Abstract
Here we characterize the genetic structure of Black Salamanders (Aneides flavipunctatus) in the Klamath Mountains of northwestern California and southwestern Oregon using mitochondrial and nuclear DNA sequences. We hypothesized that the Sacramento, Smith, Klamath, and Rogue River watersheds would represent distinct genetic populations based on prior ecological results, which suggest that Black Salamanders avoid high elevations such as the ridges that separate watersheds. Our mitochondrial results revealed two major lineages, one in the Sacramento River watershed, and another containing the Klamath, Smith, and Rogue River watersheds. Clustering analyses of our thirteen nuclear loci show the Sacramento watershed population to be genetically distinctive. Populations in the Klamath, Smith, and Rogue watersheds are also distinctive but not as differentiated and their boundaries do not correspond to watersheds. Our historical demographic analyses suggest that the Sacramento population has been isolated from the Klamath populations since the mid-Pleistocene, with negligible subsequent gene flow (2 Nm ≤ 0.1). The Smith and Rogue River watershed populations show genetic signals of recent population expansion. These results suggest that the Sacramento River and Klamath River watersheds served as Pleistocene refugia, and that the Rogue and Smith River watersheds were colonized more recently by northward range expansion from the Klamath.
1. Introduction
Genetic data are widely used for studying microevolutionary processes such as how geographical features shape current patterns of diversity and population structure [1]. This field of study, known as landscape genetics, focuses on evolutionary processes at a finer scale than is the case for typical phylogeography studies, and allows one to assess the impact of landscape features such as mountain ranges or rivers on gene flow and isolation between populations. Such an approach provides biologists with new ecological and evolutionary insights regarding their study organisms or regions of interest [2]. While population genetics compares discrete populations that have already been identified using ecological, morphological, or phylogenetic data, landscape genetics does not require that discrete populations be designated a priori. These types of studies are particularly well suited to landscapes with high heterogeneity in topography, climate, and habitat, each of which are physical aspects of the environment that can restrict gene flow and create spatial genetic structure within species or populations. Here, we focus on the Klamath Mountains of western North America, a region that contains considerable topographic, climatic, and habitat heterogeneity [3].
The Klamath Mountains of southwest Oregon and northwest California represent a unique province on the basis of geographic, geologic, or biogeographic considerations [3,4]. A complex pattern of river canyons and mountain ridges create a topographically heterogeneous landscape that contains little flat ground [5]. The Klamath Mountains consist of five major subdivided oceanic terranes (a fragment of a tectonic plate), with the easternmost Klamath terrane being the oldest [6]. The Klamath region, a raised plain that is tilted towards the west, has been heavily eroded and dissected by rivers and streams fed by heavy winter rains. Indeed, the Klamath region has some of the highest annual rainfall in California, particularly in Del Norte, Humboldt, and Shasta Counties [4,7]. Over the past 2 million years, nearly twenty ice ages have occurred, and many areas covered with forest today were covered in ice at one time or another. Although most of the Klamath Mountains remained free of glaciation during the coldest period of the Pleistocene, about 15,000 years ago the Trinity Alps region of the Klamath Mountains contained more than 30 glaciers, with most occurring above 1,500 m in elevation [3]. This glacial maximum was followed by an abrupt increase in temperature resulting in climatic conditions similar to present conditions [4]. Today, the Klamath Mountains have a wide range of vegetation zones, with moist redwood forest along the coastline that first transitions as one moves eastward into mixed patches of oak woodland and Douglas Fir-dominated forest, and ultimately transitions into pine forest in the easternmost sectors [4]. This range contains the greatest diversity of conifer species in the world, including several species found nowhere else [5]. Although redwood forests dominate the coastal portion of the Klamath Mountains today, these forests may only be 4,000 years old, which may explain why so few species are endemic to the redwood forest habitats [8].
The black salamander (Aneides flavipunctatus) is found throughout the western portions of northern California, from the Santa Cruz Mountains northward, where the distribution terminates in extreme southwestern Oregon. This terrestrial breeding, direct developing plethodontid salamander primarily occurs in regions with greater than 75 cm of annual rainfall, can live up to 15 years in captivity, and is presumed to have limited dispersal capabilities [9]. Two recent genetic analyses of the black salamander concluded that species-level divergences occur across the range. One study based on mtDNA indicated that four distinct genetic lineages are present in this complex [10]. Two of the species-level lineages identified in that study were (1) a Shasta County lineage; and (2) a “Northwestern” lineage occupying the western Klamath Mountains; these lineages were found to be sister taxa. A separate analysis that used mitochondrial and nuclear DNA examined three ecologically and morphologically distinguishable populations of black salamanders that meet at a three way contact zone in Mendocino County, and found evidence that the main portion of the range contains species-level genetic divergence [11]. Despite these recent advances in our understanding of the genetic relationships within this species, no fine-scale genetic analysis has been conducted to investigate the relationships of the populations at the northern extent of the range of A. flavipunctatus. More specifically, the genetic structure and connectivity of populations within the Klamath River watershed are not known (only two localities were included in Rissler and Apodaca, 2007), and no samples from the Smith or Rogue River watersheds have ever been analyzed.
An ecological study of Aneides flavipunctatus showed that the species distribution is highly influenced by elevation; it is most often found at elevations below 600 m, and is almost never found at elevations greater than 1,200 m [12]. Given the elevational restriction of A. flavipunctatus, high elevation mountain ridges are expected to represent barriers to gene flow between populations. The Sacramento River watershed in Shasta County is separated from the Klamath River watershed in Trinity County by the Trinity Mountains, a high elevation mountain ridge that is greater than 1,000 m in elevation at its lowest point. The Shasta County population is distinct from Klamath River salamanders in mtDNA [10] and allozymes [13], but the level of genetic isolation from populations west of the Trinity Mountains is unstudied. Additionally, populations in Shasta County are morphologically and ecologically differentiated from populations west of the Trinity Mountains, and are distinguishable by their color pattern and microhabitat preference [12,14,15] (Figure 1). The potential also exists for genetic divergence between the Rogue and Klamath watershed populations, given the topographic features (i.e., the Siskiyou Crest) that separate them. The Siskiyou Crest is a mountain ridge with elevations well over 1,000 m, and has been shown to be a geographic barrier in the Plethodon elongatus complex [16,17]. The Smith watershed is separated from the Klamath watershed to the east by the Siskiyou Mountains, which are greater than 1,200 m in elevation, but along the coast the boundary between the watersheds is only a few hundred meters above sea level.
In this study we aim to elucidate the genetic structure of Aneides flavipunctatus in the northern portion of its range. Our goal is to assess the genetic diversity of populations within the Sacramento, Smith, Klamath, and Rogue River watersheds by analyzing sequence data from multiple mitochondrial and nuclear loci. These data will be used to identify geographically distinct genetic lineages, assess the levels of gene flow between them, and to offer recommendations on the conservation status and management of A. flavipunctatus in this region.

Figure 1.
Aneides flavipunctatus from the Klamath Mountains. (a) Smith River watershed; Af89. (Photo: M. Mulks) (b) Klamath River watershed; Af106. (Photo: M. Mulks) (c) Klamath River watershed; Af28. (Photo: M. Mulks) (d) Klamath River watershed; Af26. (Photo: M. Mulks) (e) Rogue River watershed; C1. (Photo: J. Reilly) (f) Sacramento River watershed. (Photo: G. Nafis).
2. Methods
2.1. Sampling and Geographic Distribution
We sampled Black Salamanders throughout the northern extent of the range within the Klamath, Smith, Rogue, and Sacramento River watersheds (Figure 2). Our sampling within the Klamath River watershed includes 28 salamanders from 18 localities in Del Norte, Humboldt, Trinity, and Siskiyou Counties. Our sampling of the Sacramento River watershed population includes 10 salamanders from 5 localities within Shasta County, and our sampling of the Smith River watershed includes 8 salamanders from two localities in Del Norte County. Within Oregon, we sampled 12 salamanders from Jackson Co., representing 6 distinct localities, all of which are in the Rogue River watershed. While the number of samples and localities examined varies between watershed populations they are roughly proportional to the range size of each population. Our limited sampling of the Smith watershed is due to the difficulty of finding salamanders in this region, and other than the eight samples examined here we know of only one record of an A. flavipunctatus from this watershed. Whole animals or tail tips (5mm) were collected for genetic analysis, and voucher specimens and photo vouchers are located at the Museum of Vertebrate Zoology (MVZ), UC Berkeley. Sample localities can be found in Appendix 1. Genomic DNA was extracted using either a salt extraction protocol or a DNeasy kit (Qiagen, Valencia, CA, USA).
