Introduction
The phenomenon of heterogeneous redox catalysis at electrode surfaces has been studied extensively over the last twenty years. Much attention has been focused on chemically modified electrodes, including, for example, electrodes modified with electroactive polymer films [
1], adsorbed redox active dye molecules [
2], conductive oxide surfaces [
3] , and, more recently, metallic electrodes coated with adsorbed redox active monolayers which are generated via self assembly mechanisms [
4].
Redox mediation is simple in concept. In this process surface immobilized sites may be activated electrochemically via application of a voltage to the support electrode surface. The latter sites may then oxidize or reduce other redox agents located in the solution phase adjacent to the immobilized layer, for which the direct oxidation or reduction at the electrode surface is inhibited, either because of intrinsically slow heterogeneous electron transfer kinetics, or because close approach of the soluble redox species to the electrode is prevented.
In two recent papers [
5,
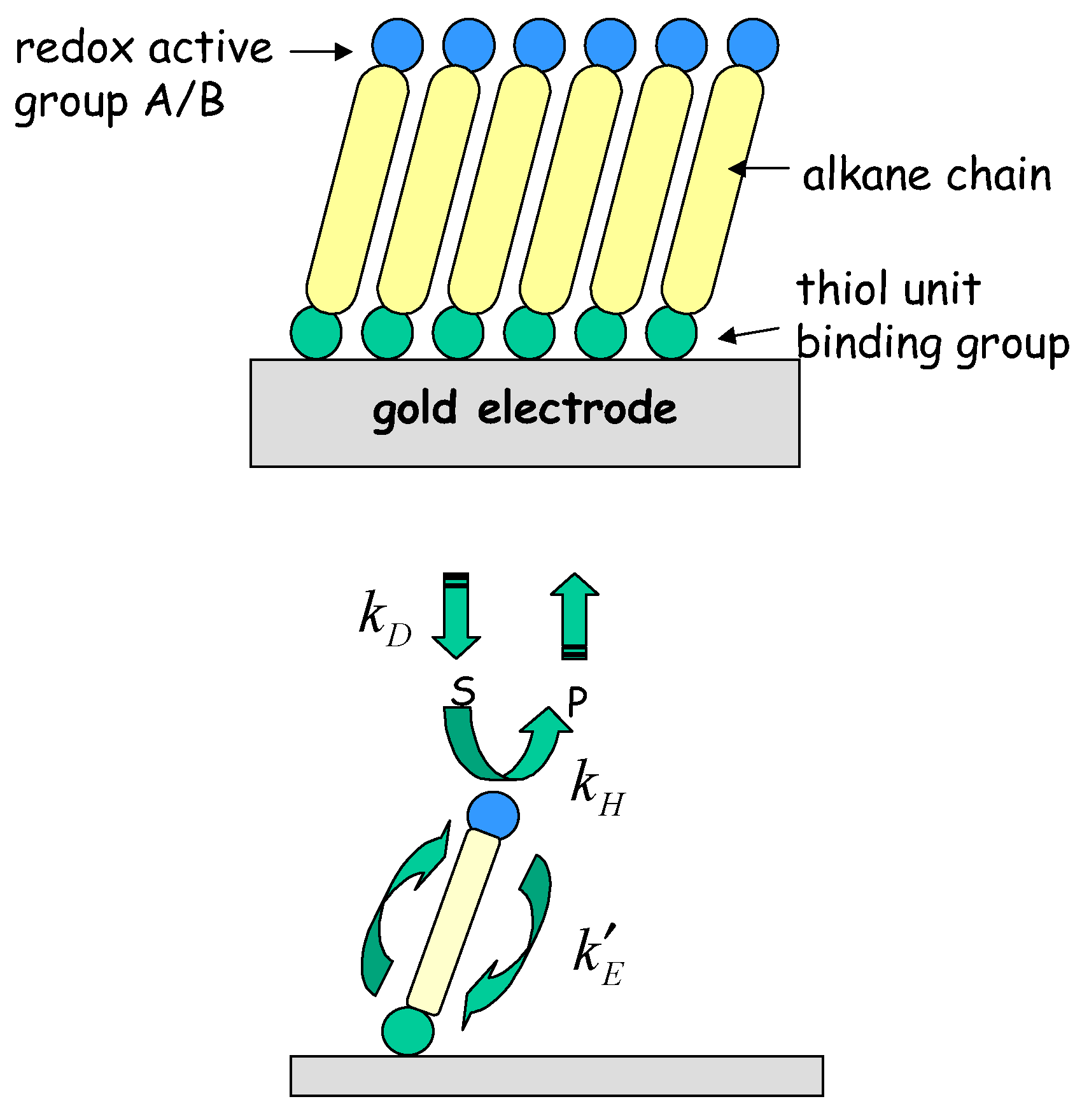
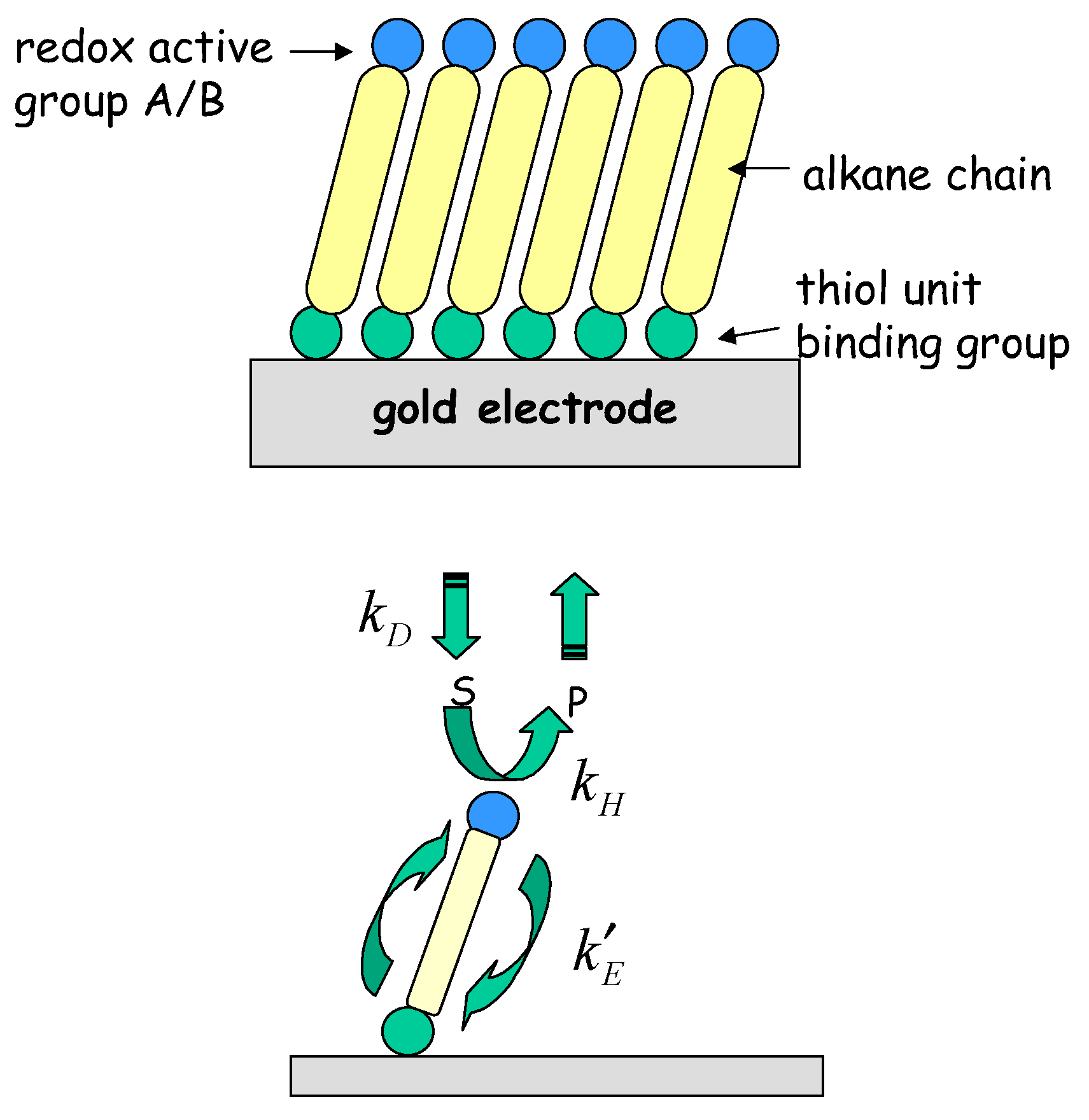
6] Creager and co-workers have developed the interesting idea that redox molecule based nanoelectrode ensembles may serve as mechanically robust model systems for individual molecular nanoelectrode systems. The former consist of redox molecule based active sites immobilized on otherwise passivated electrode surfaces. These workers visualized a redox active molecule covalently bound to a molecular connecting unit of well defined length, the end of which could be anchored to the underlying support electrode surface. A concrete manifestation of such a single molecule nanoelectrode would be a ferrocene moiety covalently attached to an alkane thiol connector. An ensemble of isolated molecular nanoelectrodes could in principle be generated on a support surface, via generation of a mixed alkane thiol based monolayer containing a fixed fraction of ferrocene containing alkane thiol units by self assembly methods. The basic idea is presented in
fig. 1.
Figure 1.
Schematic representation of mediated electron transfer at an individual molecular nanoelectrode.
Figure 1.
Schematic representation of mediated electron transfer at an individual molecular nanoelectrode.
