Synthesis and Biological Evaluation of a Novel Pentagastrin- Toxin Conjugate Designed for a Targeted Prodrug Monotherapy of Cancer

Abstract
:1. Introduction
2. Results and Discussion
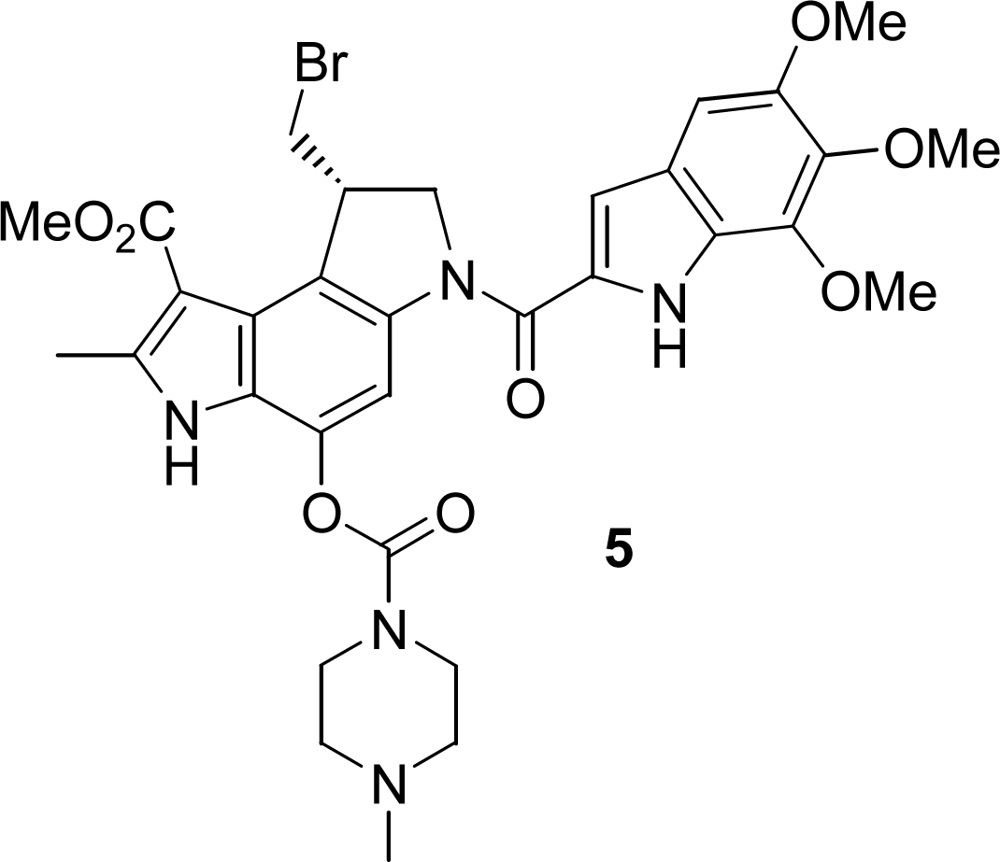
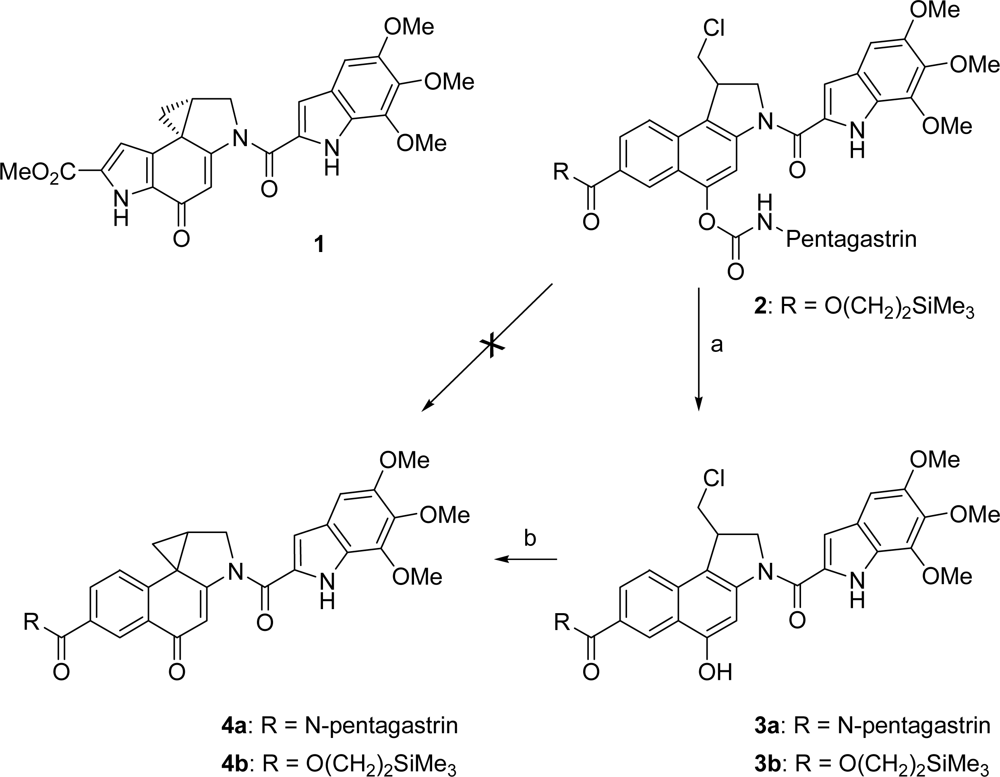
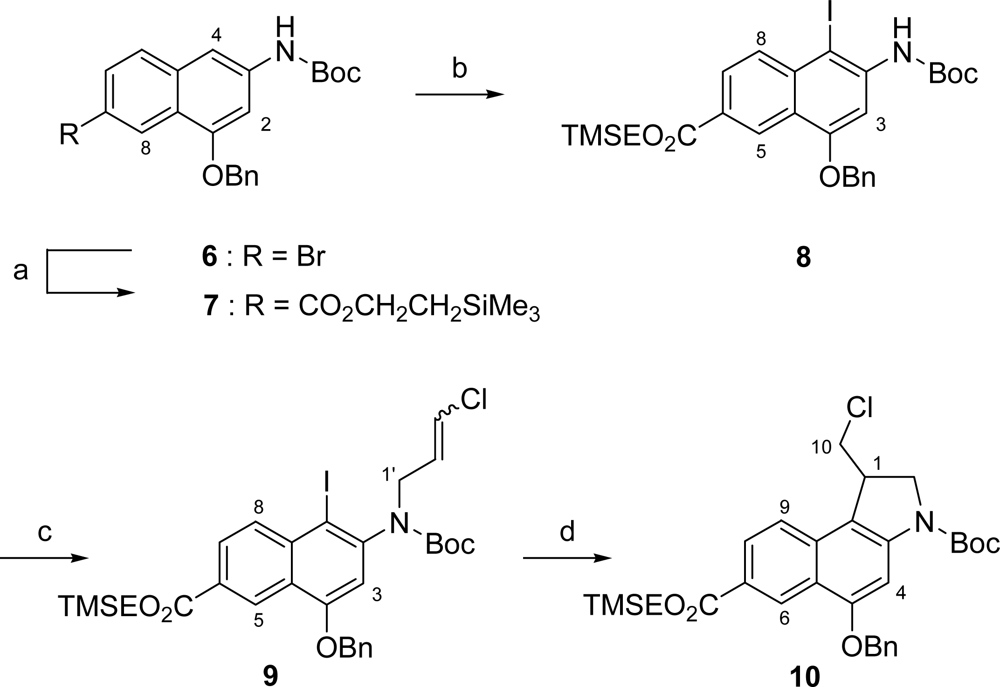
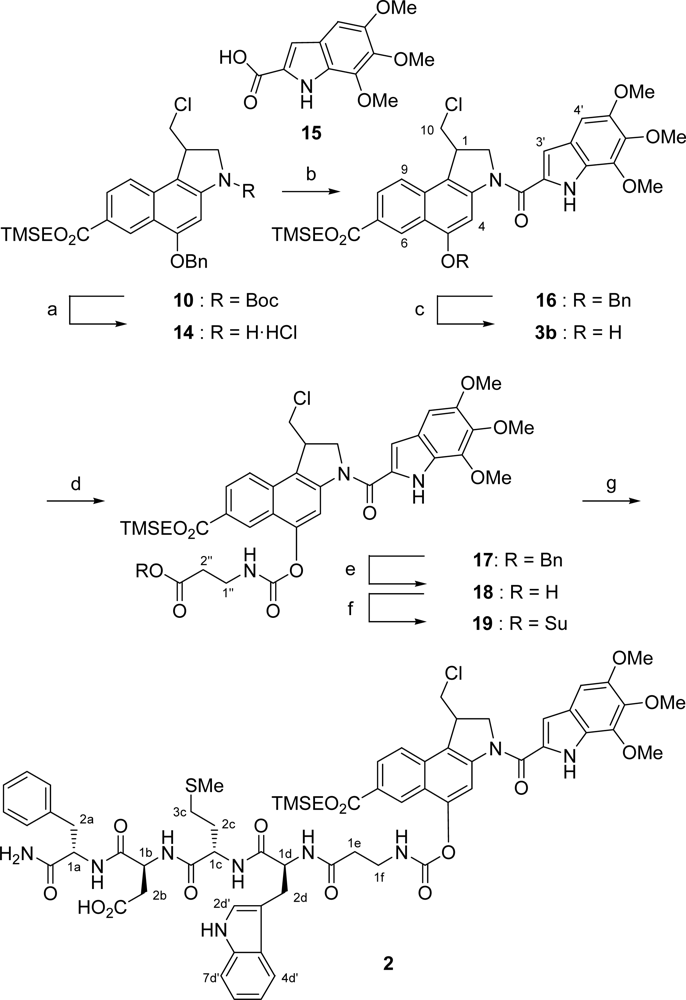
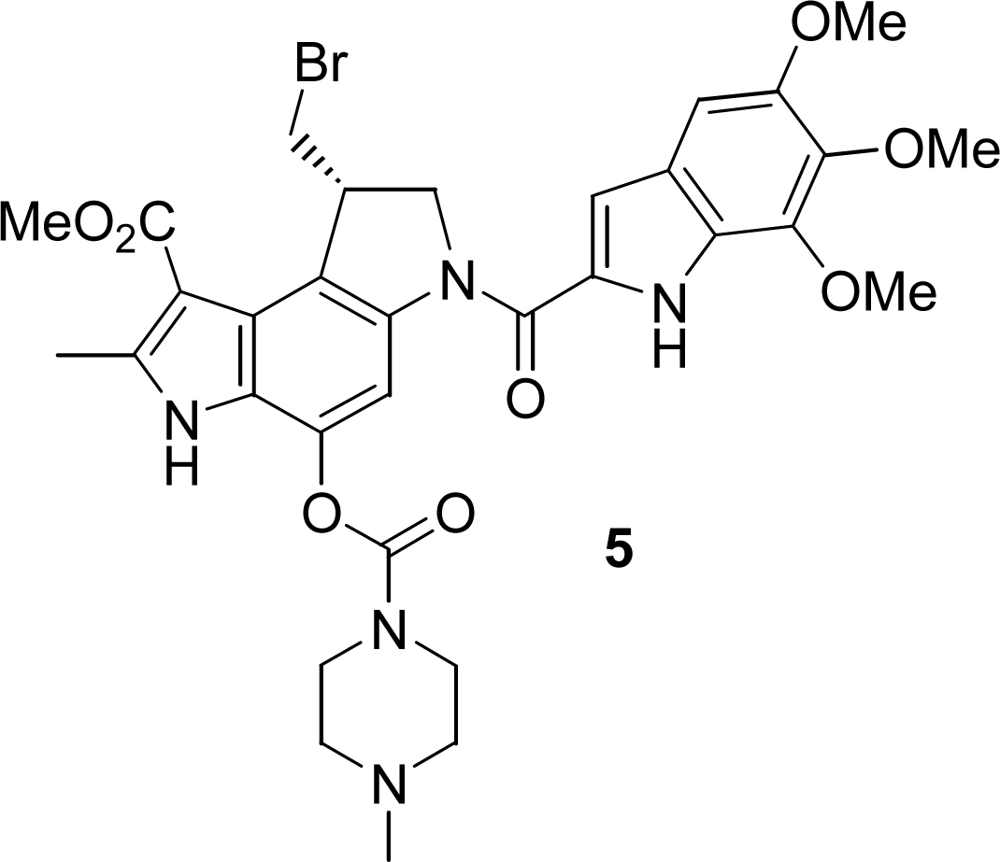
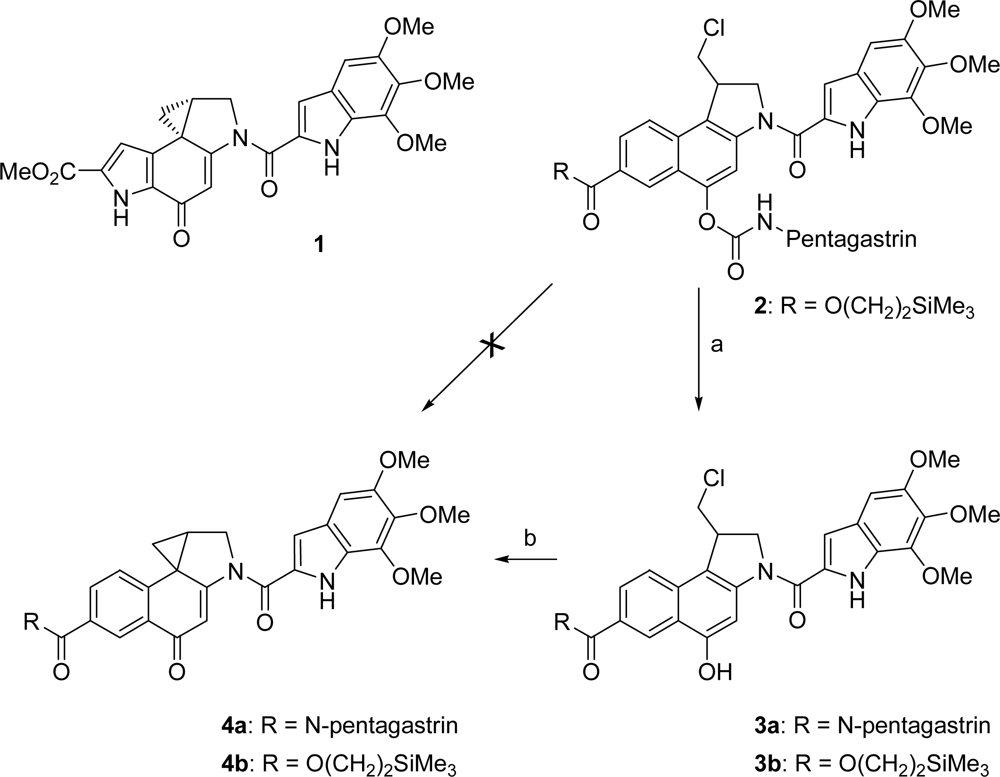
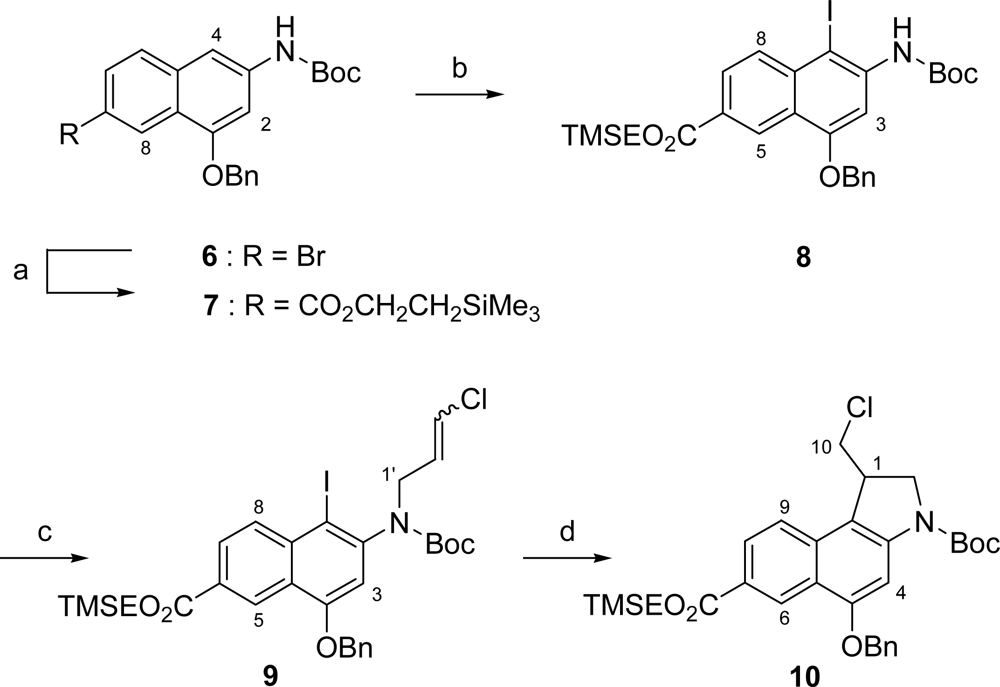
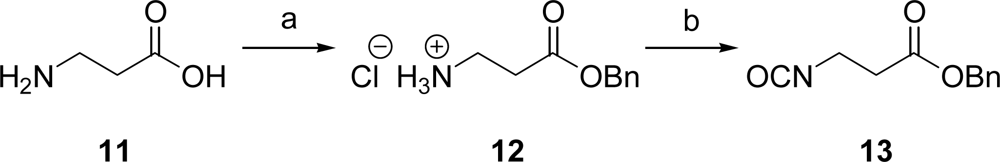
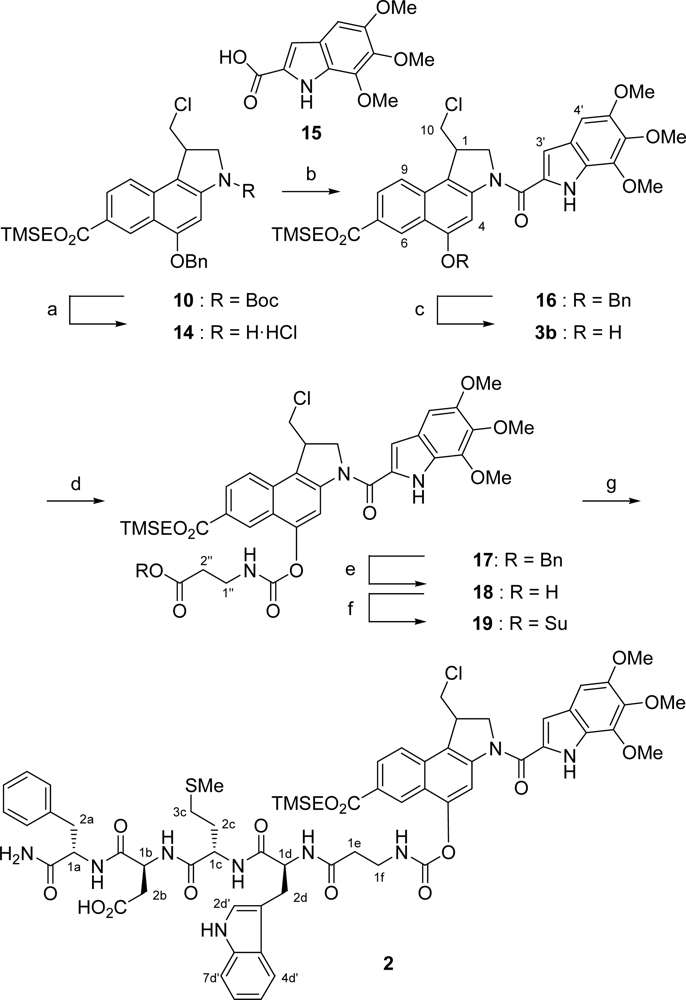
2.1. Synthesis
2.2. In vitro cytotoxicity tests
3. Experimental Section
Acknowledgments
References and Notes
- Bagshawe, KD. Antibody directed enzymes revive anti-cancer prodrugs concept. Br. J. Cancer 1987, 56, 531–532, Reviews:. [Google