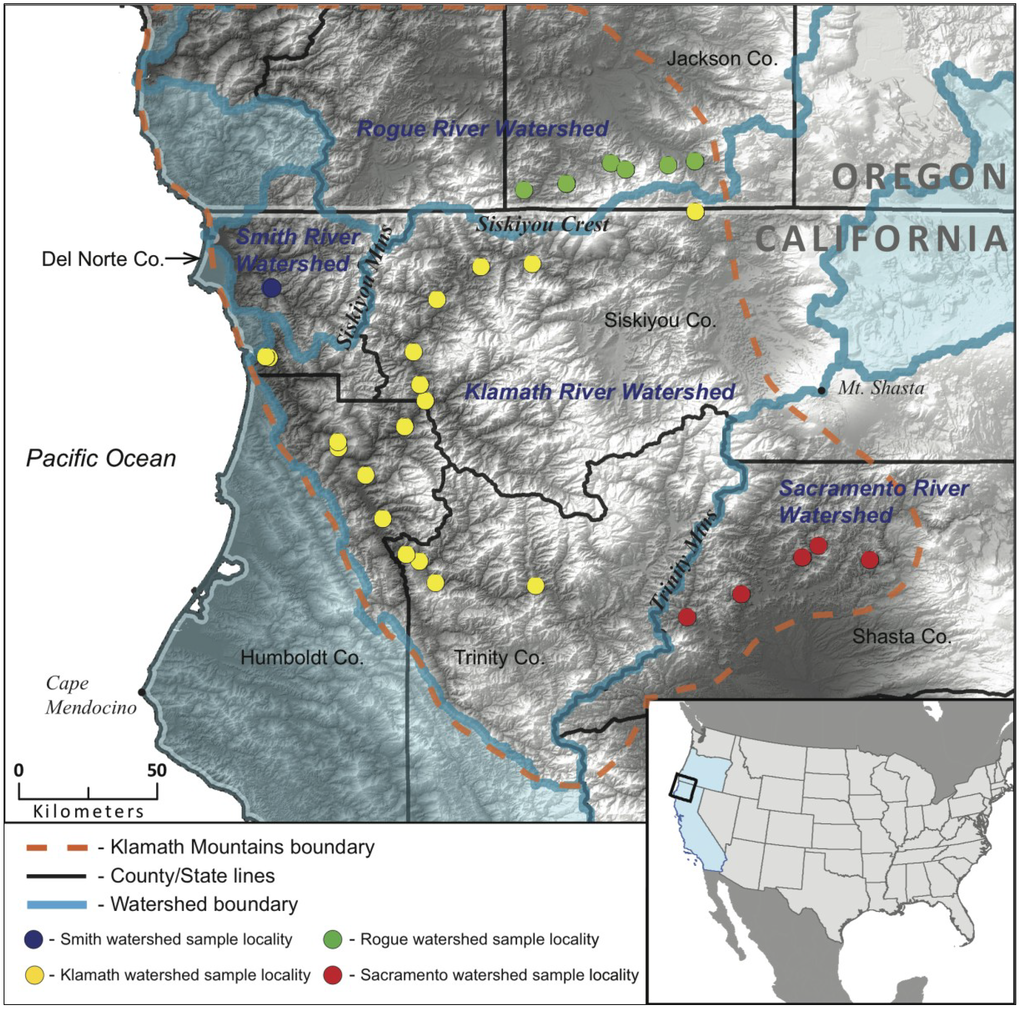
Figure 2.
Map of the four river watersheds within the Klamath Mountains inhabited by Aneides flavipunctatus, and the localities of genetic samples used in this study.
2.2. Molecular Markers
We sequenced three mitochondrial markers including the ND4, cytochrome b, and 12S genes. Primer sequences and fragment lengths of mtDNA gene regions can be found in Appendix 2. Amplification of PCR products followed standard procedures. Purified PCR products were sequenced using both forward and reverse primers on an ABI 3730 sequencer (Applied Biosystems, Forster City, CA, USA). Forward and reverse sequences were combined in CodonCode Aligner 3.5.2 (CodonCode Corporation, Dedham, MA, USA). ND4 and 12S sequences were downloaded from Genbank for the salamanders included in a previous phylogenetic study [10].
We sequenced thirteen independent nuclear loci representing one intron locus, (POMC) and twelve anonymous nuclear loci (SR1, SR2, SR4, SR7, SR8, SR9, SR12, SR15, SR16, SR17, SR19, SR20) developed specifically for Black Salamanders [10]. Amplification and sequencing of PCR products followed the same protocols as described for the mtDNA markers. Nuclear haplotype sequences were obtained for each PCR product using the software PHASE v2.1 [18,19]. For each PCR product, the most probable pair of haplotype sequences was used in our analyses. All sequences greater than 200 bp in length are deposited in GenBank (KF056387-KF056791).
2.3. Data Characteristics
The software program DNAsp [20] was used to calculate summary statistics and population genetic parameters for the populations designated in this study. For our concatenated mitochondrial dataset, we calculated watershed-specific population genetic statistics such as nucleotide diversity, Tajima’s D [21], Fu’s Fs [22], and haplotype diversity (Table 1). Additionally, the uncorrected average sequence divergence and number of fixed differences were also calculated between our a priori defined watershed populations (Table 2). Tajima’s D values can be used to detect changes in population size or signs of selection, with negative values indicating positive selection or population expansion, and positive values indicating balancing selection or population contraction [21]. Fu’s Fs values are useful for detecting genetic signatures of population growth [22]. For both Tajima’s D and Fu’s Fs, populations that have been stable are expected to have values near zero. Population-specific Tajima’s D, Fu’s Fs, and nucleotide diversity were also calculated for all nuclear loci (Table 3), and for our watershed populations (Table 4).

Table 1.
Mitochondrial loci summary statistics for each watershed population.
| Watershed | # sequences | nucleotide diversity | # haplotypes | haplotype diversity | # polymorphic sites | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|
| Rogue | 12 | 0.00064 | 5 | 0.667 | 5 | −1.29119; p > 0.10 | −1.715 |
| Klamath | 28 | 0.01097 | 11 | 0.823 | 28 | −0.15901; p > 0.10 | 0.72 |
| Sacramento | 10 | 0.00908 | 7 | 0.867 | 26 | −0.82555; p > 0.10 | 0.24 |
| Smith | 8 | 0.00046 | 4 | 0.643 | 3 | −1.44751; p > 0.10 | −1.832 |
Table 2.
Average uncorrected mtDNA sequence divergence between watersheds (lower diagonal) and number of fixed mutations (upper diagonal).
| Rogue | Klamath | Sacramento | Smith | |
|---|---|---|---|---|
| Rogue | - | 0 | 28 | 18 |
| Klamath | 0.008 | - | 20 | 0 |
| Sacramento | 0.042 | 0.047 | - | 25 |
| Smith | 0.012 | 0.013 | 0.037 | - |
Table 3.
Nuclear loci summary statistics. H = number of haplotypes, Hd = haplotype diversity.
| Locus | No. of bp | No. Sequences | h | Hd | Variable sites (not including gaps) | Parsimony informative sites | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|---|
| SR1 | 477 | 74 | 2 | 0.03 | 1 | 0 | −1.06 | −1.97 |
| SR2 | 334 | 20 | 6 | 0.84 | 16 | 14 | −0.78 | 1.46 |
| SR4 | 236 | 24 | 4 | 0.47 | 6 | 4 | −0.2 | 1.22 |
| SR7 | 564 | 32 | 9 | 0.82 | 14 | 10 | −0.46 | −0.55 |
| SR8 | 639 | 72 | 8 | 0.69 | 7 | 6 | 0.52 | −0.39 |
| SR9 | 188 | 32 | 6 | 0.70 | 6 | 4 | 0.02 | −0.37 |
| SR12 | 434 | 26 | 5 | 0.73 | 12 | 12 | 1.45 | 4.35 |
| SR15 | 525 | 30 | 8 | 0.83 | 10 | 9 | 1.37 | 0.72 |
| SR16 | 273 | 36 | 6 | 0.66 | 7 | 6 | −0.58 | −0.54 |
| SR17 | 455 | 74 | 25 | 0.75 | 24 | 17 | −0.91 | −11.23 |
| SR19 | 604 | 76 | 6 | 0.25 | 8 | 5 | −1.36 | −1.49 |
| SR20 | 577 | 68 | 9 | 0.49 | 8 | 5 | −1.24 | −4.29 |
| POMC | 481 | 98 | 4 | 0.69 | 3 | 3 | 1.46 | 1.62 |
Table 4.
