
2II-VII, 3I-VII, 6I-VII-Icosa-O-acetyl-2I-deoxy-cyclomaltoheptaose


Abstract
:
{kind=link}
{kind=link}
1. Introduction
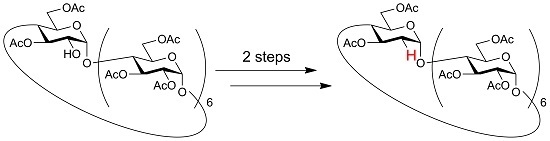
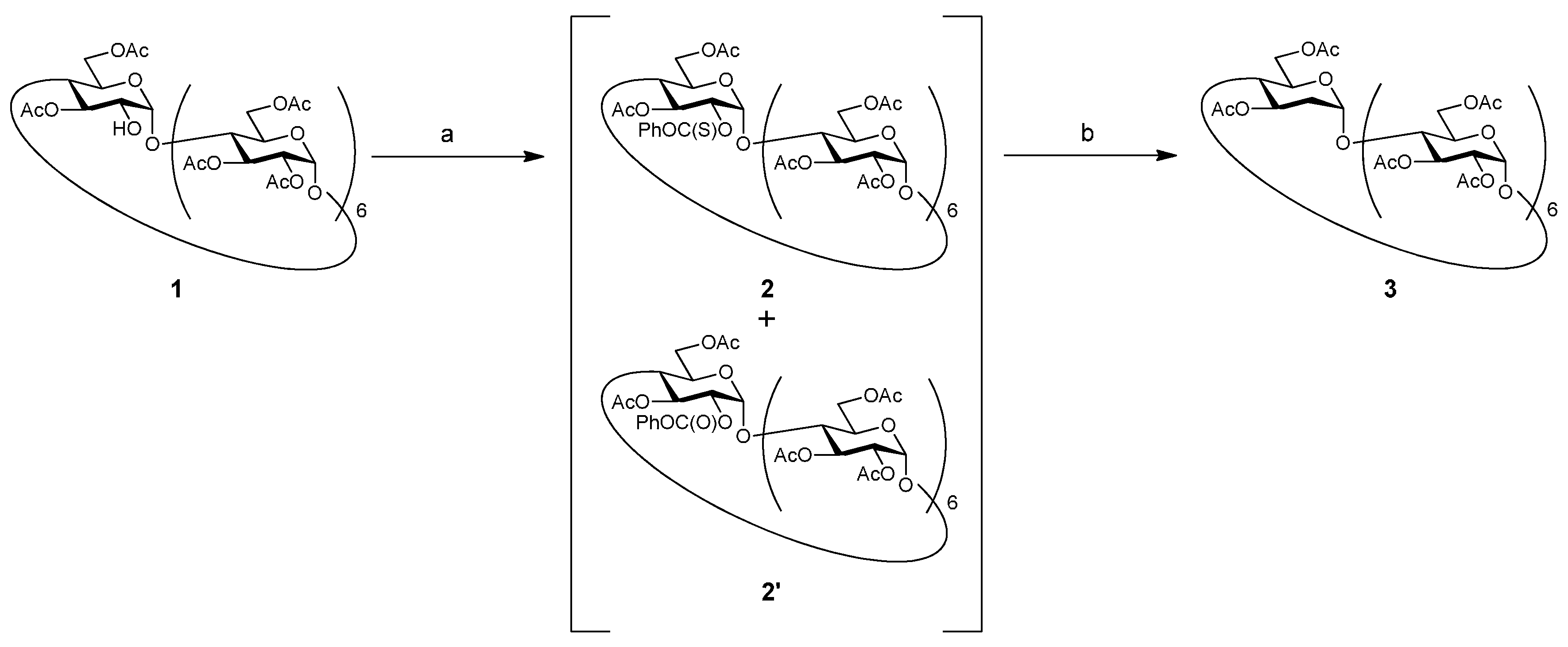
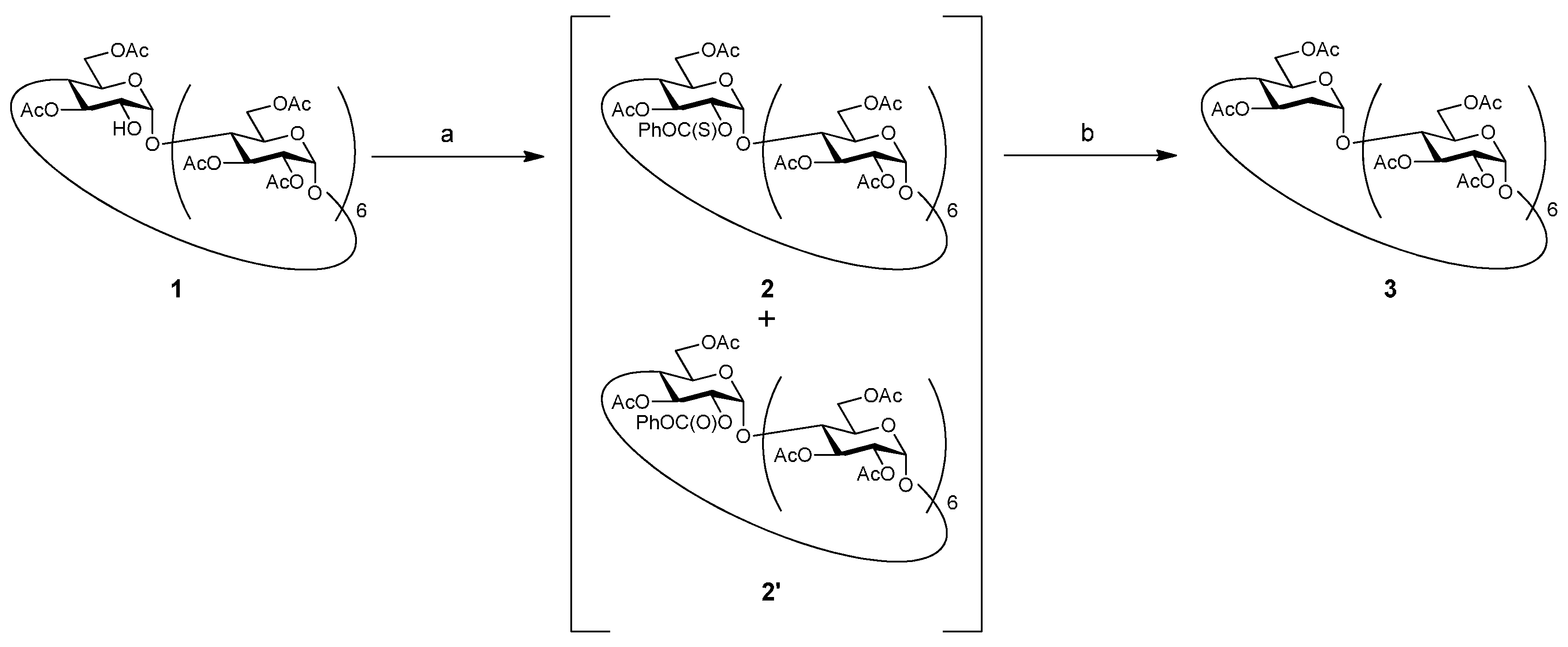
2. Results and Discussion
3. Experimental
2II-VII, 3I-VII, 6I-VII-Icosa-O-acetyl-2I-deoxy-cyclomaltoheptaose (3)
Supplementary Materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Author Contributions
Conflicts of Interest
References
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.J.; Schollmeyer, E. Applications of cyclodextrins in cosmetic products: A review. J. Cosmet. Sci. 2002, 53, 185–192. [Google Scholar] [PubMed]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS Pharm.Sci.Tech. 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Saenger, W. Cyclodextrin inclusion compounds in research and industry. Angew. Chem. Int. Ed. Engl. 1980, 19, 344–362. [Google Scholar] [CrossRef]
- Astray, G.; Gonzalez-Barreiro, C.; Mejuto, J.C.; Rial-Otero, R.; Simal-Gándara, J. A review on the use of cyclodextrins in foods. Food Hydrocoll. 2009, 23, 1631–1640. [Google Scholar] [CrossRef]
- Khan, A.R.; Forgo, P.; Stine, K.J.; D’Souza, V.T. Methods for selective modifications of cyclodextrins. Chem. Rev. 1998, 98, 1977–1996. [Google Scholar] [CrossRef] [PubMed]