Scholar]Tietze, LF; Feuerstein, T. Enzyme and Proton-Activated Prodrugs for a Selective Cancer Therapy. Curr. Pharm. Des. 2003, 9, 2155–2175. [Google Scholar]Tietze, LF; Feuerstein, T. Highly Selective Compounds for the Antibody-Directed Enzyme Prodrug Therapy of Cancer. Aust. J. Chem. 2003, 56, 841–854. [Google Scholar]Jung, M. Antibody directed enzyme prodrug therapy (ADEPT) and related approaches for anticancer therapy. Mini-Rev. Med. Chem. 2001, 1, 399–407. [Google Scholar]Syrigos, KN; Epenetos, AA. Antibody directed enzyme prodrug therapy (ADEPT): a review of the experimental and clinical considerations. Anticancer Res. 1999, 19, 605–613. [Google Scholar]Springer, CJ; Niculescu-Duvaz, I. Antibody-directed enzyme prodrug therapy (ADEPT): a review. Adv. Drug Deliv. Rev. 1997, 26, 151–172. [Google Scholar]
- Tietze, L; Major, F; Schuberth, I; Spiegl, DA; Krewer, B; Maksimenka, K; Bringmann, G; Magull, J. Selective Treatment of Cancer: Synthesis, Biological Evaluation and Structural Elucidation of Novel Analogues of the Antibiotic CC-1065 and the Duocarmycins. Chem. Eur. J. 2007, 13, 4396–4409. [Google Scholar]Tietze, LF; Major, F; Schuberth, I. Antitumor Agents: Development of Highly Potent Glycosidic Duocarmycin Analogues for Selective Cancer Therapy. Angew. Chem. 2006, 118, 6724–6727. [Google Scholar]Angew. Chem., Int. Ed. 2006, 45, 6574–6577.Tietze, LF; Krewer, B; Frauendorf, H; Major, F; Schuberth, I. Investigation of Reactivity and Selectivity of DNA-Alkylating Duocarmycin Analogues by High-Resolution Mass Spectrometry. Angew. Chem. 2006, 118, 6720–6724. [Google Scholar]Angew. Chem., Int. Ed. 2006, 45, 6570–6574.