Nuclear loci summary statistics for each watershed population.
| Watershed | Statistic | POMC | SR1 | SR2 | SR4 | SR7 | SR8 | SR9 | SR12 | SR15 | SR16 | SR17 | SR19 | SR20 | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rogue | Tajima’s D | −0.248 | N/A | N/A | N/A | N/A | −0.954 | N/A | N/A | −0.612 | N/A | 2.002 | N/A | 0.895 | 0.217 |
| Fu’s Fs | 0.23 | N/A | N/A | N/A | N/A | 1.523 | N/A | N/A | 0.172 | N/A | 5.36 | N/A | 1.116 | 1.68 | |
| Nucleotide Diversity | 0.00047 | 0 | N/A | N/A | 0 | 0.00089 | 0 | 0 | 0.00095 | 0 | 0.0045 | 0 | 0.00073 | 0.00069 | |
| Klamath | Tajima’s D | 1.885 | −1.129 | −0.1 | −1.162 | −0.038 | 0.353 | 0.313 | −0.173 | 1.421 | 0.065 | −0.358 | −1.073 | −0.915 | −0.07 |
| Fu’s Fs | 1.846 | −1.407 | 4.515 | −0.7 | 0.252 | −0.278 | −0.175 | −0.674 | 1.716 | 0.324 | −1.56 | 0.312 | −1.776 | 0.184 | |
| Nucleotide Diversity | 0.00191 | 0.00012 | 0.01168 | 0.00055 | 0.00479 | 0.0034 | 0.00965 | 0.00342 | 0.00944 | 0.00443 | 0.00793 | 0.00036 | 0.0015 | 0.00455 | |
| Sacramento | Tajima’s D | N/A | N/A | −0.304 | −1.233 | −0.933 | −1.132 | 0.85 | N/A | 1.754 | 0.839 | 0.159 | −1.28 | 0.932 | −0.035 |
| Fu’s Fs | N/A | N/A | 0.33 | 1.609 | −0.003 | 0.952 | 0.625 | N/A | 3.833 | 0.825 | −2.16 | −0.197 | 0.139 | 0.595 | |
| Nucleotide Diversity | 0 | 0 | 0.00321 | 0.00439 | 0.00062 | 0.00112 | 0.00284 | 0 | 0.00898 | 0.00666 | 0.00549 | 0.00278 | 0.00226 | 0.00295 | |
| Smith | Tajima’s D | 0.156 | N/A | N/A | N/A | −0.754 | −1.233 | −0.71 | N/A | 1.181 | N/A | 0.505 | N/A | −0.05 | −0.129 |
| Fu’s Fs | 0.551 | N/A | N/A | N/A | −0.288 | 1.609 | 1.099 | N/A | 3.146 | N/A | −4.414 | N/A | −0.427 | 0.182 | |
| Nucleotide Diversity | 0.00068 | 0 | N/A | 0 | 0.00266 | 0.00169 | 0.00585 | 0 | 0.00738 | 0 | 0.01029 | 0 | 0.00151 | 0.00251 |
2.4. Mitochondrial Relationships
Mitochondrial haplotype relationships were estimated using both Maximum Likelihood and Bayesian inference as implemented in the software programs GARLI [23] and BEAST v1.7.4 [24], respectively. For both analyses, we used several congeners (Aneides hardii, Aneides lugubris, Aneides vagrans, and Aneides ferreus) to root our trees. Both methods implemented the HKY + I + G model of sequence evolution as determined by the program jModeltest [25]. Our GARLI analysis was run with node support assessed on the basis of 500 bootstrap replicates. Our BEAST analysis consisted of two runs of 10,000,000 generations, sampling every 1000 generations for a total of 10,000 saved trees per run. A relaxed lognormal clock model was implemented using a calibration of 0.8% lineage divergence per million years, which was estimated for the salamander Taricha torosa [26]. We used a coalescent tree prior with constant population size to model population size change through time because we are analyzing an intraspecific dataset, and prior studies [11] have shown that within a region some populations may be growing while others may be declining. We assessed convergence by verifying that effective sample size (ESS) values were all greater than 200 in TRACER v1.5 [27]. If both runs were in agreement, we randomly chose the output of one run to present. The first 1,000 trees were discarded as burn-in, and the remaining trees were used to produce a maximum clade credibility tree. Node support was assessed using posterior probabilities.
We also used the program SplitsTree4 [28] to create an un-rooted phylogenetic network for the 57 Black Salamanders in our study. All mitochondrial loci were concatenated before running the analysis. The program returns a graphical representation of the relationships of each unique haplotype in space, with the distance between haplotypes representing their relative divergence from each other.
2.5. Population Structure
We used the software program TCS v1.21 [29] to estimate gene genealogies for our individual nuclear DNA loci. This cladogram estimation method, which is also known as statistical parsimony, depicted the results as networks of haplotypes. The size of each circle is proportional to the number of each haplotype recovered with colors corresponding to watershed populations.
We used the multi-locus clustering software program STRUCTURE v2.3 [30] to assign individuals to genetic populations. Individuals were only included in the analysis if they had been sequenced for five or more nuclear genes. We ran the program with two datasets: the first dataset treated all variable sites as independent loci; the second dataset was run under the linked model [31] so that all variable sites within a locus are linked with the distances between them specified. We ran the program with a 100,000 step burn-in followed by a 100,000 step run. Ten runs were completed for each specified population scheme from K = 1 to K = 6. These results were then imported into STRUCTURE HARVESTER [32] to determine the most likely number of populations using DeltaK values. The most likely population schemes from both the linked and un-linked analyses were plotted on a map of the region to visualize spatial population structure.
2.6. Historical Demography
We used the software program IMa2 [33] to analyze 13 independent nuclear loci to estimate historical demographic parameters such as effective population sizes, population divergence times, and migration rates. IMa2 requires non-recombining blocks of sequence data, so we used the program IMgc [34] to obtain the largest non-recombining block of sequence for each locus. Several runs were initially completed to find the appropriate prior values and to assess convergence. One final run was completed using the geometric heating scheme employing 20 chains and a burn-in of 5,000,000 generations followed by 5,000,000 generations sampling every 100 steps for a total of 50,000 saved steps. Parameter values were converted into demographic and time units [35]. We used an estimate of the mutation rate of non-coding DNA of 2.2 × 10−9 substitutions/site/year [36] to calculate the geometric mean of the 13-locus mutation rate (8.9315 × 10−7 substitutions/year) to convert parameters. We used a generation time of 3 years [37] to convert effective population size parameters.
3. Results and Discussion
3.1. Data Characteristics
Mitochondrial sequence data were unambiguously aligned first with MUSCLE [38], and then checked by eye. The total mtDNA alignment constituted 1,734 bp (735 bp ND4 plus associated tRNAs, 385 bp cytb, and 614 bp 12S) from 83 ingroup and 4 outgroup taxa. Of the 58 salamanders sampled from our region of interest (the Klamath Mountains), 27 unique mitochondrial haplotypes were recovered. Of the watershed populations examined, the Klamath and Sacramento watersheds displayed greater levels of nucleotide diversity and haplotype diversity than the Rogue or Smith watersheds (Table 1). Although none of the Tajima’s D values departed significantly from the null expectation of zero, both the Rogue and Smith River watersheds did have more negative values than the Klamath and Sacramento River watersheds suggesting the possibility that the former two drainages underwent recent population expansions to a greater degree than the latter two watersheds (Table 1). Moreover, the estimates of Fu’s Fs for the mtDNA is also consistent with this finding (Table 1).
The greatest uncorrected average sequence divergences (3.7%–4.7%) are between the Sacramento watershed and all other watersheds, while divergences between the Klamath, Smith and Rogue watersheds are smaller (0.8%–1.3%). The Rogue and Smith watersheds do not contain any fixed mutations when compared with the Klamath watershed, whereas salamanders from the Sacramento watershed contain 20 fixed mutations compared to the Klamath watershed (Table 2).
A total of 662 sequences were obtained from our thirteen independent nuclear loci, which contain a total of 95 parsimony informative sites (Table 3). Haplotype diversity for these loci ranged from 0.03–0.84 with an average haplotype diversity of 0.61. None of the Tajima’s D values were statistically significant. Tajima’s D, Fu’s Fs, and nucleotide diversity values for each population are presented in Table 4. The average nucleotide diversity across all thirteen loci was lowest for the Rogue watershed (0.00069); the Sacramento and Smith watersheds also showed low diversity (0.00295 and 0.00251, respectively), while the greatest diversity was found among populations from the Klamath watershed (0.00455). The average Tajima’s D and Fu’s Fs values were all close to 1; only populations from the Rogue watershed showed a positive Tajima’s D value and these populations had the most positive Fu’s Fs value (1.68).
3.2. Phylogenetic Analyses
Both Maximum Likelihood and Bayesian analyses produced phylogenetic trees with nearly identical topologies of which only the BEAST tree is presented (Figure 3a). These trees both show that the Sacramento watershed population is monophyletic with strong support (bootstrap = 99; posterior probability = 1) and is sister to all samples within the Klamath Mountains, which agrees with the findings of Rissler and Apodaca [9]. The Klamath mountains clade is anchored by a sample from the Mad River watershed, which is sister to a Klamath Mountains clade that includes all samples within the Klamath, Smith, and Rogue River watersheds, none of which form monophyletic assemblages. The basal split within the Klamath Mountains clade is between a sub-clade that includes all samples from the Smith watershed along with four samples from the mid-lower Klamath watershed, and another sub-clade that includes samples from throughout the Klamath watershed and all samples from the Rogue watershed (bootstrap = 76; posterior probability = 1).