Creager and co-workers [
5,
6] developed a simple theoretical model which enabled analytical expressions describing the current/voltage curve for mediated electron transfer at an ensemble of independent molecular nanoelectrodes to be developed. They considered possible rate limitation via bimolecular redox reaction between the active site molecule and the redox molecules in solution and by the heterogeneous redox reaction between the electrode and the active site molecule. They also used the latter expressions to interpret preliminary data for ultra sensitive electrochemical detection in flowing streams via an electrochemical amplification process which was presumed to involve redox mediation by individual analyte molecules adsorbed onto monolayer coated electrodes.
In the present paper we extend the Creager analysis and develop a comprehensive theoretical model to describe redox mediation at monolayers containing single molecule nanoelectrodes. The current response under steady state conditions will be derived and approximate analytical expressions for the current response corresponding to possible rate limitation due to bimolecular kinetics, heterogeneous electrode kinetics and reagent transport in solution will be developed. The connection between possible rate limiting situations will be presented by means of a kinetic case diagram.
Problem definition
Mediated electron transfer of solution phase species at electrode surfaces containing immobilized redox species can be examined experimentally using a number of electrochemical techniques. The technique of rotating disc voltammetry is most often applied, since in principle, the processes of reactant transport in solution, and kinetic processes at the monolayer can be cleanly separated, by conducting voltammetric experiments over a range of rotation speeds. Previous theoretical work describing mediated redox catalysis at monolayers in the context of rotating disc voltammetry has been presented by Andrieux and Saveant [
7], and Laviron [
8], More recently the analysis has been extended to a time dependent technique such as cyclic voltammetry by Aoki and co-workers [
9] and by Xie and Anson [
10,
11,
12].
In the following analysis we let A and B represent the reduced and oxidized forms of the surface immobilized redox couple, and S and P the substrate (reactant species) and product respectively. We will assume that both the heterogeneous electron transfer between the immobilized mediating site and the support electrode and the bimolecular cross exchange reaction between the substrate species S and the mediating species B are quasi reversible. The former process is quantified by the heterogeneous rate constants k’E and k’-E and the latter by the bimolecular rate constants k and k’. We also assume that solution phase material transport to the immobilized monolayer is described by a diffusional rate constant kD which is given by where Dj denotes the diffusion coefficient of species j (j = S or P) and δ denotes the diffusion layer thickness. For simplicity we assume that the diffusion coefficient of reactant species S and product species P are equal.
We consider the following general reaction scheme:
In the latter scheme the subscripts 0 and ∞ represent bulk solution and interface region respectively, and the double arrow denotes that each reaction step is microscopically reversible. Under steady state conditions we can describe the net flux (units: mol cm
-2s
-1) as follows:
where i denotes the current, n, F and A represent the number of electrons transferred, the Faraday constant and the electrode area respectively, s
∞ denotes the bulk concentration of reactant species S, s
0 is the surface concentration of reactant, p
0 is the surface concentration of product (all solution phase species having concentration units of mol cm
-3) and Γ
A , Γ
B represent the surface coverages (units: mol cm
-2) of reduced and oxidized mediator species respectively. We assume that the bulk concentration of product is zero. We also assume that the heterogeneous electrochemical rate constants are given by expressions of the Butler-Volmer form:
where k
0 denotes the standard electrochemical rate constant, α is the transfer coefficient and ξ denotes a normalized potential given by:
We also note that the total surface coverage of redox mediator species is given by:
Detailed examination of eqn.1 and eqn.4 yields:
We can also derive an expression between the rate constants for the forward and reverse step of the bimolecular cross exchange reaction between reactant species and immobilized mediator group as follows. The degree of reversibility of the cross exchange process is described in terms of the equilibrium constant K which is given by:
and so we note that
=
kHk-1.
It is also useful to note from simple thermodynamic arguments that the equilibrium constant for the cross exchange reaction is related to the standard potentials for the reactant/product transformation and the redox mediator couple via:
This identity has been used with profit by Laviron [
8] in a previously published analysis on redox catalysis at polymer coated electrode surfaces.
Eqn. 1, eqn.4, eqn.5 and eqn.6 fully define the kinetic problem, and by a suitable algebraic manipulation, one can derive a useful expression for the net reaction flux fΣ expected under steady state conditions.
The normalized master equation
To proceed further in the analysis and connect directly with experiment it is often useful to avoid the algebraic clutter and introduce normalized variables. We firstly introduce a normalized flux Ψ in which the net reaction flux f
Σ is related to the diffusion controlled flux f
D as follows:
We also find it expedient to introduce further dimensionless parameters:
The θ parameter relates the flux associated with forward heterogeneous electron transfer involving the mediator to the diffusion flux of reactant in the solution. The φ parameter relates the forward heterogeneous electron transfer flux to the flux describing the cross exchange reaction. The ζ parameter measures the degree of reversibility associated with the heterogeneous electron transfer process and the β parameter relates the flux associated with the reverse heterogeneous electron transfer step to that of reactant diffusion.
