(1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Aminobutyrate Hydrochloride

 ,
, 


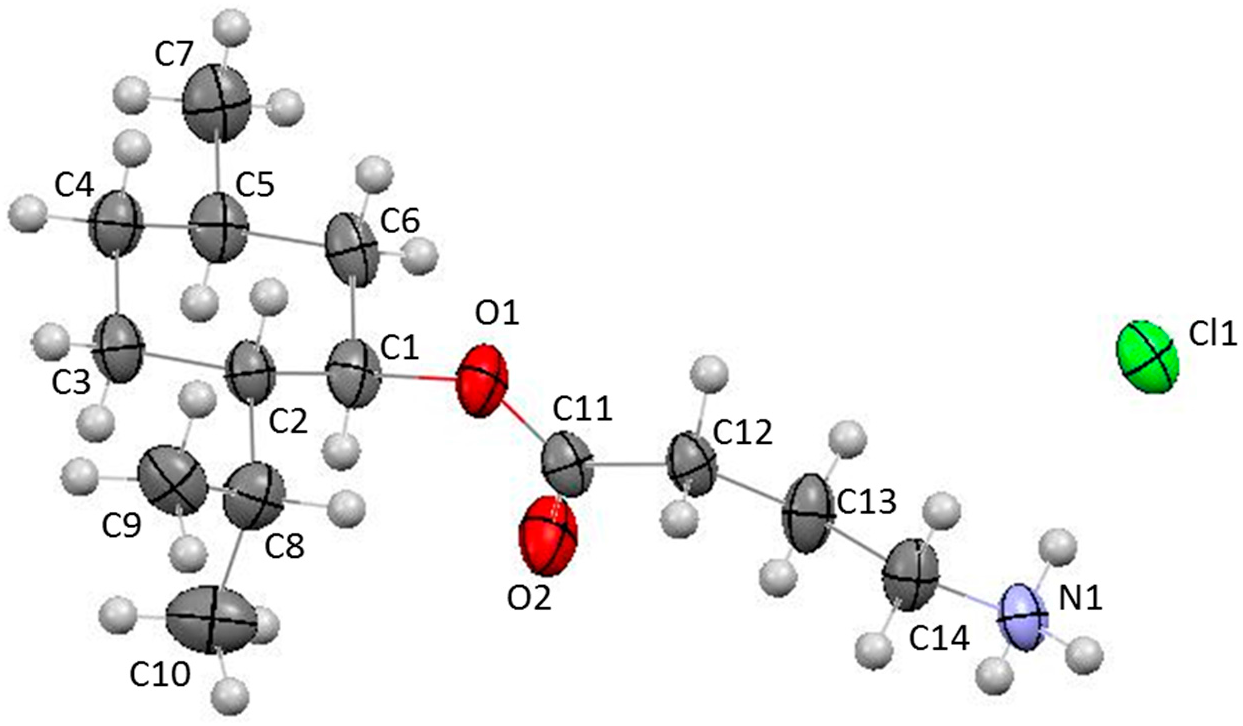{kind=link}
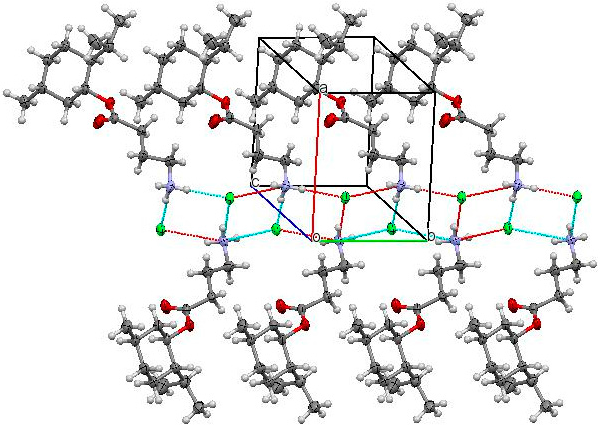{kind=link}
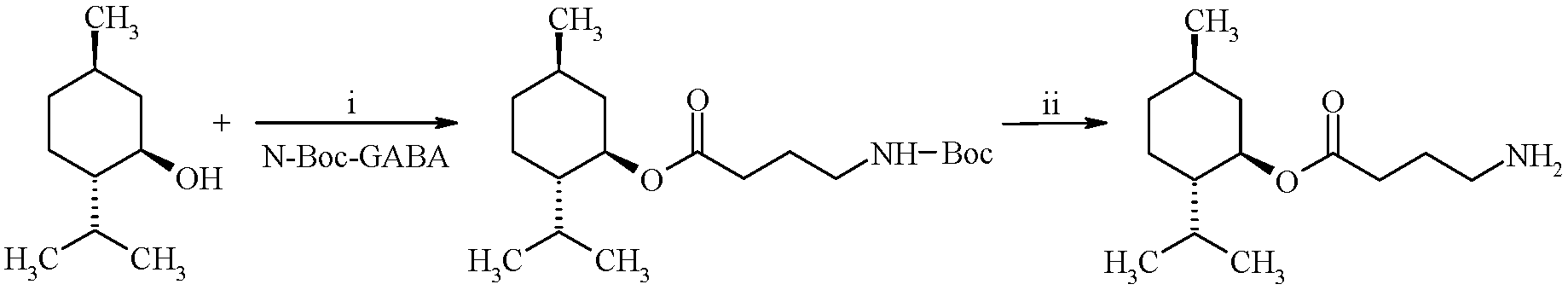{kind=link}
Abstract
:1. Introduction
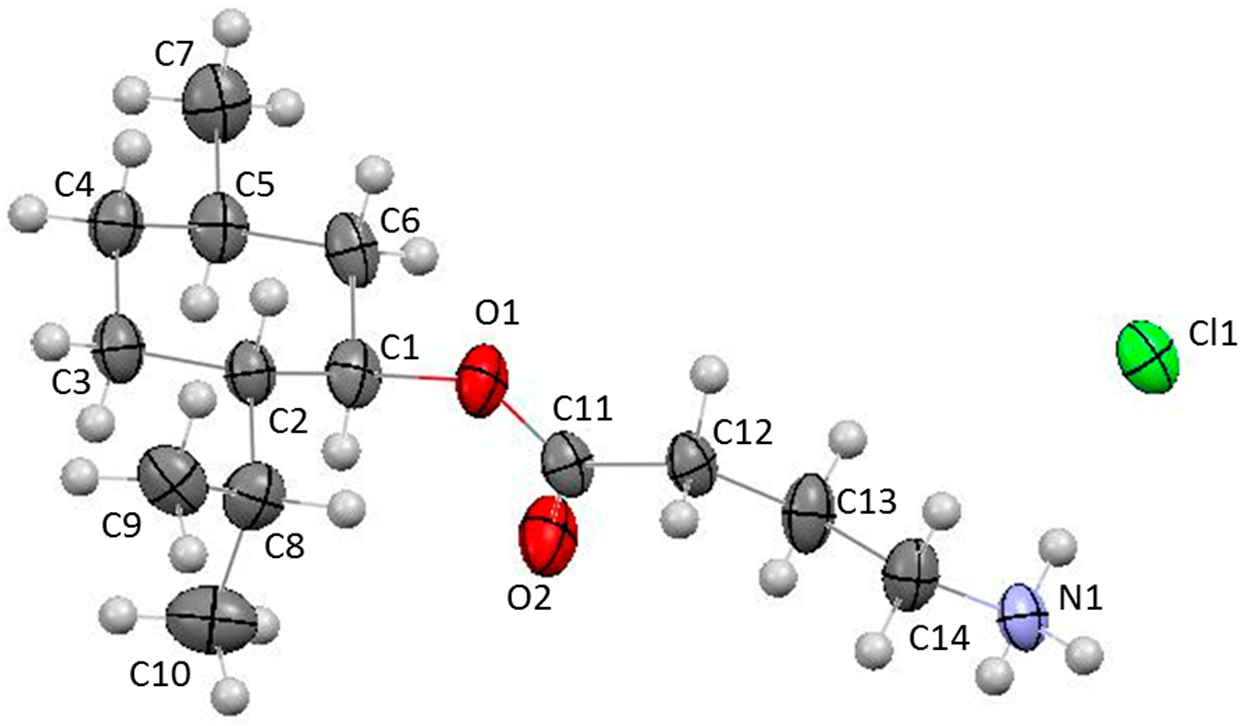
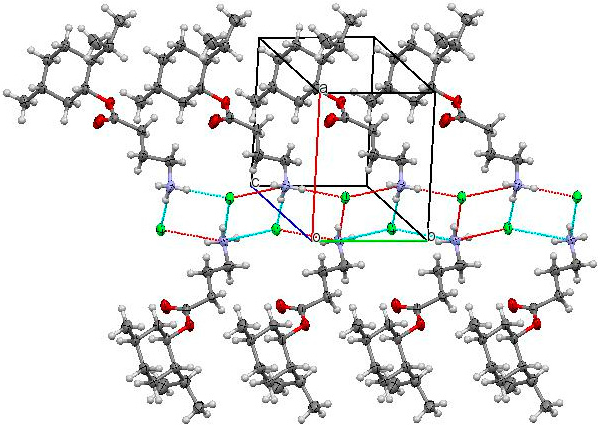
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of (1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Aminobutyrate Hydrochloride
3.3. X-ray Structural Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Galeotti, N.; Di Cesare Mannelli, L.; Mazzanti, G.; Bartolini, A.; Ghelardini, C. Menthol: A natural analgesic compound. Neurosci. Lett. 2002, 322, 145–148. [Google Scholar] [CrossRef]
- Galeotti, N.; Ghelardini, C.; Mannelli, L.; Mazzanti, G.; Baghiroli, L.; Bartolini, A. Local anaesthetic activity of (+)- and (−)-menthol. Planta Med. 2001, 67, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.G. Flavours and Fragrances: Chemistry, Bioprocessing and Sustainability; Springer: Heidelberg, Germany, 2007; pp. 1–648. [Google Scholar]