- Ganesh, T. Improved biochemical strategies for targeted delivery of taxoids. Bioorg. Med. Chem. 2007, 15, 3597–3623. [Google Scholar]Devalapally, H; Navath, RS; Yenamandra, V; Akkinepally, RR; Devarakonda, RK. Directed Enzyme Prodrug Therapy/Prodrug MonoTherapy. Arch. Pharm. Res. 2007, 30, 723–732. [Google Scholar]Alaoui, AE; Saha, N; Schmidt, F; Monneret, C; Florent, J-C. New Taxol®(paclitaxel) prodrugs designed for ADEPT and PMT strategies in cancer chemotherapy. Bioorg. Med. Chem. 2006, 14, 5012–5019. [Google Scholar]Kratz, F; Warnecke, A; Schmid, B; Chung, D-E; Gitzel, M. Prodrugs of anthracyclines in cancer chemotherapy. Curr. Med. Chem. 2006, 13, 477–523. [Google Scholar]Schmidt, F; Monneret, C. Prodrug Monotherapy: synthesis and biological evaluation of an etoposide glucuronide-prodrug. Bioorg. Med. Chem. 2003, 11, 2277–2283. [Google Scholar]Prijovich, ZM; Chen, BM; Leu, Y-L; Chern, J-W; Roffler, SR. Anti-tumour activity and toxicity of the new prodrug 9-aminocamptothecin glucuronide (9 ACG) in mice. Br. J. Cancer. 2002, 86, 1634–1638. [Google Scholar]Denny, WA. Prodrug strategies in cancer therapy. Eur. J. Med. Chem. 2001, 36, 577–595. [Google Scholar]Bosslet, K; Straub, R; Blumrich, M; Czech, J; Gerken, M; Sperker, B; Kroemer, HK; Gesson, J-P; Koch, M; Monneret, C. Elucidation of the Mechanism Enabling Tumor Selective Prodrug Monotherapy. Cancer Res. 1998, 58, 1195–1201. [Google Scholar]Bosslet, K; Czech, J; Hoffmann, D. Tumor Targeting 1995, 1, 45–50.