We can substitute eqn.10 and eqn.11 into the master expression presented in eqn.9 to obtain the following normalized master expression:
We can now simplify the master expression by considering conditions which will usually pertain experimentally. Firstly we can assume that the heterogeneous electron transfer reaction involving the mediator couple is irreversible and so the parameter ζ << 1. If we also assume that the cross exchange process involving the immobilized mediator species B and the reactant species S is thermodynamically favoured, then the equilibrium constant K >> 1. Under such circumstances we can assume that 1 + ζ ≅ 1 and
K−1 → 0 and the master expression reduces to:
We will work with this simplified expression for the remainder of the paper.
Approximate analytical expressions for the reaction flux
We now examine eqn.13 and derive a number of approximate analytical expressions for the reaction flux corresponding to specific rate limiting cases. Now a significant simplification can be made if the term
![Sensors 01 00215 i013]()
is small. Under such circumstances we note that
![Sensors 01 00215 i014]()
and so
![Sensors 01 00215 i015]()
. If the latter is substituted into eqn.13 we obtain the following approximate expression for the normalized flux:
Using eqn.10 and eqn.11 we immediately note that the net reaction flux is given by:
It is useful to invert the latter expression to obtain:
From this expression we obtain three terms on the right hand side of the expression. The first relates to rate determining heterogeneous electron transfer, and is potential dependent via the k’
E factor. The second reflects rate determining chemical reaction involving cross exchange of electrons between the immobilized mediator and the solution phase reactant. The third term reflects rate determining reactant transport. Hence the possible rate limiting steps are all cleanly separated. In
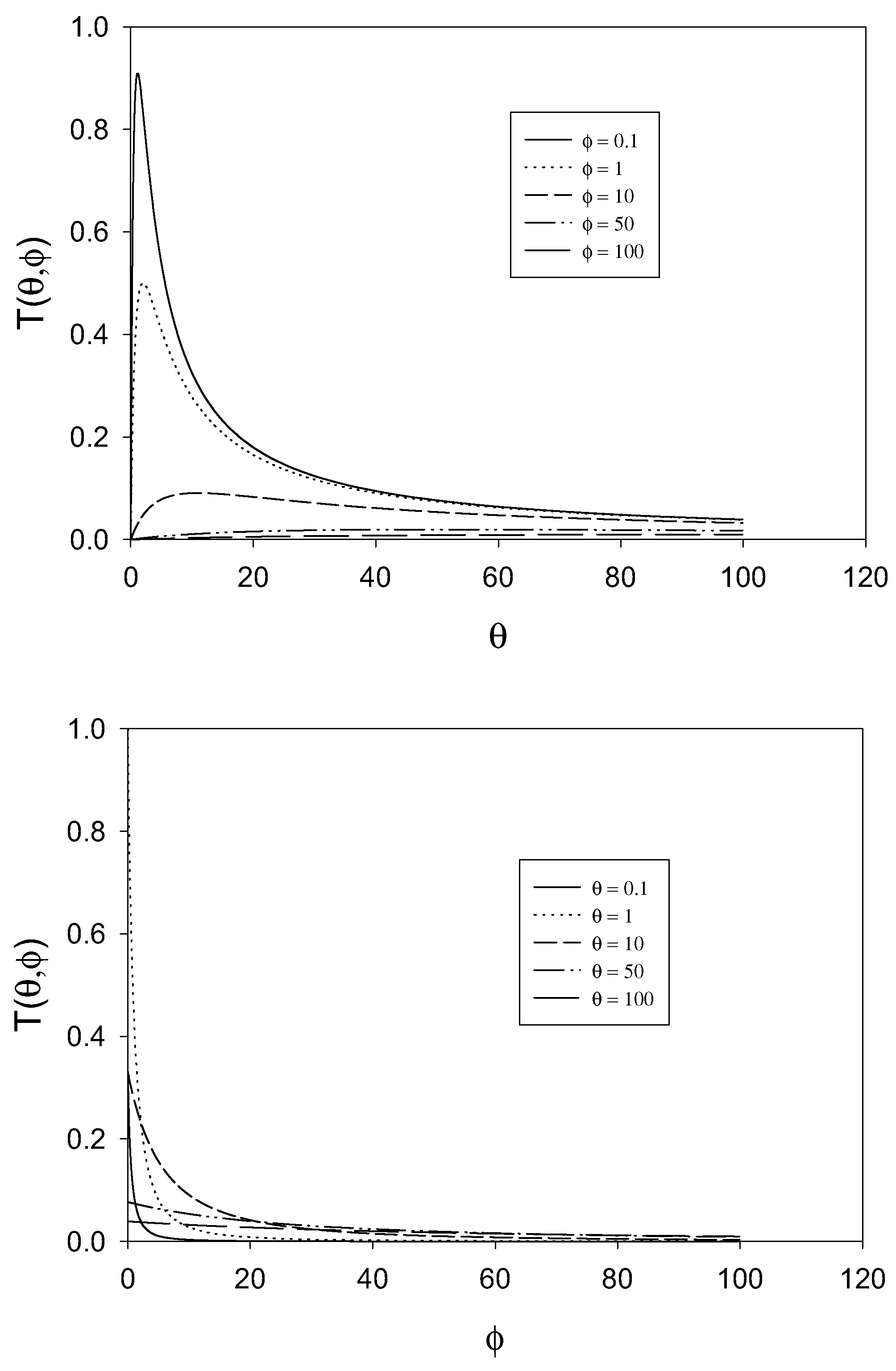
figure 2 we examine the variation of the T term as a function of the dimensionless parameters θ and φ .The approximation that the T term is small (being much less than unity) will break down when φ is small and when θ is small. The approximation is a good one when θ and φ are large, typically greater than 10.
