Electron Charge Density Distribution from X-ray Diffraction Study of the M-Nitrophenol Compound in the Monoclinic Form

Abstract
:1. Introduction
2. Methods
2.1. Spherical X-ray refinement[high order (HO)]
2.2. Charge Density Multipole Model
3. Results and Discussion
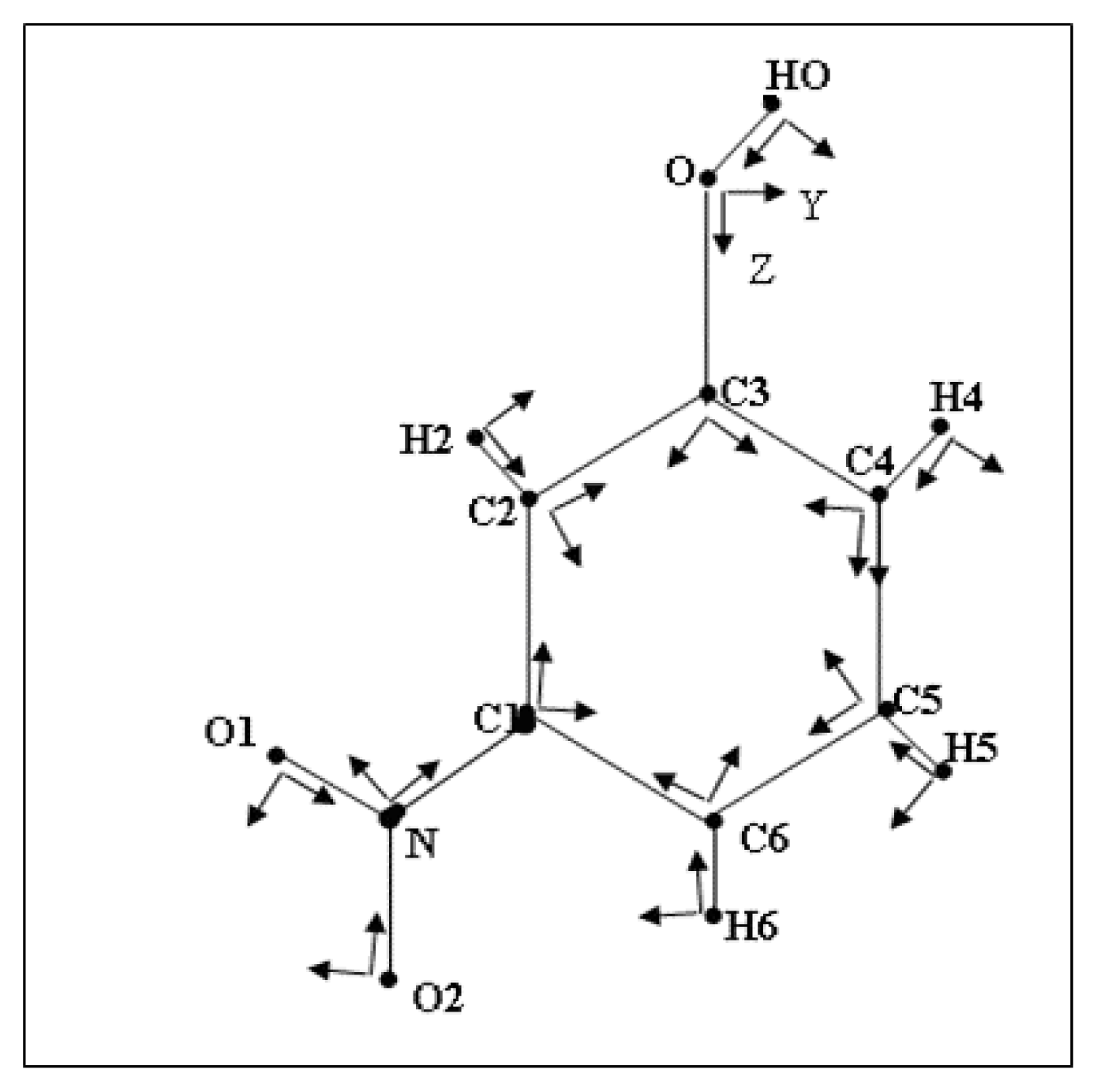
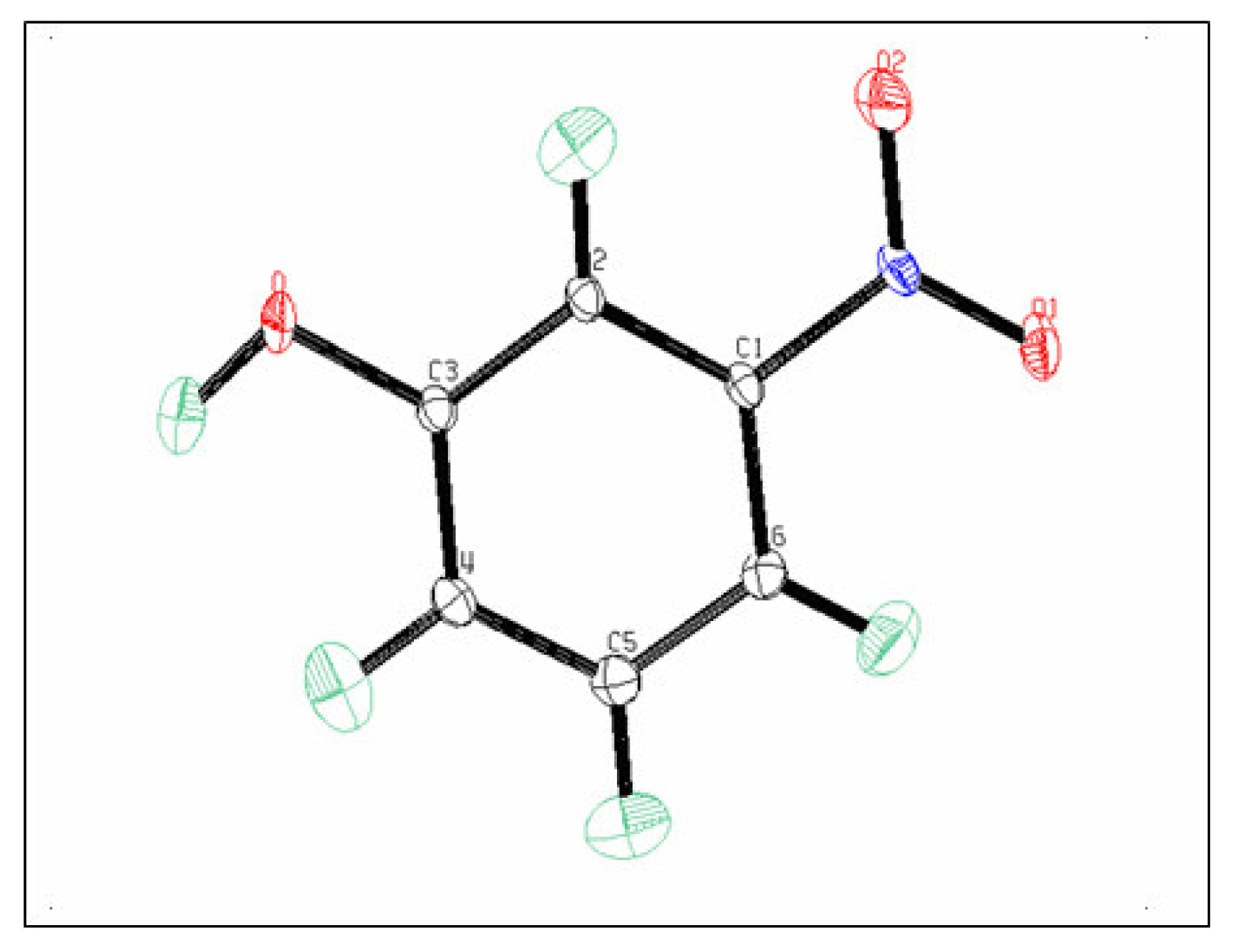
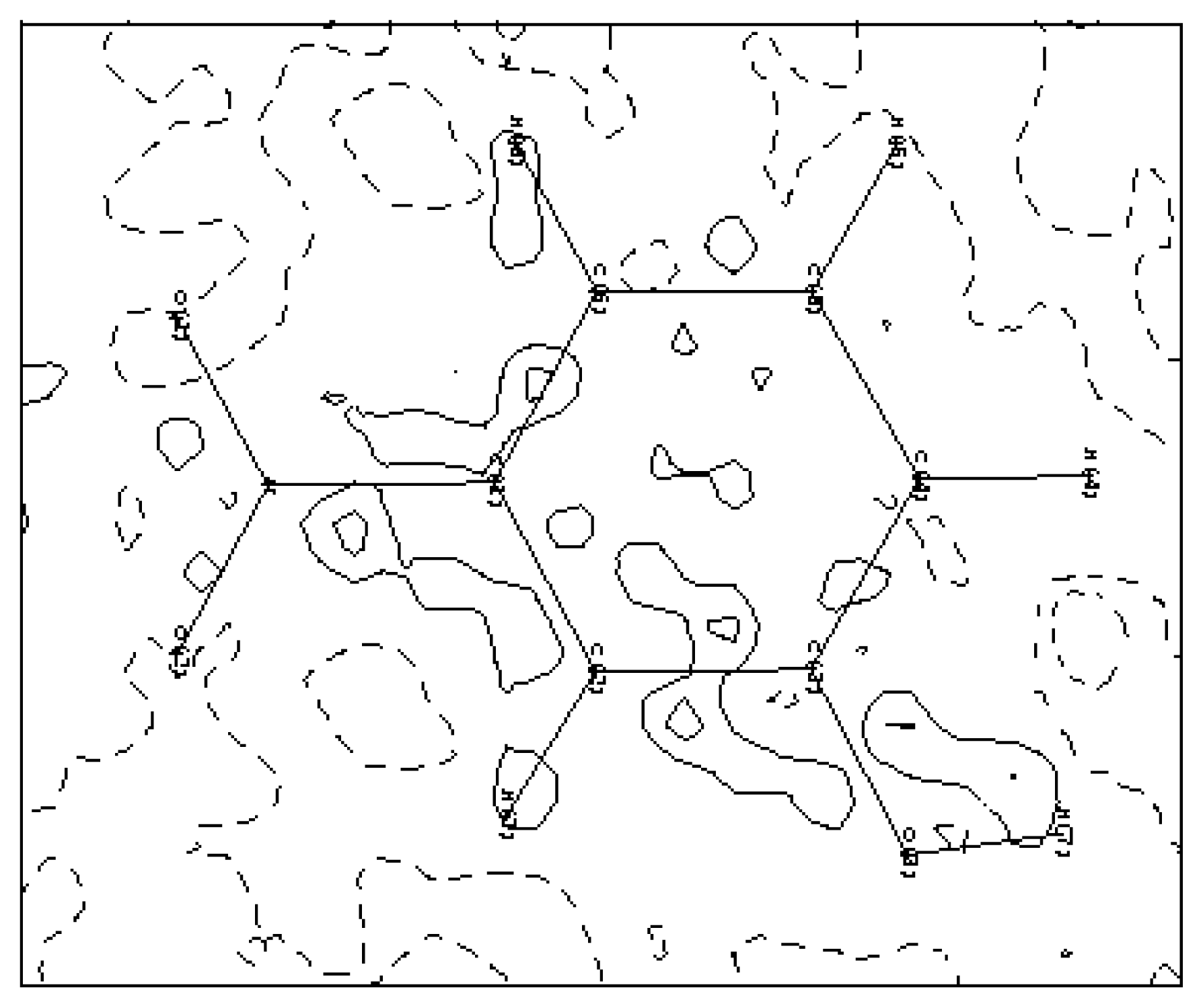
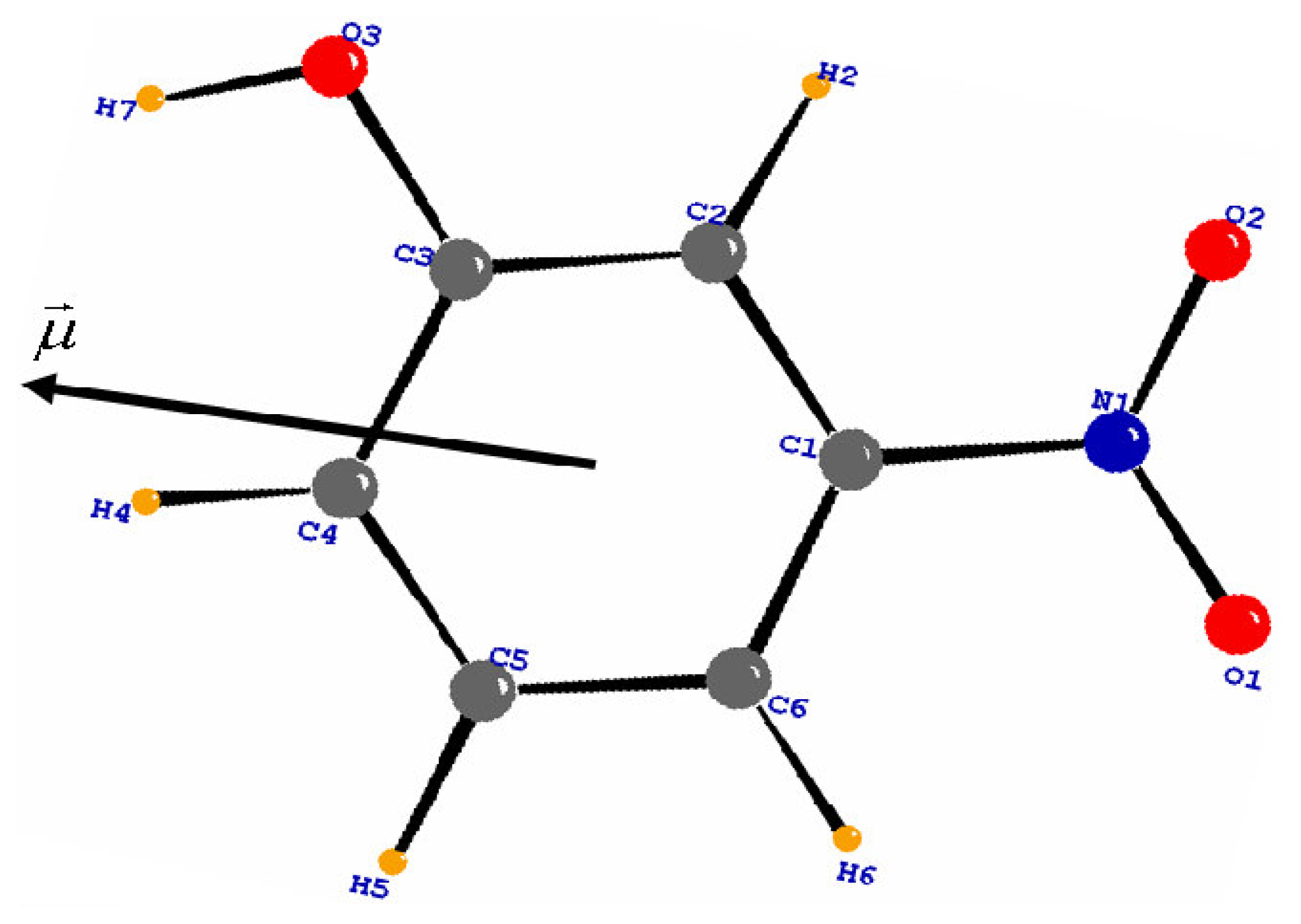