Visualization of the un-rooted phylogenetic network (Figure 3b) of only the Klamath region samples displays two major mitochondrial groups: one containing all samples from the Sacramento watershed, and another containing all samples from the Klamath, Smith, and Rogue watersheds. While the Sacramento and Klamath watersheds include many haplotypes that are divergent from one another, the samples from the Smith and Rogue watersheds tended to be clumped together, indicative of the low divergence of mitochondrial haplotypes within those populations.
Our dated BEAST tree shows that this northern assemblage split from the southern populations approximately 5.5 mya (95% CI = 4–7.2 mya), and the Sacramento watershed split from the rest of the Klamath Mountains populations nearly 3.7 mya (95% CI = 2.5–5.2 mya). The split between the Mad River sample and the Klamath populations is estimated to be 2 mya (95% CI = 1.2–3 mya), and the most basal split within the Klamath mountains clade occurs approximately 1.1 mya (95% CI = 0.7–1.6 mya).
3.3. Population Structure
The haplotype networks of our thirteen nuclear loci have a wide range of topologies (Figure 4). The Sacramento watershed population contains unique haplotypes not shared by the other watersheds in eight of the 13 networks. The Rogue watershed population is distinct from other watersheds in only one network. For 10 of the loci, the Rogue watershed individuals share common haplotypes with the Klamath watershed individuals, and for 11 of the loci, the Smith individuals share common haplotypes with the Klamath.
Clustering analysis of our 13 nuclear loci converged on two different population schemes depending on the model used. In the analysis that treated each variable site as an independent locus, the DeltaK method [39] identified three clusters: one consisting of the Sacramento watershed salamanders, a second containing salamanders from the Smith, mid-Klamath and Trinity River (southern portion of the Klamath River watershed), and a third containing salamanders from the Rogue, upper Klamath River, and the mouth of the Klamath River (Figure 5a–c).
The analysis run under the Linkage model grouped individuals into four clusters based on the DeltaK values (Figure 5d–f). In this analysis, the results are similar to the unlinked analysis except that the cluster containing the Smith, mid-Klamath, and Trinity salamanders is further split into two clusters: one containing all the Smith River salamanders along with a single mid-Klamath locality and a locality in the lower Trinity River. The other cluster contains the upper Trinity River localities along with a single mid-Klamath individual. The salamanders from the lower Klamath that group with the Smith in our four-population clustering analysis are also most closely related to Smith samples in our mtDNA gene tree, suggesting that this population shares a common evolutionary history.
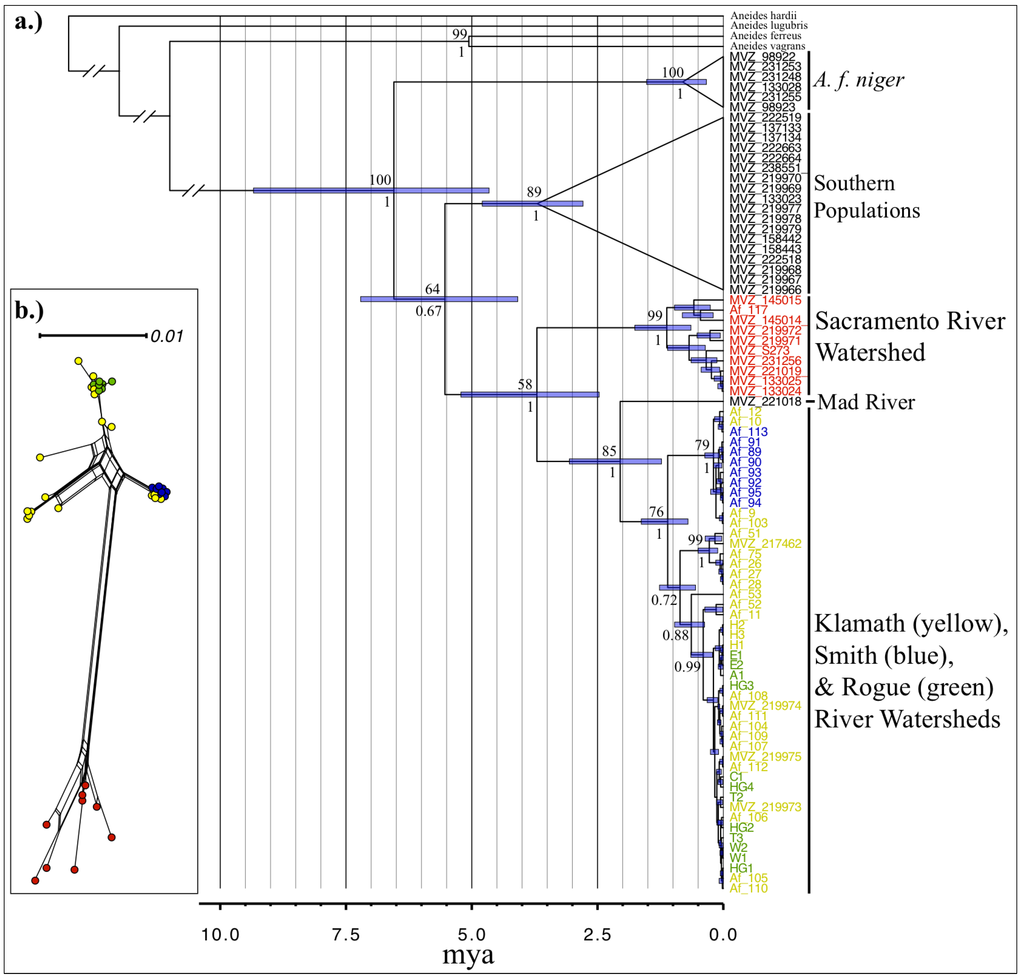
Figure 3.
(a) Time-calibrated ultrametric tree for mtDNA. Node support is represented by bootstrap (above) and posterior probability (below) values. Node bars represent 95% CI for divergence times. (b) mtDNA haplotype parsimony network of Klamath Mtns samples.
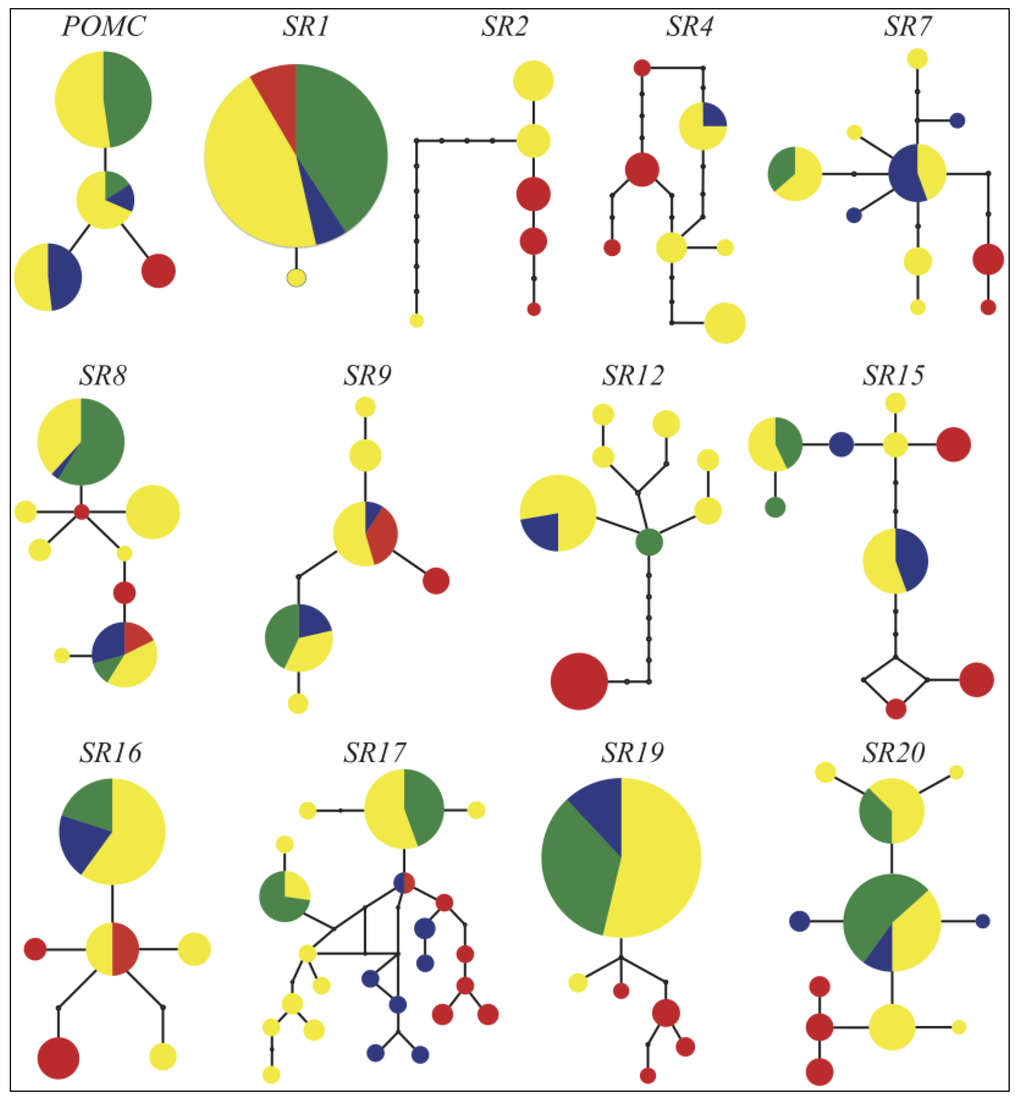
Figure 4.