Figure 2.
Variation of the T term defined as
![Sensors 01 00215 i018]()
with θ and φ.
Figure 2.
Variation of the T term defined as
![Sensors 01 00215 i018]()
with θ and φ.
We can obtain still more simple results. First consider the situation in which θ >> φ. This corresponds to the situation where
![Sensors 01 00215 i019]()
Hence
![Sensors 01 00215 i020]()
. Now θ >> 1 and so (1+
θ)
2 ≅
θ2 and so
T ≅
4 << 1. Hence the normalized flux is given by:
Hence we conclude that for θ >> φ and for θ >> 1, Ψ ≅ 1 or in terms of the net flux:
and control of the net rate is by diffusive transport of the reactant species in solution.
Conversely, if
![Sensors 01 00215 i023]()
then θ << φ and (1 +
θ +
φ)
2 ≅ (1 +
φ)
2 and
T(
θ,
φ) ≅
, which again will be small since φ is large and θ is small. Hence the expression for the normalized flux takes the form:
Hence when θ << φ and for φ >> 1, the normalized flux is Ψ ≅
, or in terms of the net reaction flux:
and the net rate will be determined by the kinetics cross exchange reaction.
Finally when the parameters θ and φ are both small, then to a good approximation (1 +
θ +
φ)
2 ≅ 1 and
T(
θ,
φ) ≅ 4
θ. Under these circumstances we note that the normalized flux expression reduces to:
Hence for small θ and φ the normalized flux is Ψ ≅
θ and transforming the latter expression it is readily shown that the net reaction flux is:
and the reaction is controlled by the heterogeneous electrode kinetics involving generation of the active form of the immobilized mediator species.
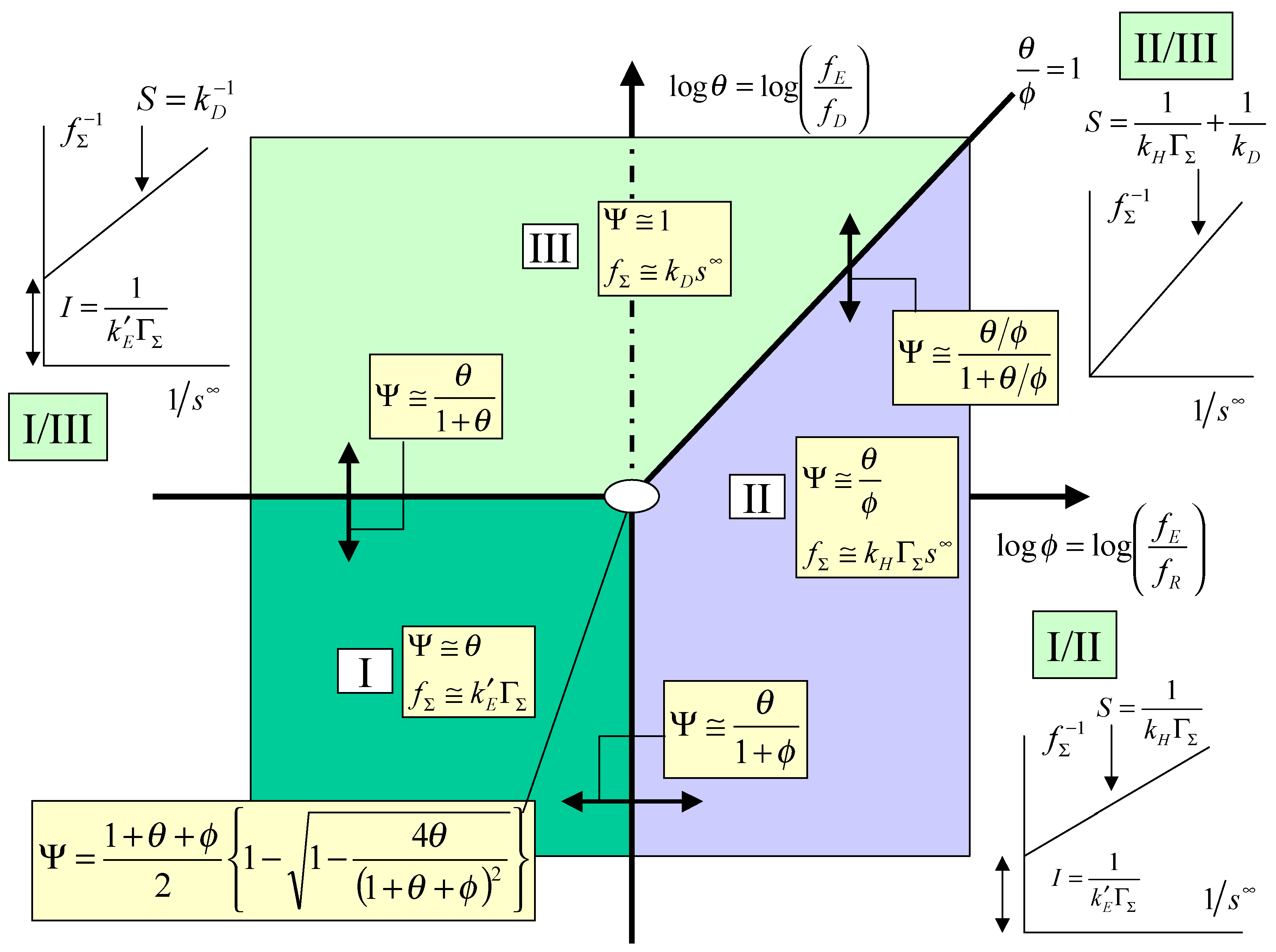
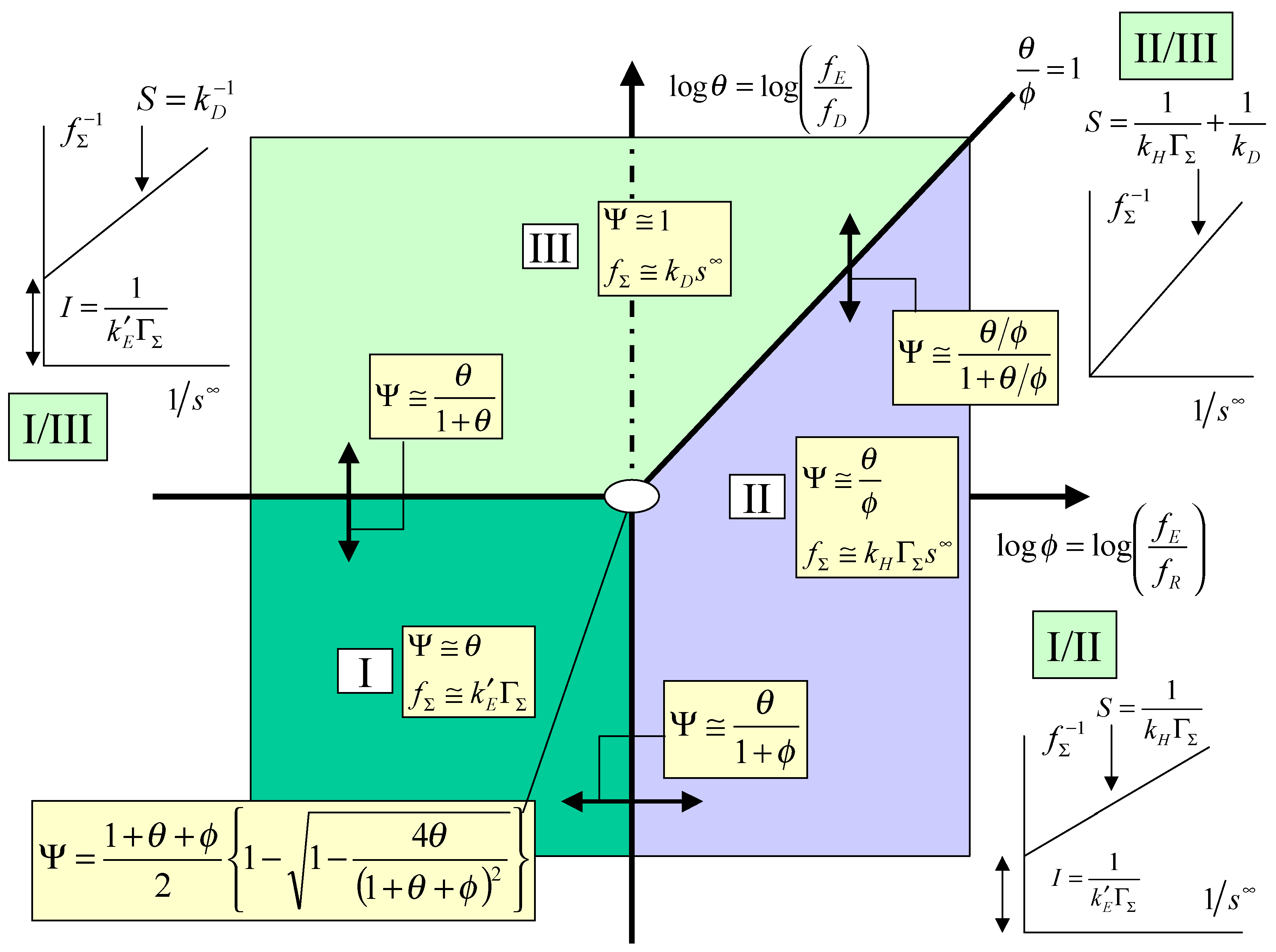
The kinetic case diagram
The kinetic analysis just presented may be conveniently summarized in terms of a kinetic case diagram. In this type of presentation the dimensionless parameters which govern the limiting behaviour of the kinetic rate equations are plotted in either a two or three dimensional format. Since in this paper we have focused on the simpler master expression outlined in eqn.13 the case diagram will be two dimensional. If instead we repeated the kinetic analysis using the more general expression outlined in eqn.12, a three dimensional case diagram would pertain. Here the pertinent parameters are log
θ = log
and log
φ = log
and these are used to define the axes of the two dimensional case diagram outlined in
figure 3, in which log φ serves as abscissa and log θ as ordinate. The locations of the limiting kinetic behaviour are clearly outlined in the diagram.