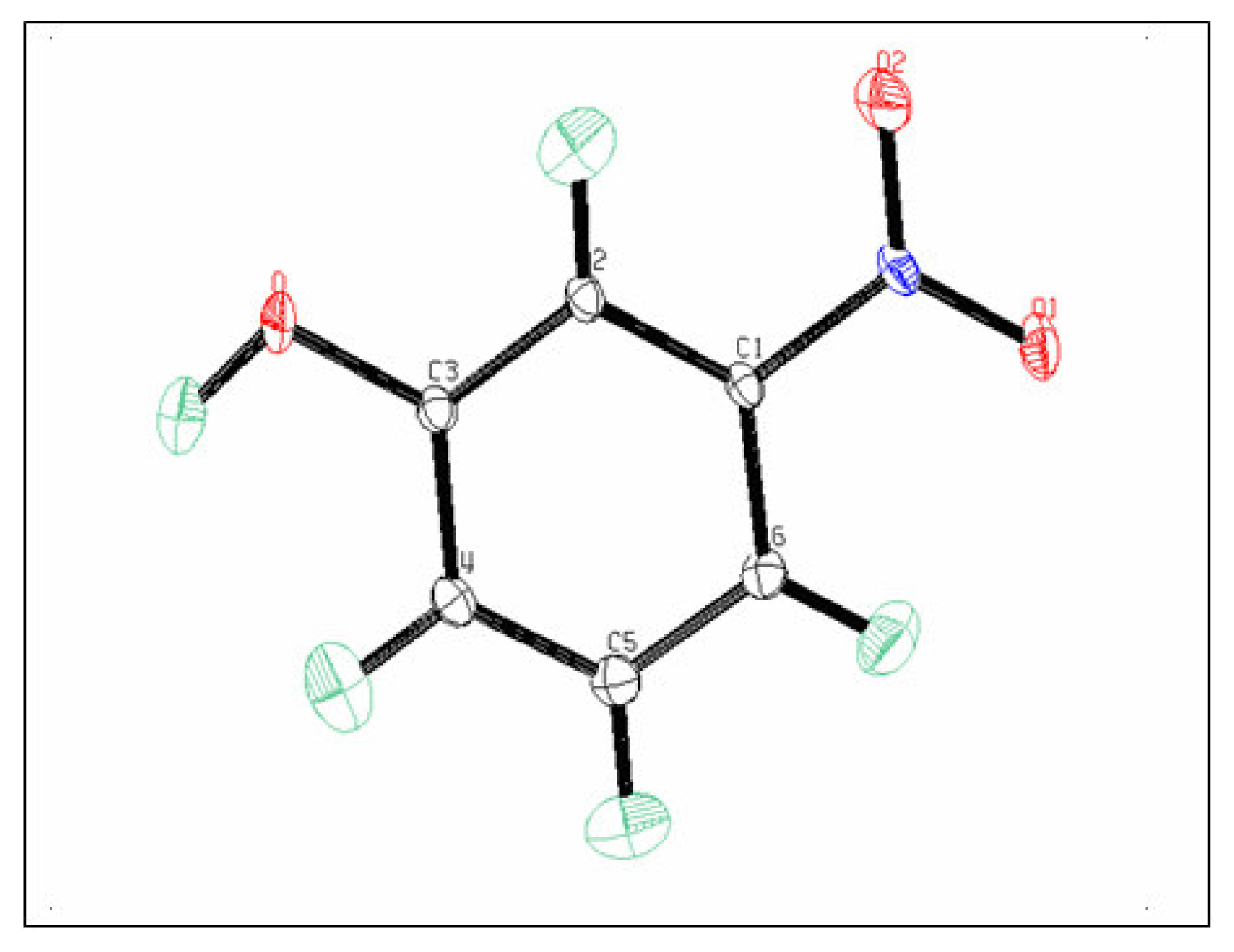
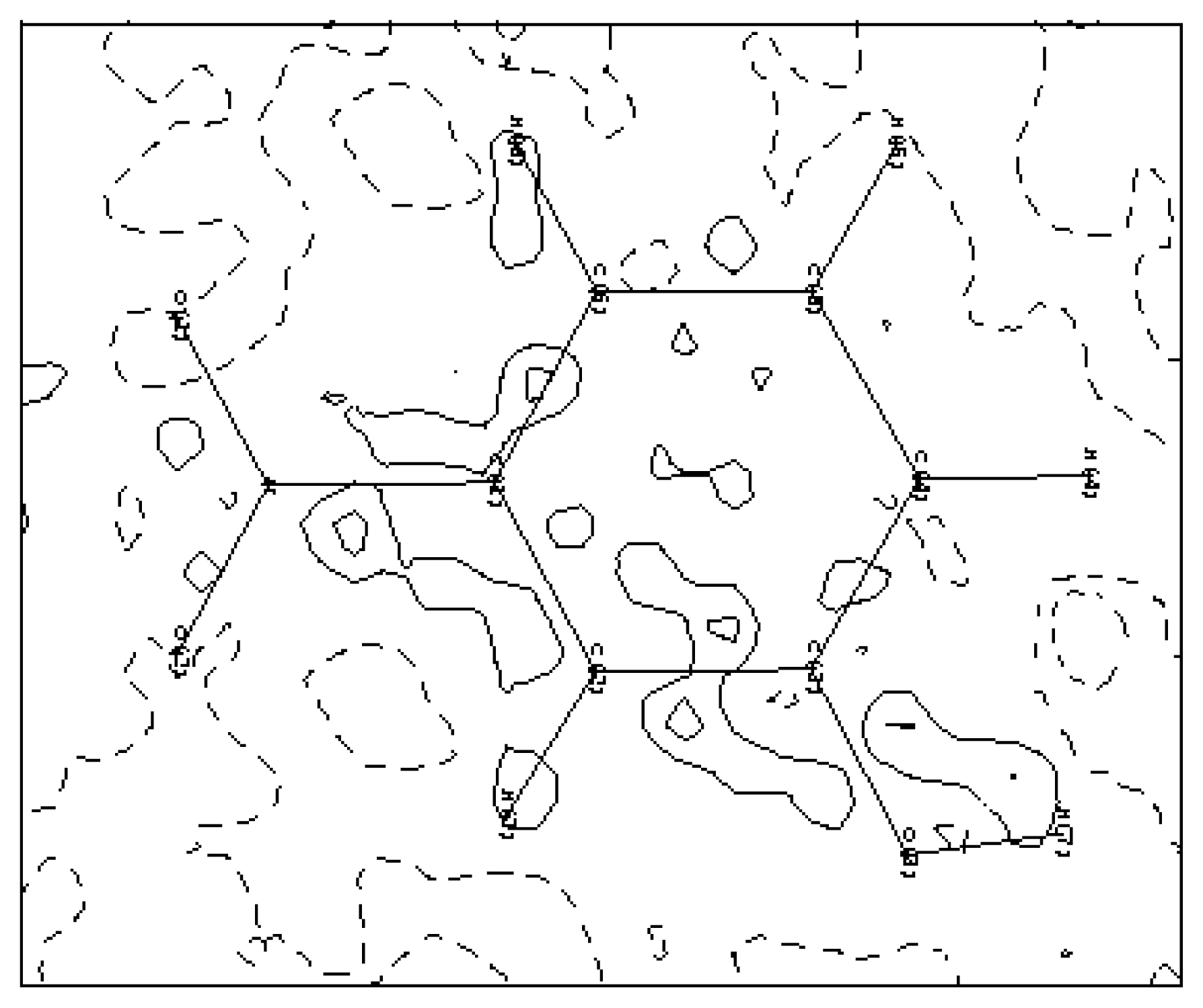
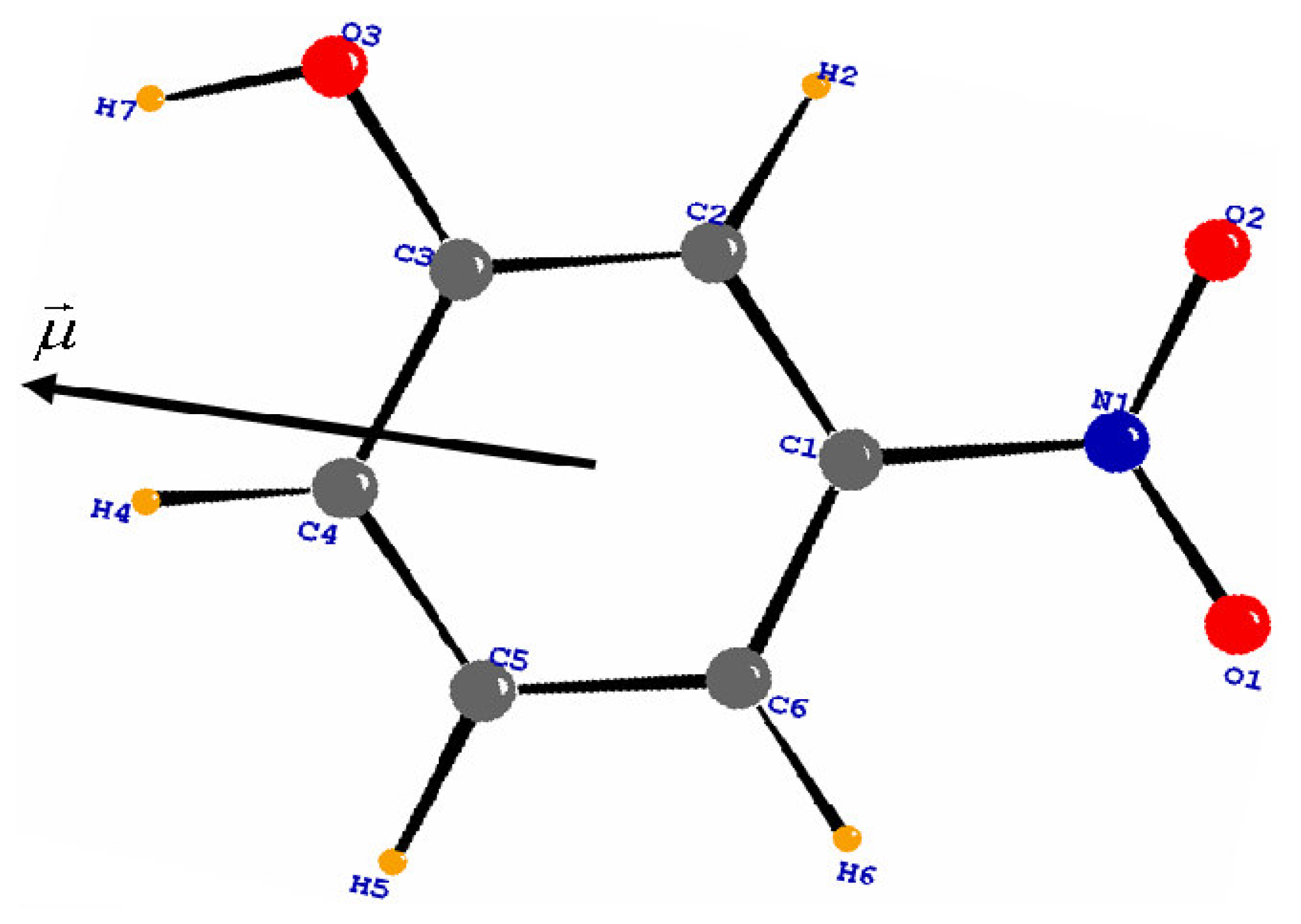
3.1. Structural and Thermal vibration analysis
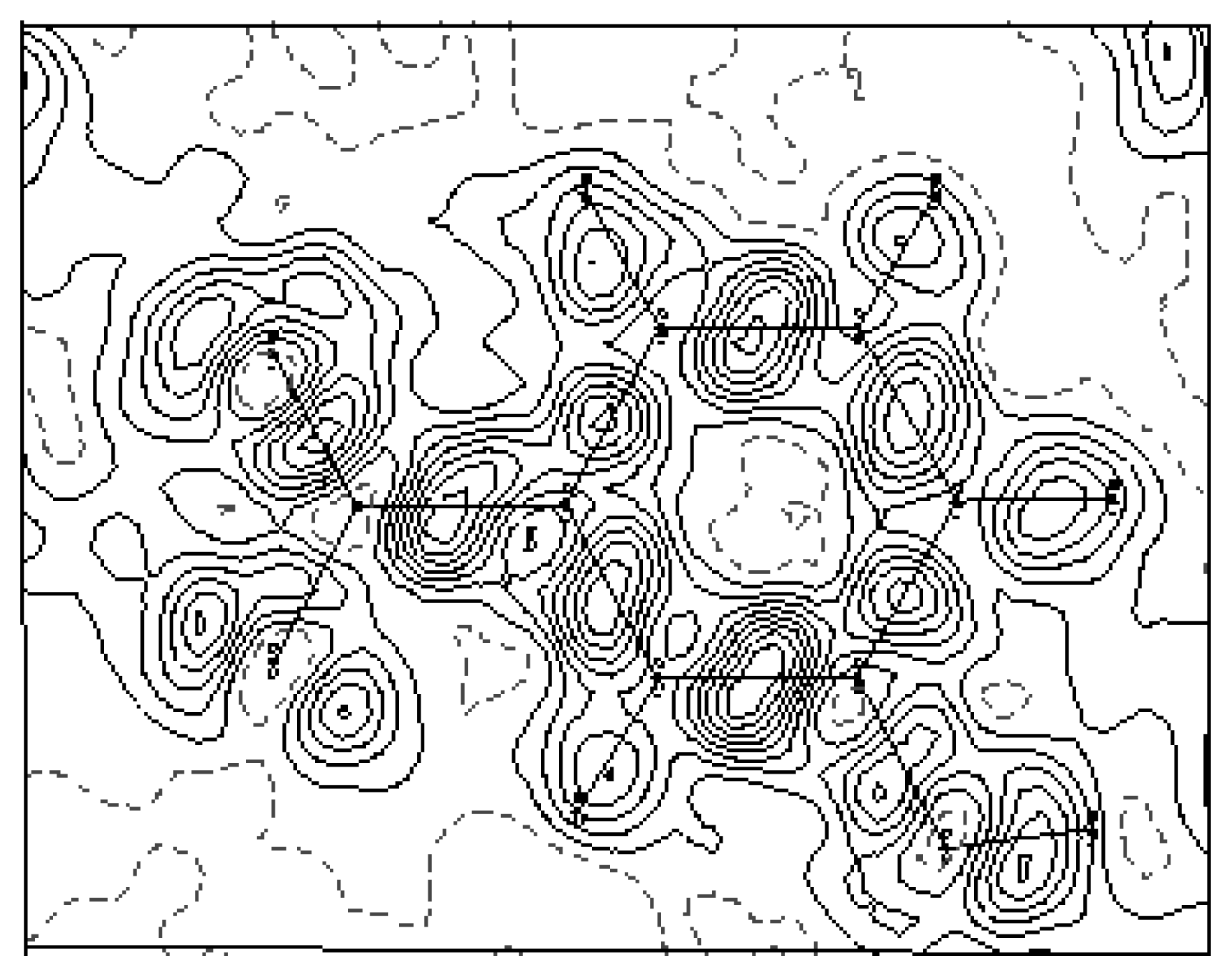
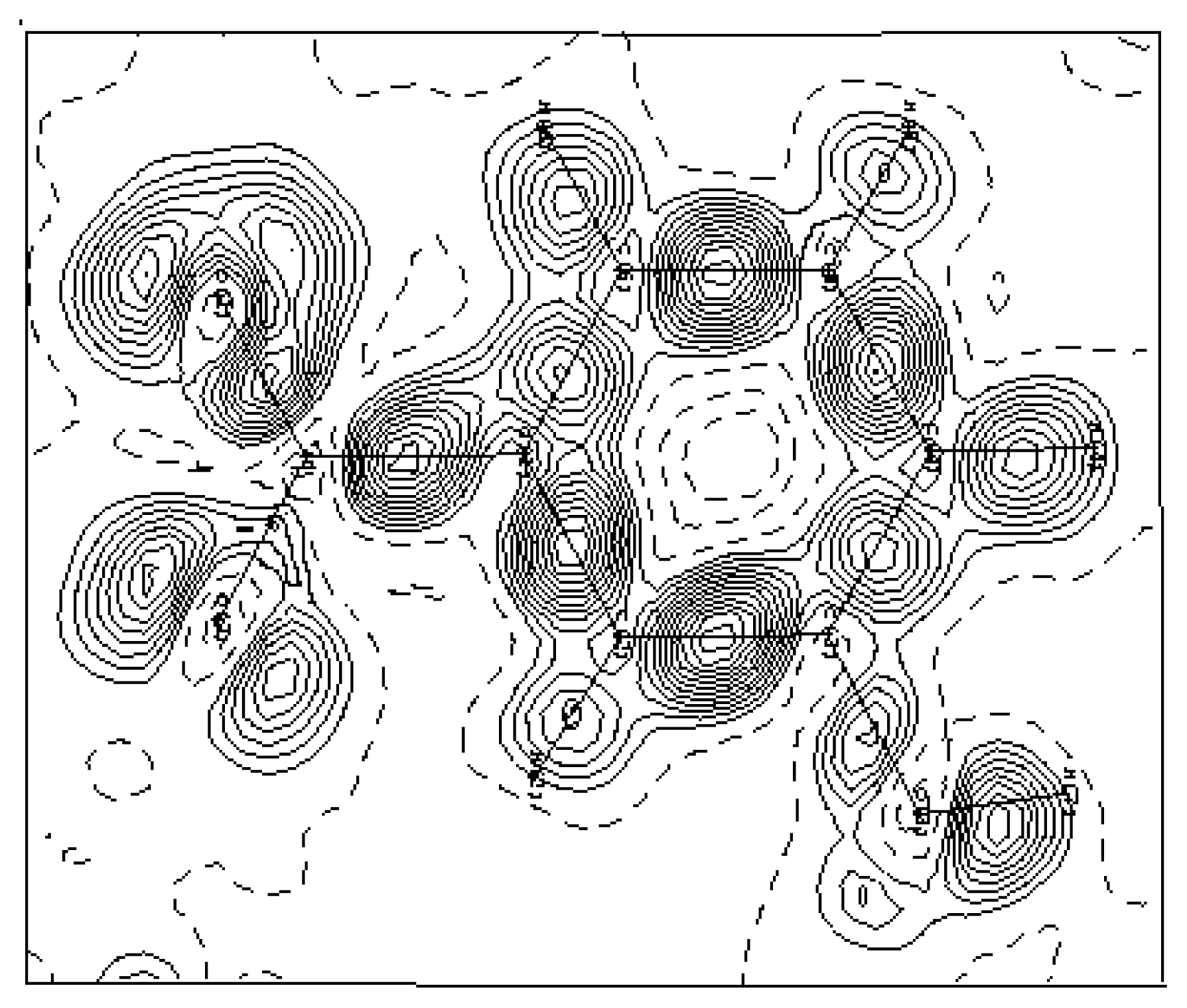
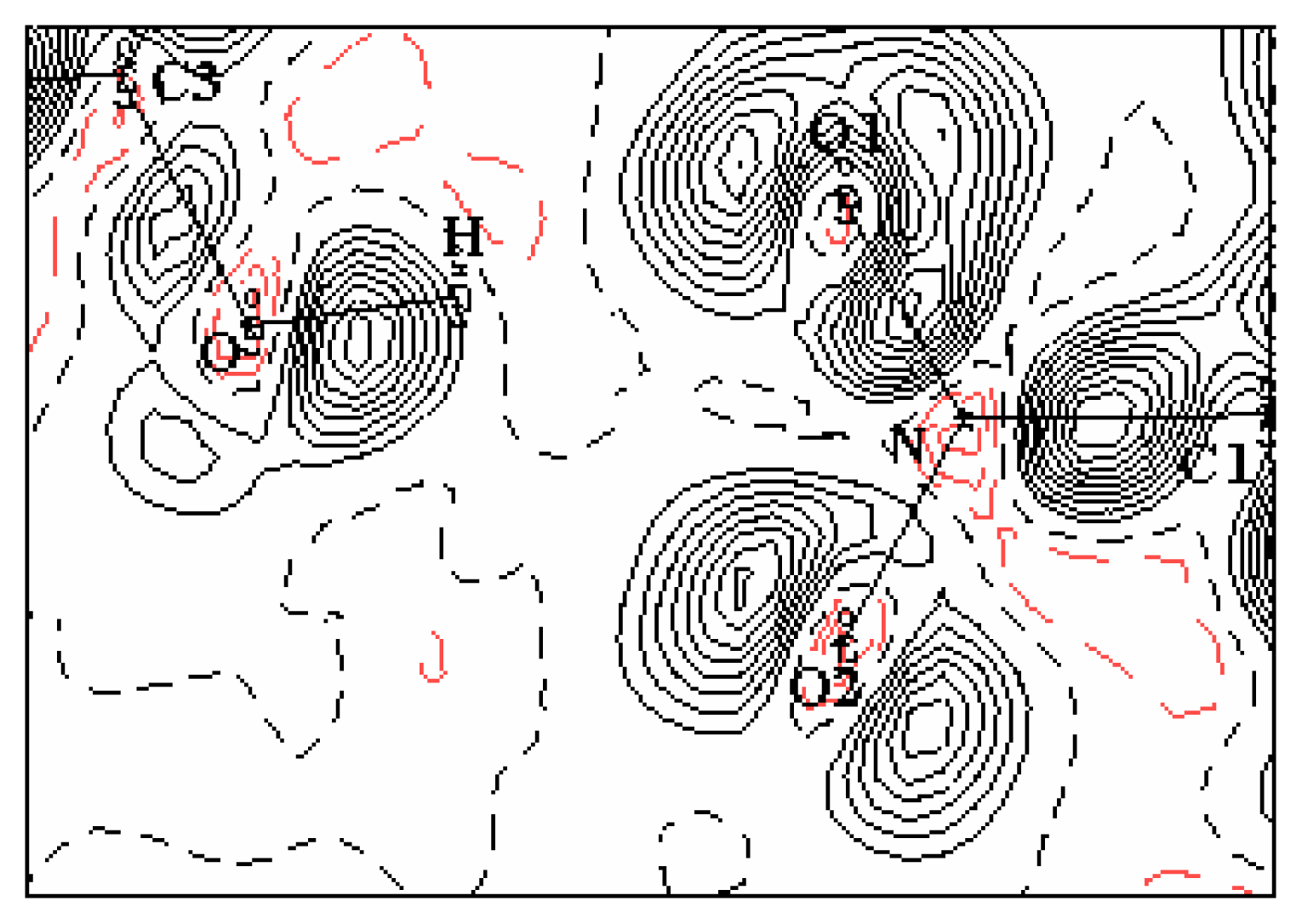
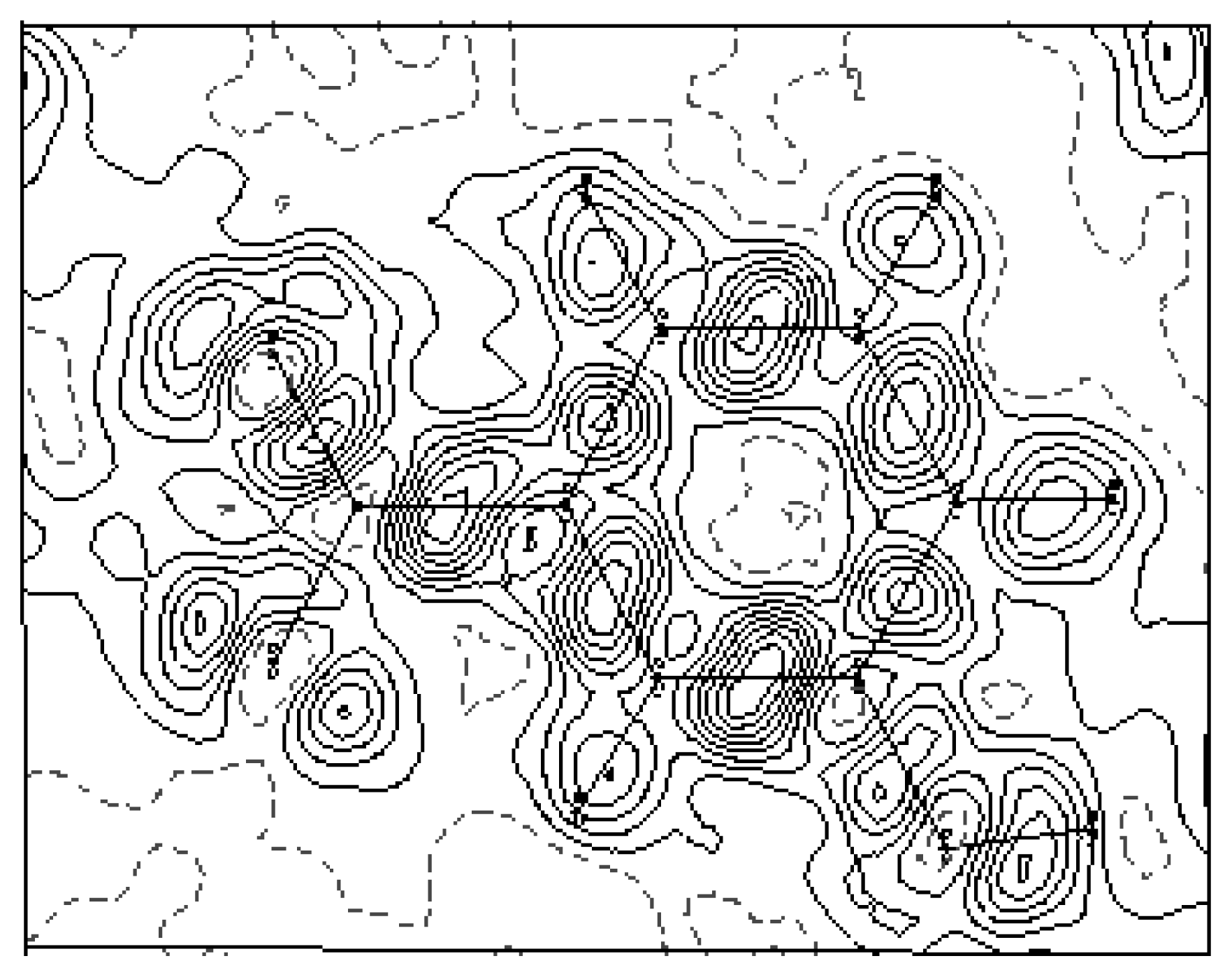
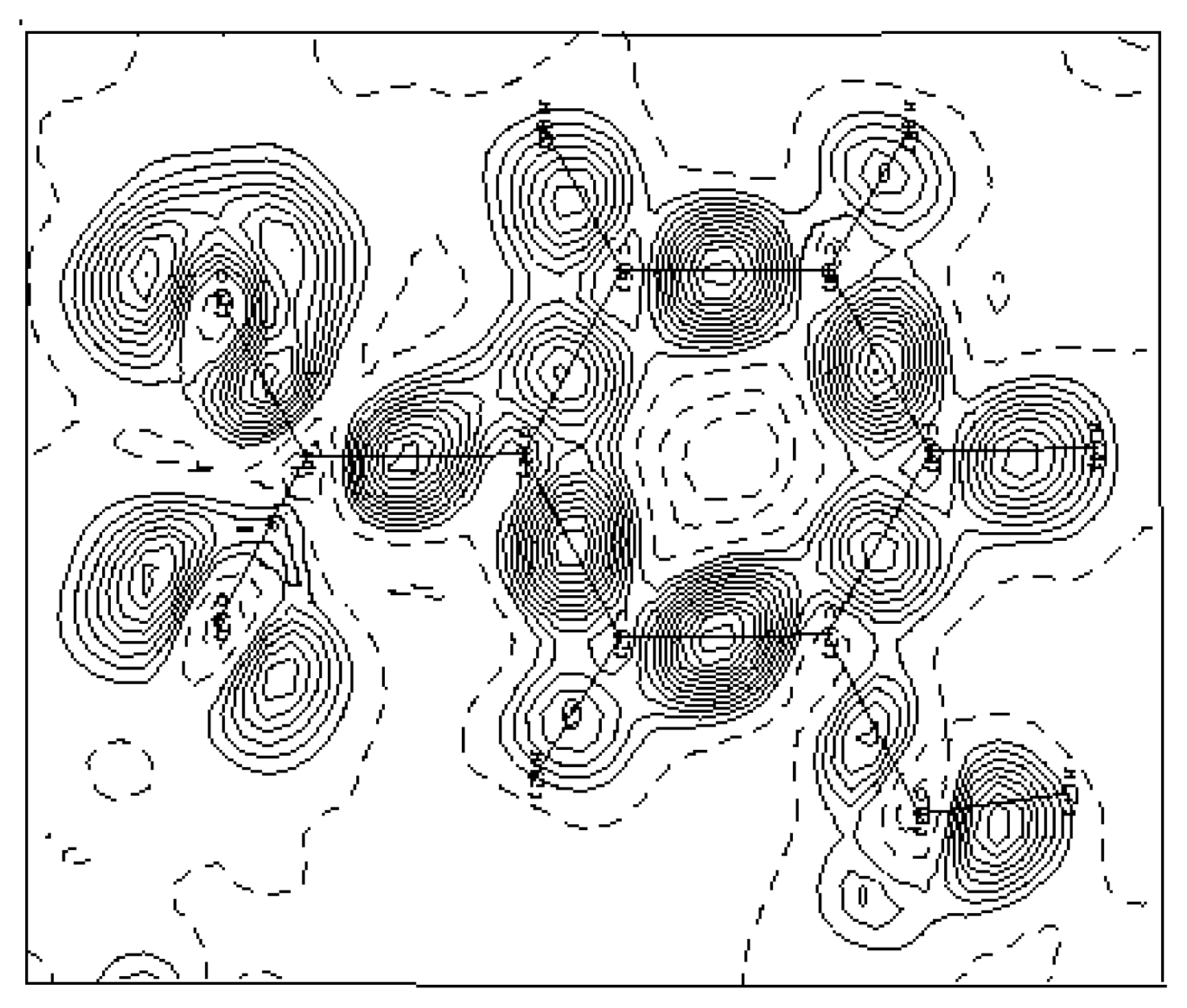
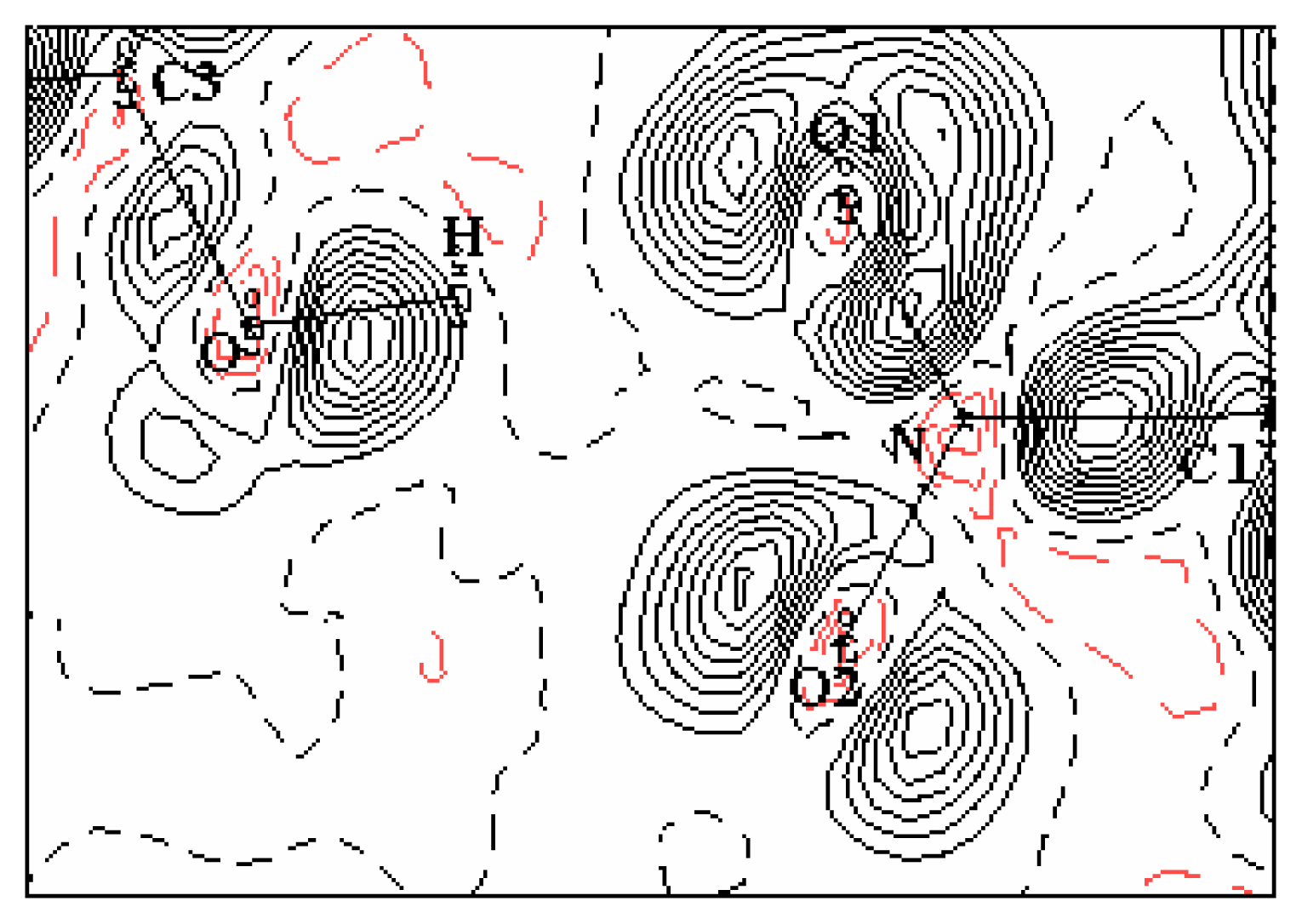
3.2. Electron-density maps