Haplotype networks for nuclear loci. Colors correspond to watershed populations as shown in Figure 2. Blue = Smith, Yellow = Klamath, Green = Rogue, Red = Sacramento.
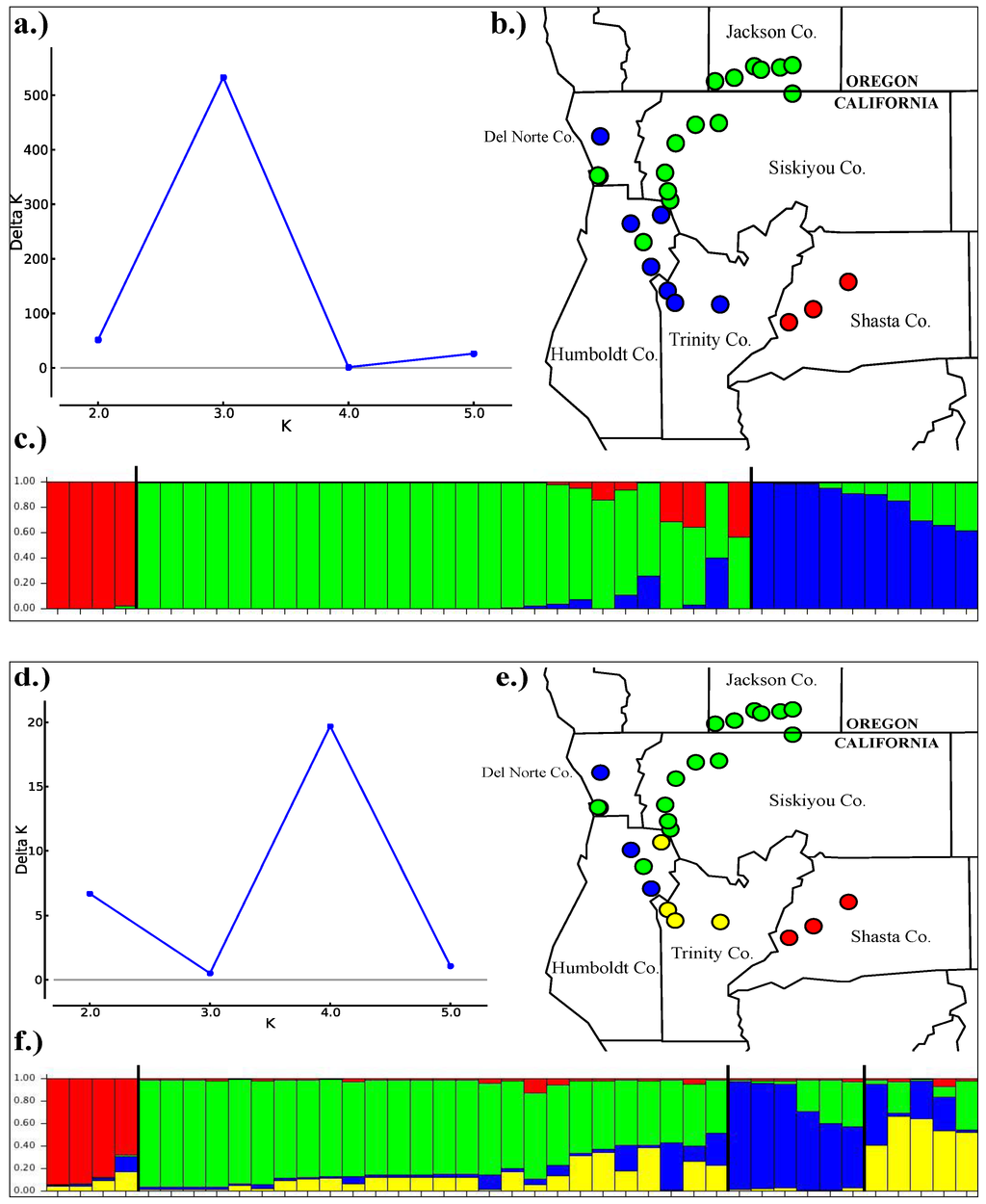
Figure 5.
(a–c) STRUCTURE results for three populations using the unlinked sites model. (a) deltaK results for the optimal value of K. (b) map of the three-population scheme. (c) three-population STRUCTURE bar graph. (d–f) STRUCTURE results for four populations using the linked sites model. (d) deltaK results for the optimal value of K. (e) map of the four-population scheme. (f) four-population STRUCTURE bar graph.
3.4. Historical Demography
Because our mitochondrial phylogeny confirmed that populations from the Sacramento watershed form a cluster that is sister to all remaining populations from the Klamath Mountains, we chose to compare the Sacramento and Klamath watershed populations in our two-population IMa2 analysis. The converted parameter values can be found in Table 5. The population divergence time between these two watershed populations is estimated to be approximately 767,000 years (95% CI = 0.6–11 mya), corresponding to a population split in the late Pleistocene. The timing of this split coincides with the lowest carbon dioxide levels ever measured from ice cores, which is correlated with lower global temperatures [40]. This significant drop in temperature may have restricted Aneides flavipunctatus to lower elevations, effectively making the Trinity Mountains a formidable barrier to dispersal and separating the populations on either side. The effective population size of the Sacramento population is estimated to be 34,000 individuals (95% CI = 19,000–69,000 individuals), and the Klamath population is estimated at 83,000 individuals (95% CI = 54,000–125,000 individuals). The ancestral population is estimated at 125,000 individuals (95% CI = 20,000–899,000 individuals), which is close to the sum of the two contemporary effective populations, suggesting that the effective population sizes have remained relatively stable since their divergence. Gene flow estimates in the form of 2 Nm values suggest that gene flow has been negligible in either direction, with 2 Nm estimates of 0.116 for gene flow into the Sacramento population from the Klamath, and 0.002 for gene flow into the Klamath from the Sacramento watershed. The upper 95% values for 2 Nm (a product of the upper 95% value for population size and half the upper 95% value for migration rate) in either direction are also less than 1. Although our IMa2 analyses of the four watershed populations and the STRUCTURE delimited populations failed to converge, we choose to explore other two-population comparisons using IMa2 (Supplementary Table S1). Most notably, our comparison of the Klamath and Rogue watersheds revealed a population divergence time of roughly 27,000 years, with a low effective population size of approximately 1,000 individuals for the Rogue population. Gene flow estimates suggest low gene flow into the Klamath watershed from the Rogue (2 Nm = 2.9), with negligible gene flow into the Rogue from the Klamath (2 Nm = 0.2). An additional run comparing the Smith and Klamath populations failed to converge on a divergence time, but suggest a small effective population size of the Smith population (approximately 5,200 individuals) with limited connectivity between the Smith and Klamath (2 Nm < 1 in either direction).
Table 5.
Converted demographic parameters from IMa2 for the Sacramento watershed and Klamath watershed population comparison.
| Population Divergence Time (years) | Effective Population Size (individuals) | Population Migration Rate (2Nm) | |||||
|---|---|---|---|---|---|---|---|
| Population Comparion | Value | t0 | Shasta (q0) | Klamath (q1) | Ancestor (q2) | m0>1 | m1>0 |
| Sacramento vs Klamath | HiPt | 766,948 | 34,055 | 82,572 | 125,492 | 0.116 | 0.002 |
| 95%Lo | 599,003 | 19,127 | 54,582 | 20,993 | 0.006 | 0.001 | |
| 95%Hi | 10,899,624 | 68,577 | 125,492 | 898,971 | 0.531 | 0.346 | |
3.5. Refugia in the Klamath Mountains
The Klamath Mountains of northwest California and southwest Oregon have been shown to be important refugia for many taxa [41]. Their rugged topography and high elevation mountain ridges appear to create sheltered corridors along the lower elevation river drainages. In this study, we hypothesized that Aneides flavipunctatus would show phylogeographic structuring with genetic breaks corresponding to high elevation ridges that separate major river watersheds. Our mitochondrial phylogeny supports this hypothesis for the Klamath and Sacramento watershed populations, which are separated by the Trinity Mountain ridge. However, our mitochondrial results show that there is minimal divergence between the Klamath and Rogue watershed populations as well as between the Klamath and Smith watershed populations. These results suggest that high elevation ridges may not always represent significant barriers to dispersal. In fact, a recent study found that another closely related salamander (Ensatina eschscholtzii) inhabits much higher elevations in the Klamath Mountains than previously thought [42], presenting the possibility that A. flavipunctatus also occurs at higher elevations than is currently documented.
