche diode arrays for fill factor recovery. Opt. Express 2014, 22, 4202–4213. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Cuenca, R.M.; Liu, B.; Malik, B.H.; Jabbour, J.M.; Maitland, K.C.; Wright, J.; Cheng, Y.-S.L.; Jo, J.A. Handheld multispectral fluorescence lifetime imaging system for in vivo applications. Biomed. Opt. Express 2014, 5, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Gorpas, D.; Ma, D.; Bec, J.; Yankelevich, D.; Marcu, L. Real-time visualization of tissue surface biochemical features derived from fluorescence lifetime measurements. IEEE Trans. Med. Imaging 2016, 35, 1802–1811. [Google Scholar] [CrossRef]
- Cosci, A.; Nogueira, M.S.; Pratavieira, S.; Takahama, A.; de Souza Azevedo, R.; Kurachi, C.; Kurachi, C. Time-resolved fluorescence spectroscopy for clinical diagnosis of actinic cheilitis. Biomed. Opt. Express 2016, 7, 4210–4219. [Google Scholar] [CrossRef]
- Lagarto, J.L.; Phipps, J.E.; Faller, L.; Ma, D.; Unger, J.; Bec, J.; Griffey, S.; Sorger, J.; Farwell, D.G.; Marcu, L. Electrocautery effects on fluorescence lifetime measurements: An in vivo study in the oral cavity. J. Photochem. Photobiol. B Biol. 2018, 185, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Poland, S.P.; Krstajić, N.; Monypenny, J.; Coelho, S.; Tyndall, D.; Walker, R.J.; Devauges, V.; Richardson, J.; Dutton, N.; Barber, P.; et al. A high speed multifocal multiphoton fluorescence lifetime imaging microscope for live-cell FRET imaging. Biomed. Opt. Express 2015, 6, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Biskup, C.; Zimmer, T.; Kelbauskas, L.; Hoffmann, B.; Becker, W.; Bergmann, A.; Benndorf, K. Multi-dimensional fluorescence lifetime and FRET measurements. Microsc Res. Tech. 2007, 451, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Grecco, H.E.; Roda-Navarro, P.; Verveer, P.J. Global analysis of time correlated single photon counting FRET-FLIM data. Opt. Express 2009, 17, 6493–6508. [Google Scholar] [CrossRef] [PubMed]
- Strat, D.; Dolp, F.; von Einem, B.; Steinmetz, C.; von Arnim, C.A.F.; Rueck, A. Spectrally resolved fluorescence lifetime imaging microscopy: Förster resonant energy transfer global analysis with a one- and two-exponential donor model. J. Biomed. Opt. 2011, 16, 026002. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Fluorophores | Tissue Measurements | |
|---|---|---|
| Gate width | 4 ns | 4 ns |
| Gate shift | 200 ps | 400 ps |
| Number of gates | 80 | 40 |
| Phantom | i | ii | iii | iv |
|---|---|---|---|---|
| Layer 1 | Water (2.0 mm) | FAD (1.0 mm) | FAD (1.0 mm) | FAD (1.0 mm) |
| Layer 2 | FAD (2.0 mm) | POPOP (2.0 mm) | Water (2.0 mm) | Water (1.5 mm) |
| Layer 3 | - | - | POPOP (2.0 mm) | Water (2.0 mm) |
| Layer 4 | - | - | - | POPOP (2.0 mm) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagarto, J.L.; Credi, C.; Villa, F.; Tisa, S.; Zappa, F.; Shcheslavskiy, V.; Pavone, F.S.; Cicchi, R. Multispectral Depth-Resolved Fluorescence Lifetime Spectroscopy Using SPAD Array Detectors and Fiber Probes. Sensors 2019, 19, 2678. https://doi.org/10.3390/s19122678
Lagarto JL, Credi C, Villa F, Tisa S, Zappa F, Shcheslavskiy V, Pavone FS, Cicchi R. Multispectral Depth-Resolved Fluorescence Lifetime Spectroscopy Using SPAD Array Detectors and Fiber Probes. Sensors. 2019; 19(12):2678. https://doi.org/10.3390/s19122678
Chicago/Turabian StyleLagarto, João L., Caterina Credi, Federica Villa, Simone Tisa, Franco Zappa, Vladislav Shcheslavskiy, Francesco Saverio Pavone, and Riccardo Cicchi. 2019. "Multispectral Depth-Resolved Fluorescence Lifetime Spectroscopy Using SPAD Array Detectors and Fiber Probes" Sensors 19, no. 12: 2678. https://doi.org/10.3390/s19122678