3.3. Molecular dipole moment
3.4. Hydrogen bonds
4 . Experimental Section
5. Conclusion
References
- Hamzaoui, F.; Baert, F.; Wojcik, G. Electron-density study of m-nitrophenol in the orthorhombic structure. Acta Cryst. B 1996, 52, 159–164. [Google Scholar]
- Wojcik, G.; Marqueton, Y. The phase transition of m-nitrophenol. Mol. Cryst. Liq. Cryst 1989, 168, 247–254. [Google Scholar]
- Wojcik, G.; Toupet, L. The inter-and intramolecular charge transfer along the polymeric chain of hydrogen-bonded molecules in two crystal forms of m-nitrophenol. Mol. Cryst. Liq. Cryst 1993, 229, 153–159. [Google Scholar]
- Panadares, F.; Ungaretti, L.; Coda, A. The crystal structure of a monoclinic phase of m-nitrophenol. Acta Cryst. B 1975, 31, 2671–2675. [Google Scholar]
- Koritsanszky, T.; Howard, S.; Richter, T.; Su, Z.; Mallinson, P.R.; Hansen, N.K. XD a Computer Program Package for Multipole Refinement and Analysis of Electron Densities from Diffraction Data; Free University of Berlin: Berlin, Germany, 2003. [Google Scholar]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Cryst A 1978, 34, 909–921. [Google Scholar]
- Volkov, A.; Abramov, Y.; Coppens, P.; Gatti, C. On the origin of topological differences between experimental and theoretical crystal charge densities. Acta Cryst. A 2000, 56, 332–339. [Google Scholar]
- Volkov, A.; Gatti, C.; Abramov, Y.; Coppens, P. Evaluation of net atomic charges and atomic and molecular electrostatic moments through topological analysis of the experimental charge density. Acta Cryst. A 2000, 56, 252–258. [Google Scholar]
- Jeffrey, G.A.; Cruickshank, D.W.J. Molecular structure determination by X-ray crystal analysis: modern methods and their accuracy. Quart. Rev. Chem.Soc 1953, 7, 335–376. [Google Scholar]
- International Tables for X-ray Crystallography; Kynoch Press: Birmingham, 1962; Volume III.
- Stewart, R.F.; Davidson, E.R.; Simpson, W.T. coherent X-Ray Scattering for the Hydrogen Atom in the Hydrogen Molecule. J. Chem, Phys 1965, 42, 3175–3187. [Google Scholar]
- Coppens, P. X-ray Charge Densities and Chemical Bonding; New York; Oxford, 1997. [Google Scholar]
- International Tables for X-ray Crystallography; Kluwer Academic Publishers, 1999; Volume C.