- Eccles, R.; Weber, O. Common Cold; Birkhäuser: Basel, Switzerland, 2009; pp. 1–354. [Google Scholar]
- Lau, B.K.; Karim, S.; Goodchild, A.K.; Vaughan, C.W.; Drew, G.M. Menthol enhances phasic and tonic GABAA receptor-mediated currents in midbrain periaqueductal grey neurons. Br. J. Pharmacol. 2014, 171, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Burgi, H.-B.; Dunitz, J.D. Structure Correlation; Wiley-VCH: Weinheim, Germany, 1994; pp. 1–936. [Google Scholar]
- Zefirov, N.S.; Palyulin, V.A.; Dashevskaya, E.E. Stereochemical studies. XXXIV. Quantitative description of ring puckering via torsional angles—The case of 6-membered rings. J. Phys. Org. Chem. 1990, 3, 147–158. [Google Scholar] [CrossRef]
- Cheedarala, R.K.; Sunkara, V.; Park, J.W. Facile synthesis of second-generation dendrons with an orthogonal functional group at the focal point. Synth. Commun. 2009, 39, 1966–1980. [Google Scholar] [CrossRef]
- Pozdnev, V.F. Activation of carboxylic acids with pyrocarbonates. Esterification of N-acylamino acids with secondary alcohols using di-tret-butylpyrocarbonate—Pyridine as the condensing reagents. Russ. J. Bioorg. Chem. 1985, 11, 725–732. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterkina, M.; Shishkina, S.; Maltsev, G.; Rakipov, I.; Kravchenko, I. (1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Aminobutyrate Hydrochloride. Molbank 2017, 2017, M956. https://doi.org/10.3390/M956
Nesterkina M, Shishkina S, Maltsev G, Rakipov I, Kravchenko I. (1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Aminobutyrate Hydrochloride. Molbank. 2017; 2017(3):M956. https://doi.org/10.3390/M956
Chicago/Turabian StyleNesterkina, Mariia, Svitlana Shishkina, Georgy Maltsev, Ildar Rakipov, and Iryna Kravchenko. 2017. "(1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Aminobutyrate Hydrochloride" Molbank 2017, no. 3: M956. https://doi.org/10.3390/M956
APA StyleNesterkina, M., Shishkina, S., Maltsev, G., Rakipov, I., & Kravchenko, I. (2017). (1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Aminobutyrate Hydrochloride. Molbank, 2017(3), M956. https://doi.org/10.3390/M956