- Dienst, E.V.V.; Snellink, B.H.; Piekartz, I.V.; Gansey, M.H.G.; Venema, F.; Feiters, M.C.; Nolte, R.J.M.; Engbersen, J.F.J.; Reinhoudt, D.N. Selective functionalization and flexible coupling of cyclodextrins at the secondary hydroxyl face. J. Org. Chem. 1995, 60, 6537–6545. [Google Scholar] [CrossRef]
- Atsumi, M.; Izumida, M.; Yuan, D.Q.; Fujita, K. Selective synthesis and structure determination of 6A, 6C, 6E-tri (O-sulfonyl)-β-cyclodextrins. Tetrahedron Lett. 2000, 41, 8117–8120. [Google Scholar] [CrossRef]
- Tian, S.; Forgo, P.; D'Souza, V.T. Selective modification at the 3-position of β-cyclodextrin. Tetrahedron Lett. 1996, 37, 8309–8312. [Google Scholar] [CrossRef]
- Takeo, K.; Mitoh, H.; Uemura, K. Selective chemical modification of cyclomalto-oligosaccharides via tert-butyldimethylsilylation. Carbohydr. Res. 1989, 187, 203–221. [Google Scholar] [CrossRef]
- Boger, J.; Corcoran, R.J.; Lehn, J.M. Cyclodextrin chemistry. Selective modification of all primary hydroxyl groups of α-and β-cyclodextrins. Helv. Chim. Acta 1978, 61, 2190–2218. [Google Scholar] [CrossRef]
- Yamamura, H.; Fujita, K. Preparation of heptakis(6-O-(p-tosyl))-β-cyclodextrin and heptakis(6-O-(p-tosyl))-2-O-(p-tosyl)-β-cyclodextrin and their conversion to heptakis(3, 6-anhydro)-β-cyclodextrin. Chem. Pharm. Bull. 1991, 39, 2505–2508. [Google Scholar] [CrossRef]
- Tian, S.; Zhu, H.; Forgo, P.; D’Souza, V.T. Selectively monomodified cyclodextrins. Synthetic strategies. J. Org. Chem. 2000, 65, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Masurier, N.; Estour, F.; Lefèvre, B.; Brasme, B.; Masson, P.; Lafont, O. Improved access to 2-O-monobenzyl ethers of β-cyclodextrin as precursors of catalysts for organophosphoryl esters hydrolysis. Carbohydr. Res. 2006, 341, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Cousin, H.; Cardinael, P.; Oulyadi, H.; Pannecoucke, X.; Combret, J.C. Synthesis of the three isomeric mono-2-, 3-, or 6-hydroxy permethylated β-cyclodextrins and unambiguous high field NMR characterisation. Tetrahedron Asymmetry 2001, 12, 81–88. [Google Scholar] [CrossRef]
- Cromer, R.; Spohr, U.; Khare, D.P.; LePendu, J.; Lemieux, R.U. Molecular recognition XII. The binding of the H human blood group determinants and congeners by a lectin of Galactia tenuiflora. Can. J. Chem. 1992, 70, 1511–1530. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyagawa, A.; Kano, K.; Yoshida, A.; Yamamura, H. 2II-VII, 3I-VII, 6I-VII-Icosa-O-acetyl-2I-deoxy-cyclomaltoheptaose. Molbank 2016, 2016, M900. https://doi.org/10.3390/M900
Miyagawa A, Kano K, Yoshida A, Yamamura H. 2II-VII, 3I-VII, 6I-VII-Icosa-O-acetyl-2I-deoxy-cyclomaltoheptaose. Molbank. 2016; 2016(2):M900. https://doi.org/10.3390/M900
Chicago/Turabian StyleMiyagawa, Atsushi, Kazuki Kano, Aya Yoshida, and Hatsuo Yamamura. 2016. "2II-VII, 3I-VII, 6I-VII-Icosa-O-acetyl-2I-deoxy-cyclomaltoheptaose" Molbank 2016, no. 2: M900. https://doi.org/10.3390/M900