- Reubi, J; Mäcke, HR; Krenning, EP. Candidates for peptide receptor radiotherapy today and in the future. J. Nucl. Med. 2005, 46, 67S–75S. [Google Scholar]Dyba, M; Tarasova, NI; Michejda, CJ. Small Molecule Toxins Targeting Tumor Receptors. Curr. Pharm. Des. 2004, 10, 2311–2334. [Google Scholar]Reubi, JC. Peptide Receptors as Molecular Targets for Cancer Diagnosis and Therapy. Endocrine Reviews 2003, 24, 389–427. [Google Scholar]Reubi, JC; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. 2003, 30, 781–793. [Google Scholar]Langer, M; Beck-Sickinger, AG. Peptides as Carrier for Tumor Diagnosis and Treatment. Curr. Med. Chem. 2001, 1, 71–93. [Google Scholar]Breeman, WAP; de Jong, M; Kwekkeboom, DJ; Valkema, R; Bakker, WH; Kooij, PPM; Visser, TJ; Krenning, EP. Somatostatin receptor-mediated imaging and therapy: basic science, current knowledge, limitations and future perspectives. Eur. J. Nucl. Med. 2001, 28, 1421–1429. [Google Scholar]Heppeler, A; Froidevaux, S; Eberle, AN; Maecke, HR. Receptor targeting for tumor localisation and therapy with radiopeptides. Curr. Med. Chem. 2000, 7, 971–994. [Google Scholar]
- Nock, BA; Maina, T; Béhé, M; Nikolopoulou, A; Gotthardt, M; Schmitt, JS; Behr, TM; Macke, HR. CCK-2/Gastrin Receptor–Targeted Tumor Imaging with99mTc-Labeled Minigastrin Analogs. J. Nucl. Med. 2005, 46, 1727–1736. [Google Scholar]Béhé, M; Behr, TM. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers (Peptide Science) 2002, 66, 399–418. [Google Scholar]Behr, TM; Jenner, N; Béhé, M; Angerstein, C; Gratz, S; Raue, F; Becker, W. Radiolabeled Peptides for Targeting Cholecystokinin-B/Gastrin Receptor-Expressing Tumors. J. Nucl. Med. 1999, 40, 1029–1044. [Google Scholar]Behr, TM; Jenner, N; Radetzky, S; Béhé, M; Gratz, S; Yücekent, S; Raue, F; Becker, W. Targeting of cholecystokinin-B/gastrin receptors in vivo: preclinical and initial clinical evaluation of the diagnostic and therapeutic potential of radiolabelled gastrin. Eur. J. Nucl. Med. 1998, 25, 424–430. [Google Scholar]
- Schmidt, BF; Hernandez, L; Rouzer, C; Czerwinski, G; Chmurny, G; Michejda, CJ. Peptide-Linked 1,3-Dialkyl-3-acyltriazenes:Gastrin Receptor Directed Antineoplastic Alkylating Agents. J. Med. Chem. 1994, 37, 3812–3818. [Google Scholar]
- Czerwinski, G; Tarasova, NI; Michejda, CJ. Cytotoxic agents directed to peptide hormone receptors: Defining the requirements for a successful drug. Proc. Natl. Acad. Sci. USA 1998, 95, 11520–11525. [Google Scholar]
- Buchholz, S; Keller, G; Schally, AV; Halmos, G; Hohla, F; Heinrich, E; Koester, F; Baker, B; Engel, JB. Therapy of ovarian cancers with targeted cytotoxic analogs of bombesin, somatostatin, and luteinizing hormone-releasing hormone and their combinations. Proc. Natl. Acad. Sci. USA 2006, 103, 10403–10407. [Google Scholar]Nagy, A; Schally, AV. Minireview. Targeting of Cytotoxic Luteinizing Hormone-Releasing Hormone Analogs to Breast, Ovarian, Endometrial, and Prostate Cancers. Biol. Reprod. 2005, 73, 851–859. [Google Scholar]Nagy, A; Schally, AV; Armatis, P; Szepesházi, K; Halmos, G; Kovács, M; Zarandi, M; Groot, K; Miyazaki, M; Jungwirth, A; Horvath, J. Cytotoxic analogs of luteinizing hormone-releasing hormone containing doxorubicin or 2-pyrrolinodoxorubicin, a derivative 500-1000 times more potent. Proc. Natl. Acad. Sci. USA 1996, 93, 7269–7273. [Google Scholar]
- Engel, JB; Keller, G; Schally, AV; Halmos, G; Hammann, B; Nagy, A. Effective Inhibition of Experimental Human Ovarian Cancers with a Targeted Cytotoxic Bombesin Analogue AN-215. Clin. Cancer Res. 2005, 11, 2408–2415. [Google Scholar]Nagy, A; Armatis, P; Cai, RZ; Szepesházi, K; Halmos, G; Schally, AV. Design, synthesis, and in vitro evaluation of cytotoxic analogs of bombesin-like peptides containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin . Proc. Natl. Acad. Sci. USA 1997, 94, 652–656. [Google Scholar]
- Szepesházi, K; Schally, AV; Halmos, G; Armatis, P; Hebert, F; Sun, B; Feil, A; Kiaris, H; Nagy, A. Targeted Cytotoxic Somatostatin Analogue AN-238 Inhibits Somatostatin Receptor-positive Experimental Colon Cancers Independently of Their p53 Status. Cancer Res. 2002, 62, 781–788. [Google Scholar]Nagy, A; Schally, AV; Halmos, G; Armatis, P; Cai, RZ; Csernus, V; Kovács, M; Koppán, M; Szepesházi, K; Kahán, Z. Synthesis and biological evaluation of cytotoxic analogs of somatostatin containing doxorubicin or its intensely potent derivative, 2-pyrrolino-doxorubicin. Proc. Natl. Acad. Sci. USA 1998, 95, 1794–1799. [Google Scholar]