- Trueblood, K.N. Program THMAI1, Department of chemistry and biochemistry; University of California: Los Angeles, 1990. [Google Scholar]
- Hirshfeld, F.L. Can X-ray data distinguish bonding effects from vibrational smearing? Acta Cryst. A 1976, 32, 239–244. [Google Scholar]
- Hirshfeld, F.L. Difference densities by least-squares refinement: fumaramic acid. Acta Cryst B 1971, 27, 769. [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor Chim Acta 1977, 44, 129. [Google Scholar]
- Hirshfeld, F.L.; Hope, H. An x-ray determination of the charge deformation density in 2- cyanoguanidine. Acta Cryst. B 1980, 36, 406. [Google Scholar]
- Johnoson, C.K. ORTEP program; Report ORNL-3794; Oak Ridge National Laboratory: Tennessee, 1965. [Google Scholar]
- Blessing, R.H. DREAD - data reduction and error analysis for single-crystal diffractometer data. J. Appl. Cryst 1989, 22, 396–397. [Google Scholar]
- Souhassou, M.; Blessing, R.H. Topological analysis of experimental electron densities. J. Appl. Cryst 1999, 32, 210–217. [Google Scholar]
- Souhassou, M.; Lecomte, C.; Blessing, R.H; Aubry, A.; Rohmer, M.M.; Wiest, R.; Benard, M.; Marraud, M. Electron distributions in peptides and related molecules.1. An experimental and theoretical study of N-acetyl-L-tryptophan methylamide. Acta Cryst. B 1991, 47, 253–266. [Google Scholar]
- Wojcik, G.; Holband, J.; Szymczak, J.J.; Roszak, S.; Leszczynski, J. Interactions in Polymorphic Crystals of m-Nitrophenol as Studied by Variable-Temperature X-ray Diffraction and Quantum Chemical Calculations. Crystal Growth & Design 2006, 6, 274–282. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O | O2 | O1 | N | C6 | C5 | C4 | C3 | C2 | |
|---|---|---|---|---|---|---|---|---|---|
| C1 | −5 | 5 | 2 | 14 | 5 | 17 | 16 | 10 | −4 |
| C2 | −5 | 27 | −9 | −1 | 7 | 7 | 8 | 11 | |
| C3 | −10 | 16 | −10 | −7 | 8 | 5 | 1 | ||
| C4 | −19 | 5 | −1 | −2 | 4 | 4 | |||
| C5 | −19 | −13 | 0 | −7 | −4 | ||||
| C6 | −18 | −17 | 5 | −3 | |||||
| N | −4 | −15 | −7 | ||||||
| O1 | 2 | 5 | |||||||
| O2 | −40 |
| ATOM | Pv | q |
|---|---|---|
| C1 | 4.153 | −0.153 |
| C2 | 4.270 | −0.270 |
| C3 | 3.971 | 0.028 |
| C4 | 4.395 | −0.395 |
| C5 | 4.462 | −0.462 |
| C6 | 4.268 | −0.268 |
| N | 4.353 | 0.646 |
| O1 | 6.233 | −0.233 |
| O2 | 6.218 | −0.218 |
| O | 6.290 | −0.290 |
| H2 | 0.681 | 0.318 |
| H4 | 0.783 | 0.216 |
| H5 | 0.749 | 0.250 |
| H6 | 0.711 | 0.288 |
| HO | 0.671 | 0.328 |
| Atoms | Pml | d2 | d3 | q1 | q3 | q4 | o1 | o3 | o4 | o7 |
|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 4.1536 | −0.003 | 0.082 | 0.185 | −0.001 | −0.056 | 0.212 | −0.082 | 0.175 | −0.135 |
| C2 | 4.2703 | 0.131 | 0.354 | 0.032 | −0.027 | −0.154 | 0.265 | −0.124 | 0.192 | −0.054 |
| C3 | 3.9710 | 0.018 | 0.197 | 0.224 | 0.044 | −0.119 | 0.181 | −0.019 | 0.153 | −0.091 |
| C4 | 4.3953 | −0.105 | 0.114 | 0.287 | 0.118 | −0.126 | 0.130 | 0.040 | 0.112 | 0.004 |
| C5 | 4.4621 | −0.037 | 0.062 | 0.141 | 0.025 | −0.246 | 0.165 | 0.096 | 0.140 | −0.009 |
| C6 | 4.2681 | −0.033 | 0.008 | −0.039 | −0.279 | 0.162 | 0.224 | 0.059 | 0.186 | −0.031 |
| N | 4.3534 | −0.015 | 0.012 | 0.050 | −0.081 | −0.176 | 0.177 | −0.006 | 0.134 | 0.033 |
| O1 | 6.2335 | −0.128 | −0.055 | −0.001 | 0.003 | −0.207 | 0.056 | 0.048 | 0.046 | −0.103 |
| O2 | 6.2179 | 0.078 | −0.073 | −0.092 | 0.111 | −0.251 | 0.068 | 0.021 | 0.040 | 0.098 |
| O | 6.2901 | −0.036 | −0.033 | 0.068 | 0.004 | −0.052 | 0.092 | −0.018 | 0.100 | 0.011 |
| H2 | 0.6813 | 0.227 | ||||||||
| H4 | 0.7835 | 0.281 | ||||||||
| H5 | 0.7492 | 0.234 | ||||||||
| H6 | 0.7115 | 0.196 | ||||||||
| HO | 0.6711 | 0.126 |
| μx | μy | μz | μ (Debye) |
|---|---|---|---|
| −0.032 | −0.032 | −0.636 | 5.81 (20) |
| D — H ...A | D — H...A (°) | D — H (Å) | D…A (Å) | H….A (Å) |
|---|---|---|---|---|
| C2—H2…..O2 (a) | 95.26 (8) | 1.105 (2) | 2.717 (3) | 2.382 (3) |
| C6—H6..…O1 (a) | 91.79 (9) | 1.093 (2) | 2.727 (3) | 2.464 (2) |
| C4—H4…..O1 (b) | 129.70 (9) | 1.094 (2) | 3.386 (4) | 2.581 (3) |
| O —HO…..N (b) | 155.47 (8) | 1.029 (1) | 3.495 (4) | 2.532 (3) |
| O —HO…..O1 (b) | 178.03 (8) | 1.029 (1) | 2.908 (3) | 1.880 (2) |
| O —HO......O2 (b) | 127.49 (8) | 1.029 (1) | 3.315 (4) | 2.586 (3) |
| C5—H5…..O1 (c) | 124.62 (9) | 1.109 (2) | 3.693 (5) | 2.949 (4) |
| C5—H5…..O2 (d) | 140.67 (9) | 1.109 (9) | 3.338 (3) | 2.405 (2) |
| C5—H5…..O (e) | 131.87 (9) | 1.109 (2) | 3.471 (3) | 2.631 (2) |
| Crystal data | ||||
| Chemical formula | C6H5NO3 | |||
| Chemical formula weight | 139.11 | |||
| Cell setting | Monoclinic | |||
| Space group | P21/n | |||
| a (Å) | 11.026(4) | |||
| b (Å) | 6.736(1) | |||
| c (Å) | 8.119(21) | |||
| β (°) | 97.73 (2) | |||
| V (Å3) | 597.50 | |||
| Z | 4 | |||
| Dx(mg m−3) | 1.54 | |||
| Radiation type | Mo Kα | |||
| Wavelength (Å) | 0.71073 | |||
| No. of reflections for cell parameters | 25 | |||
| θ range (°) | 11 – 25 | |||
| μ (mm−1) | 0.127 | |||
| Temperature (K) | 122 (1) | |||
| Crystal form | Prism | |||
| Crystal size | 0.10 × 0.27 × 0.32 | |||
| Crystal color | Colorless | |||
| Data collection | ||||
| Diffractometer | Nonius CAD-4 | |||
| Data collection method | θ − 2θ | |||
| 2θmax | 117.71 | |||
| No. of measured reflections | 3148 | |||
| No. of independent reflections | 1736 | |||
| No. of observed reflections | 1412 | |||
| Criterion for observed reflections | I ≥ 3σ (I ) | |||
| Rint | 0.021 | |||
| Range of h, k, l | −25 → h → 23 | |||
| 0 → k → 15 | ||||
| 0 → l → 18 | ||||
| No. of standard reflections | 3 | |||
| Frequency of standard reflections | Every 120 min | |||
| Refinement | ||||
| N | R | wR | S | |
| Spherical refinement | 92 | 0.038 | 0.040 | 1.02 |
| Multipole refinement | 221 | 0.022 | 0.035 | 1.12 |
| N is the number of refined parameters and M is the number of observations. | ||||
| Source of atomic scattering factors | International Tables for X-ray Crystallography (1999, Vol. C) | |||
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Hamzaoui, F.; Drissi, M.; Chouaih, A.; Lagant, P.; Vergoten, G. Electron Charge Density Distribution from X-ray Diffraction Study of the M-Nitrophenol Compound in the Monoclinic Form. Int. J. Mol. Sci. 2007, 8, 103-115. https://doi.org/10.3390/i8020103
Hamzaoui F, Drissi M, Chouaih A, Lagant P, Vergoten G. Electron Charge Density Distribution from X-ray Diffraction Study of the M-Nitrophenol Compound in the Monoclinic Form. International Journal of Molecular Sciences. 2007; 8(2):103-115. https://doi.org/10.3390/i8020103
Chicago/Turabian StyleHamzaoui, Fodil, Mokhtaria Drissi, Abdelkader Chouaih, Philippe Lagant, and Gérard Vergoten. 2007. "Electron Charge Density Distribution from X-ray Diffraction Study of the M-Nitrophenol Compound in the Monoclinic Form" International Journal of Molecular Sciences 8, no. 2: 103-115. https://doi.org/10.3390/i8020103
APA StyleHamzaoui, F., Drissi, M., Chouaih, A., Lagant, P., & Vergoten, G. (2007). Electron Charge Density Distribution from X-ray Diffraction Study of the M-Nitrophenol Compound in the Monoclinic Form. International Journal of Molecular Sciences, 8(2), 103-115. https://doi.org/10.3390/i8020103