Case I which is located in the lower left hand quadrant of the case diagram defines the region where the net flux or current is controlled by the heterogeneous electron transfer kinetics involving mediator generation. Here the cross exchange reaction between mediator and solution phase reactant and the diffusive transport of the reactant will both be rapid. The pertinent expression for the flux is given by eqn.22. In this region the current will exhibit a marked potential dependence due to the fact that the flux is directly proportional to the heterogeneous electrochemical rate constant k’
E, which is described by the Butler-Volmer equation. Case I will be valid when both θ and φ are small. One would expect a Tafel behaviour to be evident in the current/potential curve. Creager and co-workers [
5] have developed a similar type of limiting kinetic expression (see eqn.7 and
figure 3 in [
5]). In this region the current response is expected to be independent of the bulk concentration of the substrate and independent of the magnitude of the rate constant quantifying the cross exchange kinetics.
Case II is located in the lower right quadrant and also extends into a section of the upper right hand quadrant . The region is delineated by the lines φ = 1 and θ/φ = 1. In case II the net flux or current is controlled by the kinetics of the cross exchange reaction between the immobilized mediator and the solution phase reactant. Both mediator generation and diffusive transport of reactant will be relatively rapid. The current response will be well described by eqn.20. Here the current will be proportional to both the mediator surface coverage and the bulk concentration of the substrate. It will be independent of applied electrode potential. We also note that the boundary between case I and case II is given by the expression:
Transforming this expression into the net flux we obtain that:
Hence we note that as the normalized potential ξ (where the latter is defined by eqn.3) is increased we cross from case I to case II. We also note from eqn.24 that a plot of inverse flux or inverse current versus inverse substrate concentration is linear, with a slope given by
S =
, and an intercept given by
I =
. This intercept should be potential dependent. For a given surface coverage the intercept should decrease in magnitude with increasing potential. This useful diagnostic plot is also included in the relevant section of the kinetic case diagram. Indeed Creager and co-workers [
5] indicated that variation of reactant concentration should prove to be a useful mechanistic indicator and the analysis suggested in the present paper supplements this comment.
We finally consider case III. This region is located in the top left quadrant of the case diagram and also extends into the the top right quadrant but is bounded there by the line θ/φ = 1. Hence case III is bounded by the lines θ = 1 and θ/φ = 1. In this region the normalized flux or current will be determined by diffusive transport of reaction in the solution. The heterogeneous and cross exchange kinetics will both be rapid. The steady state current response will be given by eqn.18. In this region the current will not depend on the applied electrode potential. In their work Creager and co-workers [
5] did not consider specifically this case. The current will depend linearly with the bulk concentration of reactant. It will also depend on the diffusion rate constant k
D.
Figure 3.
Kinetic case diagram for mediated electron transfer at a redox active monolayer.
Figure 3.
Kinetic case diagram for mediated electron transfer at a redox active monolayer.
We note that the equation relating the case I/case III regions takes the form:
and reflects the joint control by heterogeneous electron transfer and reactant diffusion. Transforming this expression into the net flux we obtain:
We note from eqn.26 that the steady state current / potential curve should be sigmoidal and exhibit a potential independent plateau region reflecting the onset of diffusion control. Inversion of eqn.26 indicates that a plot of inverse flux or inverse current is linear with inverse bulk reactant concentration, with a slope S given by
S =
and an intercept I given by
I =
. The intercept will be potential dependent and will decrease in magnitude with increasing surface coverage of mediator and with increasing potential. This characteristic case I/III plot is presented in
figure 3.
We finally note that the case II/ case III boundary is defined by the following expression:
which reflects joint rate control by the kinetics of the cross exchange reaction and by reactant diffusion in solution. We can readily show that the net flux is given by:
In this case the net current should be independent of electrode potential and depend both on the bulk reactant concentration and mediator surface coverage. Inversion of eqn.28 suggests that a plot of the inverse flux versus inverse reactant concentration is linear, with a zero intercept and a slope S given by
S =
+
. This diagnostic plot is outlined in
figure 3.