- Langer, M; Kratz, F; Rothen-Rutishauser, B; Wunderli-Allenspach, H; Beck-Sickinger, AG. Novel Peptide Conjugates for Tumor-Specific Chemotherapy. J. Med. Chem. 2001, 44, 1341–1348. [Google Scholar]
- Tietze, LF; Panknin, O; Major, F; Krewer, B. Synthesis of a Novel Pentagastrin-Drug Conjugate for a Targeted Tumor Therapy. Chem. Eur. J. 2008, 14, 2811–2818. [Google Scholar]
- Boger, DL; Johnson, DS. CC-1065 and the Duocarmycins: Understanding their Biological Function through Mechanistic Studies. Angew. Chem. 1996, 108, 1542–1580. [Google Scholar]Angew. Chem. Int. Ed. Engl. 1996, 35, 1438–1474.Ichimura, M; Ogawa, T; Katsumata, S; Takahashi, K; Takahashi, I; Nakano, H. Duocarmycins, new antitumor antibiotics produced by streptomyces; producing organisms and improved production. J. Antibiot. 1991, 44, 1045–1053. [Google Scholar]Ichimura, M; Ogawa, T; Takahashi, K; Kobayashi, E; Kawamoto, I; Yasuzawa, T; Takahashi, I; Nakano, H. Duocarmycin, a new antitumor antibiotic from streptomyces sp. J. Antibiot. 1990, 43, 1037–1038. [Google Scholar]
- Boger, DL; Bollinger, B; Hertzog, DL; Johnson, DS; Cai, H; Mésini, P; Garbaccio, RM; Jin, Q; Kitos, PA. Reversed and Sandwiched Analogs of Duocarmycin SA: Establishment of the Origin of the Sequence-Selective Alkylation of DNA and New Insights into the Source of Catalysis. J. Am. Chem. Soc. 1997, 119, 4987–4998. [Google Scholar]Boger, DL; Johnson, DS. CC-1065 and the Duocarmycins: Understanding their Biological Function through Mechanistic Studies. Angew. Chem. 1996, 108, 1542–1580. [Google Scholar]Angew. Chem. Int. Ed. Engl. 1996, 35, 1438–1474.Hurley, LH; Needham-VanDevanter, DR. Covalent Binding of Antitumor Antibiotics in the Minor Groove of DNA. Mechanism of Action of CC-1065 and the Pyrrolo(1,4) benzodiazepines. Acc. Chem. Res. 1986, 19, 230–237. [Google Scholar]
- Tietze, LF; Haunert, F; Feuerstein, T; Herzig, T. A Concise and Efficient Synthesis of seco-Duocarmycin SA. Eur. J. Org. Chem. 2003, 3, 562–566. [Google Scholar]Baird, R; Winstein, S. Neighboring Carbon and Hydrogen. Li. Dienones from Ar1[UNK]-3 Participation. Isolation and Behavior of Spiro(2,5)octa-1,4-diene-3-one. J. Am. Chem. Soc. 1963, 85, 567–578. [Google Scholar]
- Morley, JS; Tracy, HJ; Gregory, RA. Structure–Function Relationships in the Active C-Terminal Tetrapeptide Sequence of Gastrin. Nature 1965, 1356–1359. [Google Scholar]Tracy, HJ; Gregory, RA. The antral Hormone Gastrin: Physiological Properties of a Series of Synthetic Peptides structurally related to Gastrin I. Nature 1964, 204, 935–938. [Google Scholar]
- Alberts, SR; Erlichman, C; Reid, JM; Sloan, JA; Ames, MM; Richardson, RL; Goldberg, RM. Phase I study of the duocarmycin semisynthetic derivative KW-2189 given daily for five days every six weeks. Clin. Cancer Res. 1998, 4, 2111–2117. [Google Scholar]Nagamura, S; Kobayashi, E; Gomi, K; Saito, H. Studies on the active metabolite (DU-86) of KW-2189, a novel derivative of duocarmycin. Bioorg. Med. Chem. Lett. 1996, 6, 2147–2150. [Google Scholar]Nagamura, S; Kobayashi, E; Gomi, K; Saito, H. Synthesis and antitumor activity of duocarmycin derivatives: A-ring pyrrole analogues of duocarmycin B2. Bioorg. Med. Chem. 1996, 4, 1379–1391. [Google Scholar]Nagamura, S; Asai, A; Kanda, Y; Kobayashi, E; Gomi, K; Saito, H. Synthesis and Antitumor Activity of Duocarmycin Derivatives: Modification of Segment A of Duocarmycin B2. Chem. Pharm. Bull. 1996, 44, 1723–1730. [Google Scholar]Yasuzawa, T; Muroi, K; Ichimura, M; Takahashi, I; Ogawa, T; Takahashi, K; Sano, H; Saitoh, Y. Duocarmycins, Potent Antitumor Antibiotics Produced by Streptomyces sp. Structures and Chemistry. Chem. Pharm. Bull. 1995, 43, 378–391. [Google Scholar]Asai, A; Nagamura, S; Saito, S. A Novel Property of Duocarmycin and Its Analogs for Covalent Reaction with DNA. J. Am. Chem. Soc. 1994, 116, 4171–4177. [Google Scholar]
- Ray, S; Chaturvedi, D. Application of organic carbamates in drug design. Part 1: Anti-cancer agents-recent reports. Drugs Fut. 2004, 29, 343–357. [Google Scholar]
- Kaiser, N-F; Hallberg, A; Larhed, M. In situ Generation of Carbon Monoxide from Solid Molybdenum Hexacarbonyl. A Convenient and Fast Route to Palladium-Catalyzed Carbonylation Reactions. J. Comb. Chem. 2002, 4, 109–111. [Google Scholar]
- Boger, DL; Han, N; Tarby, CM; Boyce, CW; Cai, H; Jin, Q; Kitos, PA. Synthesis, Chemical Properties, and Preliminary Evaluation of Substituted CBI Analogs of CC-1065 and the Duocarmycins Incorporating the 7-Cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one Alkylation Subunit: Hammett Quantitation of the Magnitude of Electronic Effects on Functional Reactivity. J. Org. Chem. 1996, 61, 4894–4912. [Google Scholar]
- Boger, DL; Brunette, SR; Garbaccio, RM. Synthesis and Evaluation of a Series of C3-Substituted CBI Analogues of CC-1065 and the Duocarmycins. J. Org. Chem. 2001, 66, 5163–5173. [Google Scholar]Boger, DL; McKie, JA; Cai, H; Cacciari, B; Baraldi, PG. Synthesis and Properties of Substituted CBI Analogs of CC-1065 and the Duocarmycins Incorporating the 7-Methoxy-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (MCBI) Alkylation Subunit: Magnitude of Electronic Effects on the Functional Reactivity. J. Org. Chem. 1996, 61, 1710–1729. [Google Scholar]
- Boger, DL; Boyce, CW; Garbaccio, RM; Searcey, M. Synthesis of CC-1065 and duocarmycin analogs via intramolecular aryl radical cyclization of a tethered vinyl chloride. Tetrahedron Lett. 1998, 39, 2227–2230. [Google Scholar]Patel, VF; Andis, SL; Enkema, JK; Johnson, DA; Kennedy, JH; Mohamadi, F; Schultz, RM; Soose, DJ; Spees, MM. Total Synthesis of Seco (+)- and ent-(−)-Oxaduocarmycin SA: Construction of the (Chloromethyl)indoline Alkylating Subunit by a Novel Intramolecular Aryl Radical Cyclization onto a Vinyl Chloride. J. Org. Chem. 1997, 62, 8868–8874. [Google Scholar]Boger, DL; McKie, JA. An Efficient Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one CBI: An Enhanced and Simplified Analog of the CC-1065 and Duocarmycin Alkylation Subunits. J. Org. Chem. 1995, 60, 1271–1275. [Google Scholar]
- Studer, A; Amrein, S. Silylated Cyclohexadienes: New Alternatives to Tributyltin Hydride in Free Radical Chemistry. Angew. Chem. 2000, 112, 3196–3198. [Google Scholar]Angew. Chem. Int. Ed. 2000, 39, 3080–3082.Chatgilialoglu, C. Organosilanes as radical-based reducing agents in synthesis. Acc. Chem. Res. 1992, 25, 188. [Google Scholar]
- Belshaw, PJ; Mzengeza, S; Lajoie, GA. Chlorotrimethylsilane Mediated Formation of ω-Allyl Esters of Aspartic And Glutamic Acids. Synth. Commun. 1990, 20, 3157–3160. [Google Scholar]
- Mehta, A; Jaouhari, R; Benson, TJ; Douglas, KT. Improved efficiency and selectivity in peptide synthesis: use of triethylsilane as a carbocation scavenger in deprotection of t-butyl esters and t-butoxycarbonyl-protected sites. Tetrahedron Lett. 1992, 33, 5441–5444. [Google Scholar]
- Ram, S; Spicer, LD. Rapid debenzylation of N-benzylamino derivatives to amino-derivatives using ammonium formate as catalytic hydrogen transfer agent. Tetrahedron Lett. 1987, 28, 515–516. [Google Scholar]Bieg, T; Szeja, W. Removal of-Benzyl Protective Groups by Catalytic Transfer Hydrogenation. Synthesis 1985, 76–77. [Google Scholar]
- Tetragastrin (20) was purchased from Bachem.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIA PaCa-2 IC50 [nm] | A549 IC50 [nm] |
|---|---|---|
| 2 | 0.31 | 0.11 |
| 3b | 0.31 | 0.14 |
Share and Cite
Tietze, L.F.; Panknin, O.; Krewer, B.; Major, F.; Schuberth, I. Synthesis and Biological Evaluation of a Novel Pentagastrin- Toxin Conjugate Designed for a Targeted Prodrug Monotherapy of Cancer. Int. J. Mol. Sci. 2008, 9, 821-837. https://doi.org/10.3390/ijms9050821
Tietze LF, Panknin O, Krewer B, Major F, Schuberth I. Synthesis and Biological Evaluation of a Novel Pentagastrin- Toxin Conjugate Designed for a Targeted Prodrug Monotherapy of Cancer. International Journal of Molecular Sciences. 2008; 9(5):821-837. https://doi.org/10.3390/ijms9050821
Chicago/Turabian StyleTietze, Lutz F., Olaf Panknin, Birgit Krewer, Felix Major, and Ingrid Schuberth. 2008. "Synthesis and Biological Evaluation of a Novel Pentagastrin- Toxin Conjugate Designed for a Targeted Prodrug Monotherapy of Cancer" International Journal of Molecular Sciences 9, no. 5: 821-837. https://doi.org/10.3390/ijms9050821