The greatest genetic diversity in mitochondrial DNA was found to be in the Klamath and Sacramento watersheds, suggesting that they may have been refugia, while the lowest genetic diversity was found in the Rogue and Smith watersheds, suggesting that they have been more recently colonized from the Klamath population. Although the Siskiyou crest (which separates the Klamath from the Rogue) is not associated with a sharp genetic break it likely is a barrier to gene flow given our 2 Nm estimates, and with enough time the Rogue population could become genetically distinct. The low genetic diversity in both mtDNA and nDNA of the Rogue population is likely a result of colonization by a handful of individuals from the northern periphery of the species range. Interestingly, our mtDNA tree suggests the possibility that the Rogue watershed population is the result of at least two independent colonization events, and this hypothesis requires further testing. The eastern Rogue samples (A1, E1, and E2) form a monophyletic clade with samples from the neighboring Hilt region (samples H1-3) of the upper Klamath, while the western Rogue samples are more closely related to Klamath samples from the Seiad Valley region. Due to the low levels of genetic variation in the Rogue population, which may limit their ability to adapt changing environmental conditions or disease, we suggest that this population continue to be monitored.
The Smith watershed is separated from the Klamath by the Siskiyou Mountains, which form a high elevation ridge (>1200 m) along the inland portion of the Smith watershed, but along the coast the Smith River is separated from the Klamath River by a low elevation, forested ridge. Our genetic results suggest a recent colonization of the Smith River, and this colonization event likely occurred close to the coast where there is more continuous low elevation forested habitat. Despite being separated by low elevation forested mountains along the coast, our estimates of gene flow suggest restricted migration between the Smith and Klamath watersheds. However, our low estimate of mtDNA nucleotide diversity for the Smith population may be influenced by our sampling, as we were only able to obtain genetic samples from two nearby localities. The nucleotide diversity values of nDNA for the Smith population were almost as high as for the Sacramento population, and this finding suggests the possibility that the Smith watershed was a refugium, and that this population has more recently re-colonized the lower portion of the Klamath watershed. Another possibility for the high nDNA diversity within the Smith is a colonization scenario involving a large founding population, or that gene exchange with the lower Klamath populations is higher than estimated here, given that our gene flow parameters between these two populations did not fully converge.
Our clustering analysis of 13 nuclear loci revealed that the Sacramento watershed population is distinct, a finding that is not surprising given the topology of our reconstructed haplotype networks which showed the Sacramento watershed to rarely share haplotypes with the rest of the Klamath Mountains. Within the western Klamath Mountains, our clustering results identified two or three major populations (depending on the model used in STRUCTURE). These results show one major population (found in both analyses) to occupy the upper portion of the Klamath watershed (Siskiyou Co., CA, USA) and the Rogue watershed. The other population occupies the Trinity River, Lower Klamath River, and Smith River, and this population is further subdivided in our linked-model STRUCTURE analysis. Given that the linked-model is more appropriate for our dataset, which contains linked mutations within a gene, we believe that the four-population scheme is better supported. Since the long-term persistence of a species partly depends on the preservation of genetic diversity, and these populations each harbor distinct gene pools, we suggest that the populations recovered from our linked-model clustering analysis be treated as distinct management units for conservation purposes. Additionally, maintaining connectivity of these populations at their contact zones will be important for allowing the exchange of adaptively advantageous genes that will give these populations a better chance for long-term persistence.
Our results suggest that there are likely two or three major refugia in the Klamath Mountains for Aneides flavipunctatus: one in the Sacramento River watershed, one in the Klamath River watershed, and possibly one in the Smith watershed. Our mtDNA tree also suggests a possible refugium in the Mad River watershed, which is just outside the boundaries of the Klamath Mountains; this population requires further examination. The Klamath and Siskiyou Mountains have been shown to be Pleistocene refugia for many other species [41]. Other amphibians that show genetic signals of refugia in the Klamath Mountains include Western Toad (Anaxyrus boreas; [43]), Tailed Frog (Ascaphus truei [44,45]), Long-toed Salamander (Ambystoma macrodactylum [46]), Pacific Giant Salamander (Dicamptodon tenebrosus [47]), Rough-skinned Newt (Taricha granulosa [48]), Ensatina (Ensatina eschscholtzii [49]), Del Norte Salamander (Plethodon elongatus [16]), Siskiyou Mountains Salamander (Plethodon stormi [16]), and the Scott Bar Salamander (Plethodon asupak [16]). Other taxa that show genetic signatures of Klamath refugia include the Foxtail Pine (Pinus balfouriana [50]), Rockcress (genus Boechera [51]), and the Ariodin Slug (Prophysaon coeruleum [52]). The long held belief by many biogeographers and botanists is that the environment of the Klamath-Siskiyou region has been buffered since the Tertiary because of the region’s proximity to the coast and rugged topography [53]. Because of this long period of relative stability in the Klamath-Siskiyou region, it is now considered one of the most significant refuges for temperate north American plants and animals [53], and is recognized as a hotspot of amphibian endemism in the United States, second only to the Appalachian Mountains [54,55,56,57].
3.6. Should the Sacramento Watershed Population be Considered a Distinct Species?
Our mitochondrial phylogeny showed that salamanders from the Sacramento River watershed population (specifically in Shasta Co., CA, USA) form a monophyletic clade that diverged from the rest of the Klamath mountains salamanders nearly 3 million years ago. The uncorrected sequence divergence between Sacramento watershed salamanders and Klamath watershed salamanders is over 4%. The monophyly of the Sacramento watershed population and the 4.5% divergence estimated here are in agreement with the findings of Rissler and Apodaca [10]. Our estimate of population divergence time based on 13 nuclear loci is approximately 750,000 years, and gene flow since that time has effectively been zero. The discrepancy between our dated mtDNA tree and our coalescent-based IMa2 results can be explained by the fundamental differences of the analyses used, and the different molecular markers used for each analysis [58]. Our mtDNA tree is estimating the gene-divergence event, which typically occurs before population divergence. Our coalescent population divergence time estimate takes into account incomplete lineage sorting and gene flow, and is thought to give an estimate of the actual time that populations became physically separated. The nDNA haplotype networks show a clear pattern of genetic differentiation of the Sacramento watershed cluster compared to the rest of the Klamath Mountains. An earlier allozyme study revealed that the Shasta County population is one of the most genetically divergent populations in the Aneides flavipunctatus complex, and that it diverged from the rest of the range approximately 2.6 million years ago [13]. These combined results suggest that this population has been isolated for a substantial amount of time, and that connectivity with the rest of the range has been lost.
Previous studies have documented the ecological and morphological divergence of the Shasta County population from populations west of the Trinity Mountains. While salamanders in the Klamath watershed average 17 vertebrae, Shasta County salamanders average only 16 vertebrae [12]. Additionally, salamanders in the Klamath watershed are considered paedomorphic, in the sense that they retain the juvenile xanthophore pigments into adulthood giving the salamanders a gold/gray frosted coloration [12,14]. Shasta County salamanders do not retain this pigmentation and are solid black with small white specks concentrated on the body (Figure 1). From an ecological perspective, the Shasta salamanders prefer shaded streamside habitat while Klamath watershed salamanders prefer low elevation rock talus piles with some southerly exposure [12,13,15]. While we believe that the Shasta population represents an independent evolving lineage, elevation of this lineage to species status would render the rest of the A. flavipunctatus complex paraphyletic. Studies are currently in progress on the entire A. flavipunctatus complex, and future publications will include detailed phylogeographic studies with much greater sampling density than in prior studies, and a comprehensive taxonomic revision.
4. Conclusions
Our genetic analysis of Black Salamanders from the Klamath Mountains has revealed two major mitochondrial clades that are separated by the Trinity Mountains ridge that marks the boundary between the Sacramento River watershed and the Klamath River watershed. These two lineages were estimated to have been physically separated since the mid-Pleistocene, and have had very limited connectivity since then. Our clustering analysis of our 13 nuclear loci supported three distinct populations in the Klamath, Rogue, and Smith River watersheds. The boundaries of these populations do not correspond to watershed boundaries however, and these three populations all meet in northeastern Humboldt County in the region where the Trinity River meets the Klamath River. Salamanders in the Rogue and Smith River watersheds have low genetic diversity, and are most closely related to salamanders in the neighboring parts of the Klamath River watershed. We interpret these results as supporting the existence of Pleistocene refugia in the Sacramento and Klamath River watersheds, with more recent colonization of the Rogue and Smith River watersheds by northward range expansion from the Klamath. The population in Shasta County should be given special conservation consideration, as it is the most distinct population genetically, and may represent a species distinct from Aneides flavipunctatus. The three populations recovered from our linked model clustering analysis, found in the Klamath region west of Shasta County, should also be managed as distinct conservation units.
Acknowledgments
We thank Andrew Gottscho for assistance with field collections, Jimmy McGuire for helpful discussion and comments, Lydia Smith for laboratory support, Carol Spencer for accessioning voucher specimens, and Michelle Koo for map figure assistance. Collection permits were granted by the CDFG (# 9761) and the ODFW (# 048-10), and animal handling was governed by the Humboldt State University IACUC (07/08.B.34.A) and the UC Berkeley IACUC (R279-0114). Funding was provided by a grant from the Save-the-Redwoods League and the Medford Bureau of Land Management.
Conflict of Interest
The authors declare no conflict of interest.
References
- Manel, S.; Schwartz, M.K.; Luikart, G.; Taberlet, P. Landscape genetics: Combining landscape ecology and population genetics. Trends Ecol. Evol. 2003, 18, 189–197. [Google Scholar] [CrossRef]
- Storfer, A.; Murphy, M.A.; Spear, S.F.; Holderegger, R.; Waits, L.P. Landscape genetics: Where are we now? Mol. Ecol. 2010, 19, 3496–3514. [Google Scholar] [CrossRef]
- Orr, W.N.; Orr, E.L. Geology of the Pacific Northwest; Waveland Press, Inc.: Long Grove, IL, USA, 2002. [Google Scholar]
- Agee, J.K. Steward’s Fork: A Sustainable Future for the Klamath Mountains; University of California Press: Berkeley, CA, USA, 2007. [Google Scholar]
- Sawyer, J.O. Northwest California: A Natural History; University of California Press: Berkeley, CA, USA, 2006. [Google Scholar]
- Harden, D.R. California Geology; Prentice Hall: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Oregon Climate Service (OCS). Available online: http://www.ocs.orst.edu (accessed on 15 April 2013).
- Noss, R.F. The Redwood Forest: History, Ecology, and Conservation of the Coast Redwoods; Island Press: Washington, DC, USA, 2000. [Google Scholar]
- AmphibiaWeb: Information on Amphibian Biology and Conservation. Berkeley, CA, USA, 2013. Available online: http://amphibiaweb.org/ (accessed on 20 June 2013).
- Rissler, L.J.; Apodaca, J.J. Adding more ecology into species delimitation: ecological niche models and phylogeography help define cryptic species in the Black Salamander (Aneides flavipunctatus). Syst. Biol. 2007, 56, 924–942. [Google Scholar] [CrossRef]
- Reilly, S.B.; Marks, S.B.; Jennings, W.B. Defining evolutionary boundaries across parapatric ecomorphs of Black Salamanders (Aneides flavipunctatus) with conservation implications. Mol. Ecol. 2012, 21, 5745–5761. [Google Scholar] [CrossRef]
- Lynch, J.F. Patterns of ontogenetic and geographic variation in the black salamander, Aneides flavipunctatus. Smithson. Contrib. Zool. 1981, 324, 1–53. [Google Scholar] [CrossRef]
- Larson, A. Paedomorphosis in relation to rates of morphological and molecular evolution in the salamander Aneides flavipunctatus. Evolution 1980, 34, 1–17. [Google Scholar] [CrossRef]
- Lowe, C.H. Speciation and Ecology in Salamanders of the Genus Aneides. Ph.D. Thesis, University of California, Los Angeles, CA, USA, 1950. [Google Scholar]
- Nauman, R.S.; Olson, D.H. Surveys for terrestrial amphibians in Shasta County, California, with notes on the distribution of Shasta salamanders (Hydromantes shastae). Northwest. Nat. 2004, 85, 35–38. [Google Scholar] [CrossRef]
- Mahoney, M. Molecular systematics and phylogeography of the Plethodon elongatus species group: Combining phylogenetic and population genetic methods to investigate species history. Mol. Ecol. 2004, 13, 149–166. [Google Scholar] [CrossRef]
- Mead, L.S.; Clayton, D.R.; Nauman, R.S.; Olson, D.H.; Pfrender, M.E. Characterization of newly discovered populations of Plethodon from Siskiyou County, California. Herpetologica 2005, 61, 158–177. [Google Scholar] [CrossRef]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Stephens, M.; Scheet, P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data inputation. Am. J. Hum. Genet. 2005, 76, 449–462. [Google Scholar] [CrossRef]
- Rozas, J.; Sanchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Zwickl, D.J. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. Thesis, The University of Texas, Austin, TX, USA, 2006. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Tan, A.M.; Wake, D.B. MtDNA phylogeography of the California newt, Taricha torosa (Caudata, Salamandridae). Mol. Phylogenet. Evol. 1995, 4, 383–394. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer v1.5.0. 2009. Available online: http://beast.bio.ed.ac.uk/ (accessed on 10 Aug 2011).
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1660. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure: Extensions to linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Hey, J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 2010, 27, 905–920. [Google Scholar] [CrossRef]
- Woerner, A.E.; Cox, M.P.; Hammer, M.F. Recombination-filtered genomic datasets by information maximization. Bioinformatics 2007, 23, 1851–1853. [Google Scholar] [CrossRef]
- Hey, J. On the number of New World founders: A population genetic portrait of the peopling of the Americas. Plos Biol. 2005, 3, 965–975. [Google Scholar]
- Kumar, S.; Subramanian, S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 803–808. [Google Scholar] [CrossRef]
- Anderson, P.K. Ecology and evolution in island populations of salamanders in the San Francisco Bay region. Ecol. Monogr. 1960, 30, 359–386. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Luthi, D.; Le Floch, M.; Bereiter, B.; Blunier, T.; Barnola, J.; Siegenthaler, U.; Raynaud, D.; Jouzel, J.; Fischer, H.; Kawamura, K.; et al. High-resolution carbon dioxide concentration record 650,000–800,000 years before present. Nature 2008, 453, 379–382. [Google Scholar] [CrossRef]
- Shafer, A.B.A.; Cullingham, C.I.; Cote, S.D.; Coltman, D.W. Of glaciers and refugia: A decade of study sheds new light on the phylogeography of northwestern North America. Mol. Ecol. 2010, 19, 4589–4621. [Google Scholar] [CrossRef]
- Reilly, S.B.; Gottscho, A.D.; Garwood, J.M.; Jennings, W.B. Phylogenetic analysis of common garter snake (Thamnophis sirtalis) stomach contents detects cryptic range of a secretive salamander (Ensatina eschscholtzii oregonensis). Herpetol. Conserv. Biol. 2010, 5, 395–402. [Google Scholar]
- Goebel, A.M.; Ranker, T.A.; Corn, P.S.; Olmstead, R.G. Mitochondrial DNA evolution in the Anaxyrus boreas species group. Mol. Phylogenet. Evol. 2009, 50, 209–225. [Google Scholar] [CrossRef]
- Nielson, M.; Lohman, K.; Sullivan, J. Phylogeography of the tailed frog (Ascaphus truei): Implications for biogeography of the Pacific Northwest. Evolution 2001, 55, 147–160. [Google Scholar]
- Nielson, M.; Lohman, K.; Daugherty, C.H.; Allendorf, F.W.; Knudsen, K.L.; Sullivan, J. Allozyme and mitochondrial DNA variation in the tailed frog (Anura: Ascaphus): The influence of geography and gene flow. Herpetologica 2006, 62, 235–258. [Google Scholar] [CrossRef]
- Thompson, M.D.; Russell, A.P. Glacial Retreat and Its fluence on Migration of Mitochondrial Genes in the Long-Toed Salamander (Ambystoma macrodactylum) in Western North America. In Migration of Organisms; Elewa, A.M.T., Ed.; Springer: Berlin Heidelberg, Germany, 2005; pp. 205–246. [Google Scholar]
- Steele, C.A.; Storfer, A. Coalescent-based hypothesis testing supports multiple Pleistocene refugia in the Pacific Northwest for the Pacific giant salamander (Dicamptodon tenebrosus). Mol. Ecol. 2006, 15, 2477–2487. [Google Scholar] [CrossRef]
- Kuchta, S.R.; Tan, A. Isolation by distance and post-glacial range expansion in the rough-skinned newt, Taricha granulosa. Mol. Ecol. 2005, 14, 225–244. [Google Scholar] [CrossRef]
- Kuchta, S.R.; Parks, D.S.; Mueller, R.L.; Wake, D.B. Closing the ring: Historical biogeography of the salamander ring species Ensatina eschscholtzii. J. Biogeogr. 2009, 36, 982–995. [Google Scholar] [CrossRef]
- Eckert, A.J.; Tearse, B.R.; Hall, B.D. A phylogeographical analysis of the range disjunction for foxtail pine (Pinus balfouriana, Pinaceae): The role of Pleistocene glaciation. Mol. Ecol. 2008, 17, 1983–1997. [Google Scholar] [CrossRef]
- Kiefer, C.; Dobes, C.; Sharbel, T.F.; Koch, M.A. Phylogeographic structure of the chloroplast DNA gene pool in North American Boechera- A genus and continent-wide perspective. Mol. Phylogenet. Evol. 2009, 52, 303–311. [Google Scholar] [CrossRef]
- Wilke, T.; Duncan, N. Phylogeographic patterns in the American Pacific Northwest: Lessons from the arionid slug Prophysaon coeruleum. Mol. Ecol. 2004, 13, 2303–2315. [Google Scholar] [CrossRef]
- Harrison, S.P. Plant and Animal Endemism in California; University of California Press: Berkeley, CA, USA, 2013. [Google Scholar]
- Ricketts, T.H.; Dinerstein, E.; Olson, D.; Loucks, C.; Eichbaum, W. Terrestrial Ecoregions of North America: A Conservation Assessment; Island Press: Washington, DC, USA, 1999. [Google Scholar]
- Stein, B.A.; Kutner, L.S.; Adams, J.S. Precious Heritage: The Status of Biodiversity in the United States; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Conservation International. Biodiversity Hotspots: California Floristic Provence. Available online: www.biodiversityhotspots.org/xp/Hotspots/california_floristic/ (accessed on 2 May 2013).
- California Department of Fish and Wildlife (CDFW), Atlas of the Biodiversity of California; California Department of Fish and Wildlife: Sacramento, CA, USA, 2003.
- Edwards, S.V.; Beerli, P. Perspective: Gene divergence, population divergence, and the variance in coalescent time in phylogeographic studies. Evolution 2000, 54, 1839–1854. [Google Scholar]
Appendix
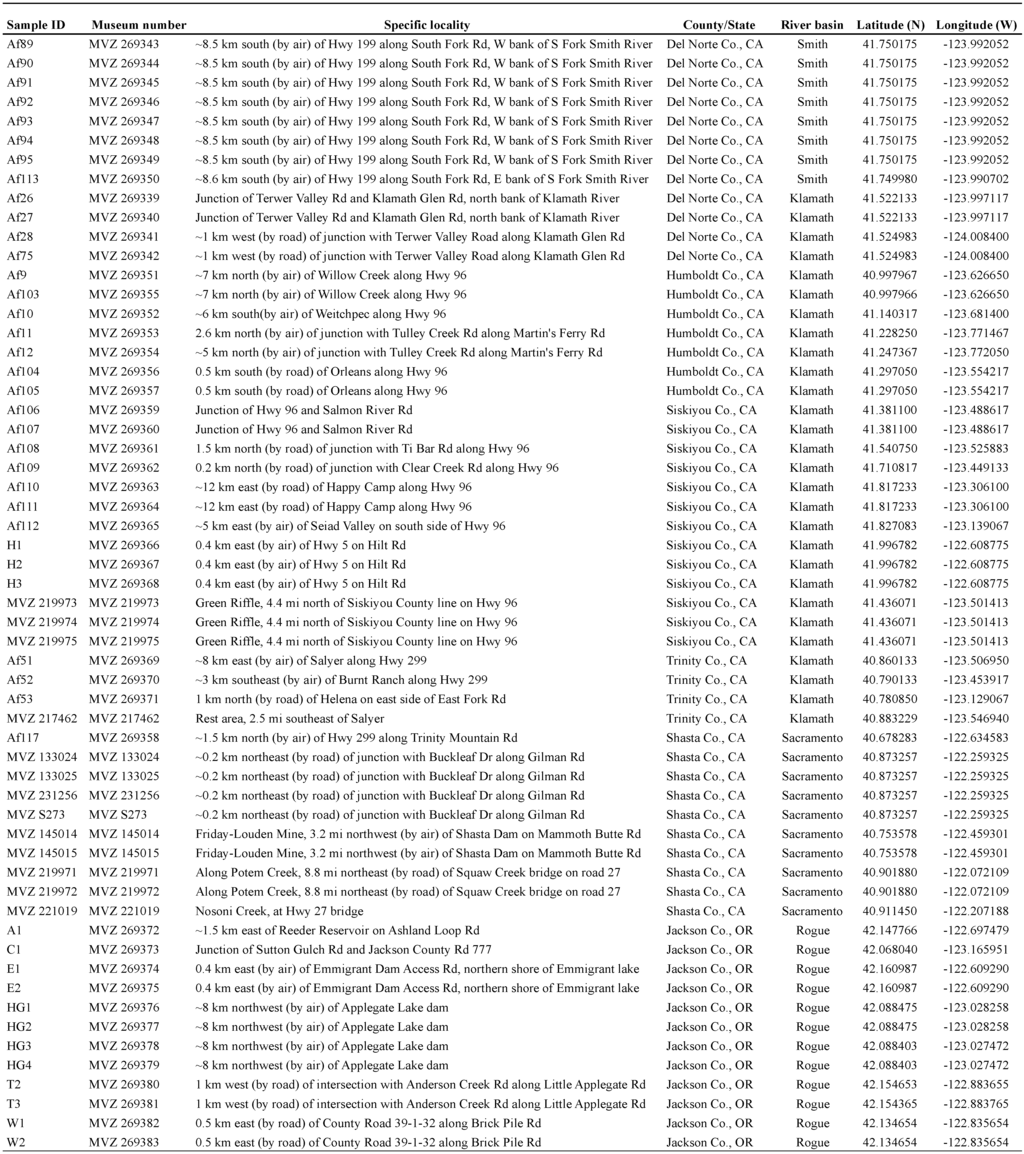
Appendix 1.
Museum catalog numbers and locality information for samples used in genetic analyses.
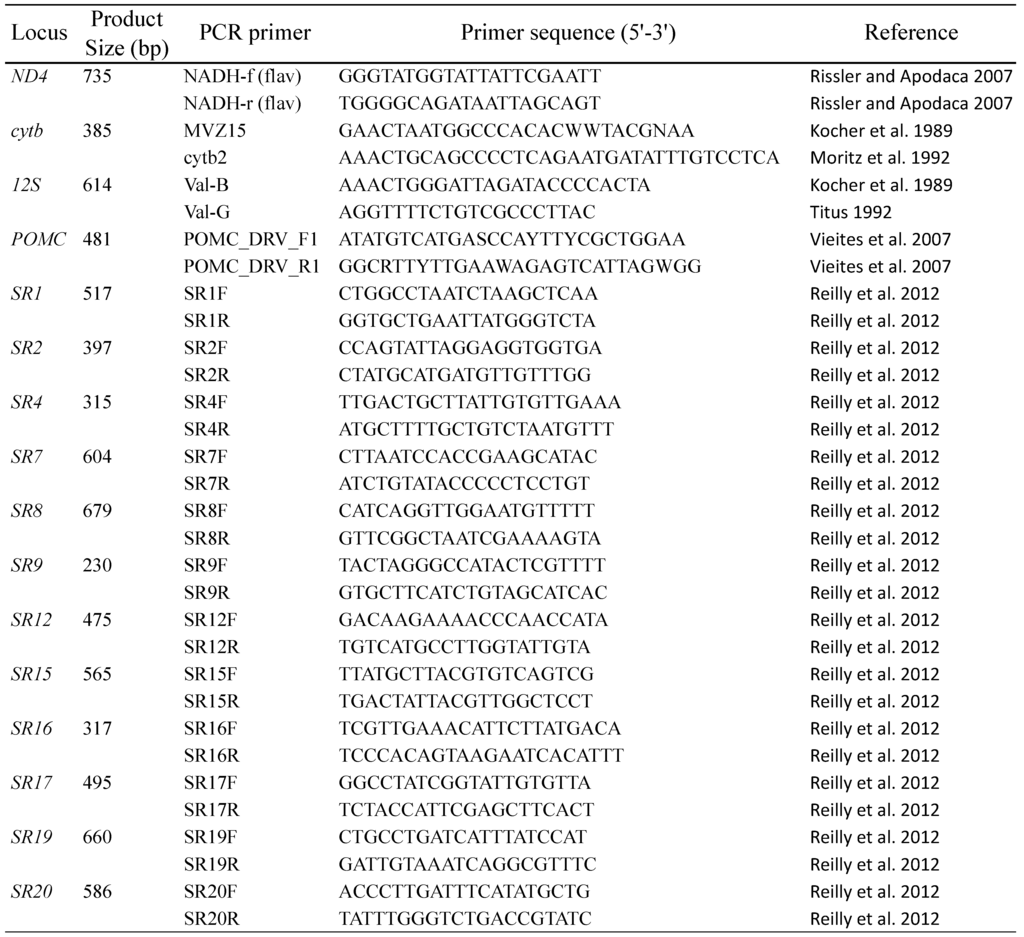
Appendix 2.
Primer information for the 16 genes sequenced in this study.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).