{kind=link}
{kind=link}
{kind=link}












 is small. Under such circumstances we note that
is small. Under such circumstances we note that  and so
and so  . If the latter is substituted into eqn.13 we obtain the following approximate expression for the normalized flux:
. If the latter is substituted into eqn.13 we obtain the following approximate expression for the normalized flux:


 with θ and φ.
with θ and φ.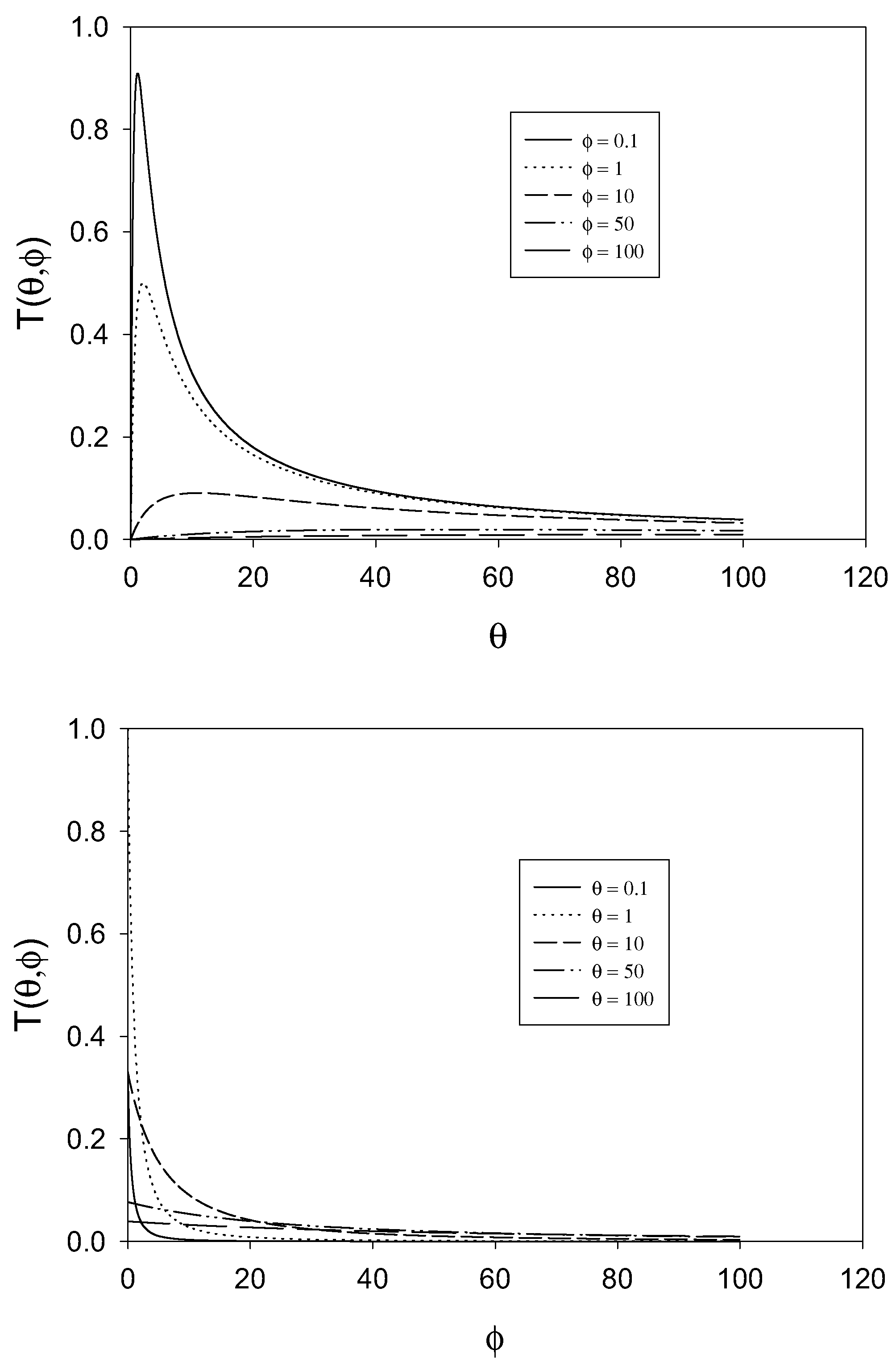
 Hence
Hence  . Now θ >> 1 and so (1+θ)2 ≅ θ2 and so T ≅ 4 << 1. Hence the normalized flux is given by:
. Now θ >> 1 and so (1+θ)2 ≅ θ2 and so T ≅ 4 << 1. Hence the normalized flux is given by:


 then θ << φ and (1 + θ + φ)2 ≅ (1 + φ)2 and T(θ, φ) ≅ , which again will be small since φ is large and θ is small. Hence the expression for the normalized flux takes the form:
then θ << φ and (1 + θ + φ)2 ≅ (1 + φ)2 and T(θ, φ) ≅ , which again will be small since φ is large and θ is small. Hence the expression for the normalized flux takes the form: