1. Introduction
Great effort has been expended to explore self-assembled supramolecular complexes due to their intriguing network topologies and potential applications in molecular recognition, magnetic materials, medicine, nonlinear optical devices and catalysts [
1–
9]. The study of metal–organic coordination polymers has currently provoked significant temporary interest in chemistry and materials science not only for their potential applications as functional solid state materials, but also for their intrinsic aesthetic appeal [
10,
11].
Of particular interest is the crystal engineering of helical metal compounds [
12–
14]. Helices are undoubtedly the most celebrated creations of nature, and also represent aesthetically pleasing and highly ordered systems. Recently, Venkataraman and co-workers [
15] have identified the geometries of the molecular modules that are meant to undergo helical self-assembly, and demonstrated the viability of achieving the self-assembly into helices. In chemistry or biochemistry helicity is present in various systems [
16]. Deoxyribonucleic acid (DNA) exists as a double helix where the two strands are linked by hydrogen bonding between complementary bases [
17]. α-Amylose is a macromolecule with a helical structure that contains about six glucose units per helical turn [
18]. As another example, peptides can also adopt an α-helical structure or form larger helical arrays as, for example, found in the collagene triple helices [
19]. In artificial supramolecular architectures, helicity can be introduced by conformational restrictions of macromolecules [
20], inter- or intramolecular hydrogen bonds [
21,
22], or coordination to metal ions [
23–
25]. It has been shown that by appropriate combination of metal centres and multidentate ligands, systems ranging from discrete helicates [
26] to coordination polymers containing infinite helices [
27] can be prepared. There are a large variety of coordination compounds whose molecular structures may be loosely described as helical. Pseudotetrahedral complexes possessing two unsymmetrical bidentate AB-type ligands coordinated to a central metal ion [M(AB)
2] or pseudooctahedral complexes [M(AB)
3] correspond to helical complexes [
12–
14].
A type of simple flexible N-donor bipyridyl ligands Py
2O [
28] and Py
2S [
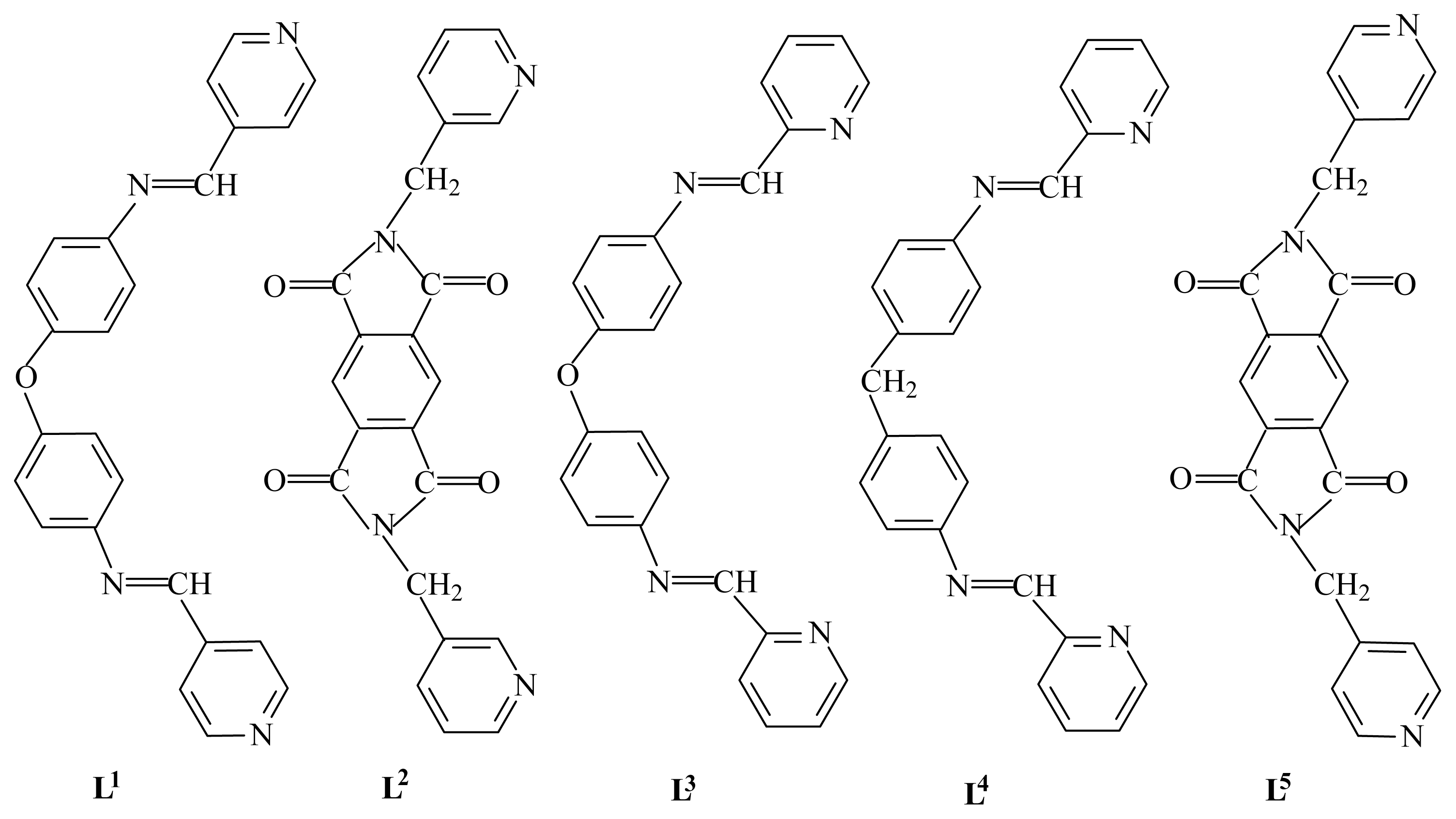
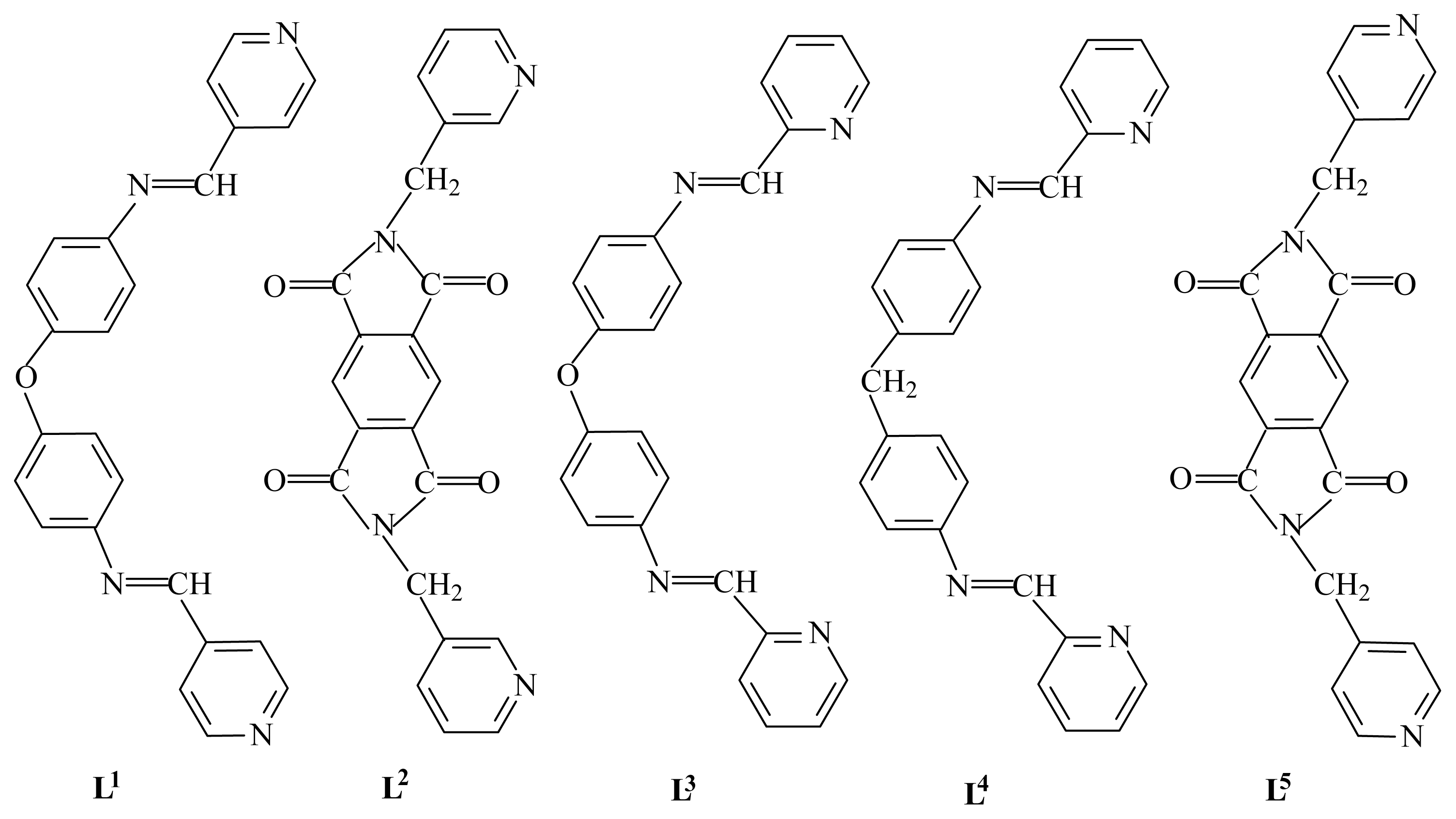
29] with angular spacers O or S has been recently reported in the formation of cyclindrical helices with Ag(I) salts. Conceivably, by replacing the O or S atom by other large noncoordinating spacers, the adjusted and versatile flexibility of the ligands could lead to different coordination modes and precise topological structures, and sophisticated metal complexes can be constructed. To this end, the rational design and preparation of ligands of required geometry in the spacer seem extremely important to the construction of new polymeric architectures. On the other hand, to the best of our knowledge, the Hg(II) coordination polymers are relatively rare. Here we report the formation of a helical network based on the use of the organic ligand L
1 (L
1 = bis[4-(4-pyridylmethyleneamino)phenyl] ether) and an one-dimensional zigzag chain structure based on the organic ligand L
2 (L
2 =
N,
N′-bis(3-pyridylmethyl)-diphthalic diimide) (
Chart 1). As part of our studies of metal–organic coordination polymers derived from bis-monodentate ligands [
30–
32]
via self-assembly, we report in this paper a polymeric organometallic double helix, using weak hydrogen bonding between I atom and H atom of the phenyl unit to crosslink the individual helical chains.
2. Results and Discussion
The ligand L
1 is prepared by simply refluxing a THF solution containing pyridine-4-carbaldehyde and bis(4-aminophenyl) ether. The ease of synthesis and high yield in a single-step reaction from commercial, inexpensive reagents make this an attractive ligand system. The ligand L
2 was prepared as described in a previous publication [
33].
Dealing with the metallic component, since the ligand L
1 is neutral, we believed that a combination of a dicationic metal centre allowing linear bridging and two strongly coordinating anions would be interesting because it would result in a binary system. For that reason HgI
2 was chosen to generate the supramolecular architecture. Upon slow diffusion at room temperature of a CH
3OH solution (10 ml) containing HgI
2 (12 mg) into a DMF solution (3 ml) of L
1 (12 mg), colourless crystals were obtained after several days and analysed by X-ray diffraction on a single crystal
1, [HgI
2(L
1)·0.5H
2O]
∞. The crystal (orthorhombic, space group
Pcca) is composed of L
1 and HgI
2. As expected, the mutual interconnection between the organic ligand L
1 and HgI
2 leads to a helical strand (
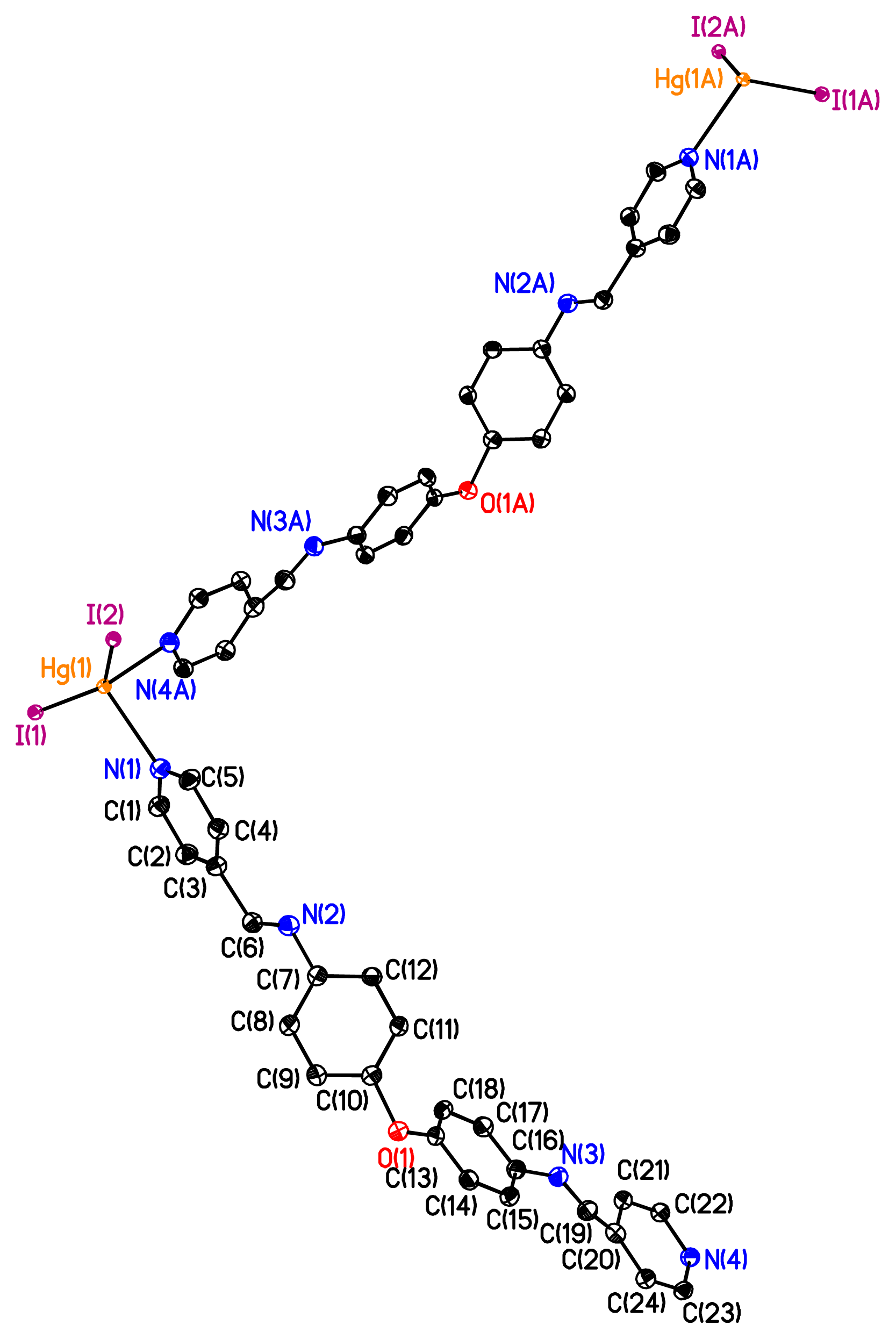
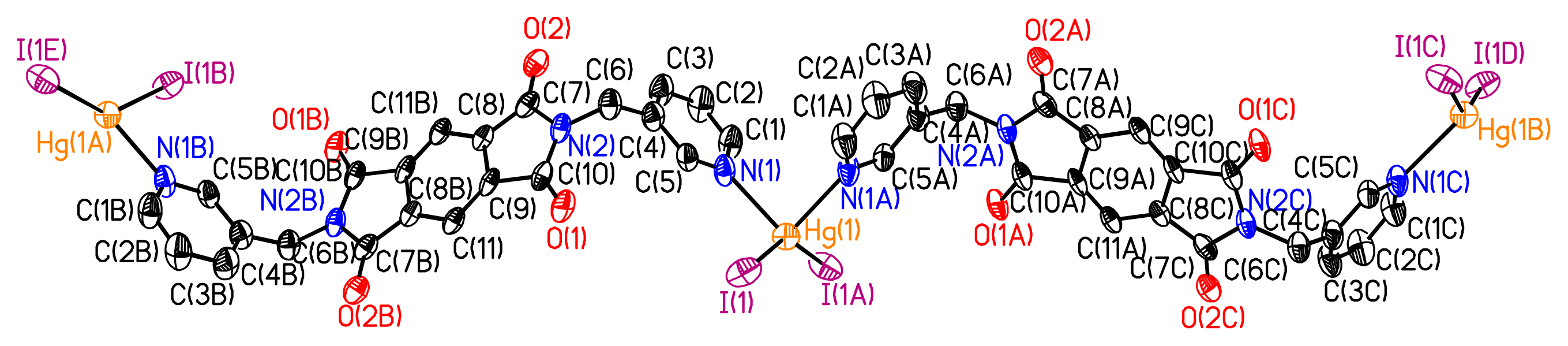
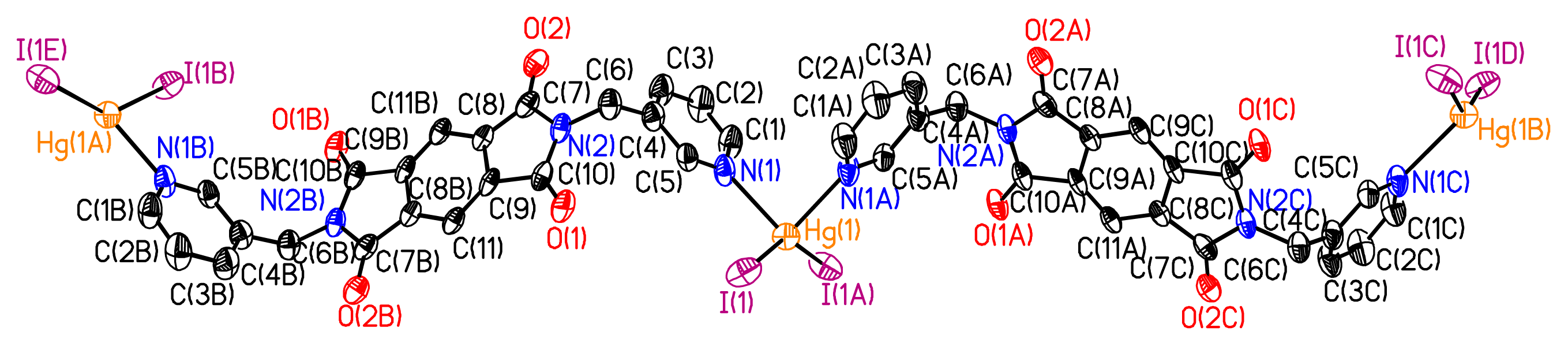
Figure 1). For the organic moiety L
1, the two pyridine units connected to the Schiff-base C=N backbone through bisphenyl ether junctions (
dC–O = 1.33 Å,
θC-O-C = 121.6° and
dC=N = 1.15 Å) are almost parallel and divergently oriented towards the concave face of the bicyclic unit. The pyridine unit is connected to phenyl unit through the Schiff-base C=N backbone in two ways: one way is that the pyridine unit and phenyl unit are almost parallel to each other, and another way is that there is 104.2° of dihedral angle between the pyridine unit and phenyl unit. The bridging of the organic ligand L
1 by HgI
2 units generates an infinite 1-D coordination network with helical geometry. The Hg
2+ cation adopts a tetrahedral geometry with the two I anions (
dHg–I = 2.65 Å) and the two N atoms (
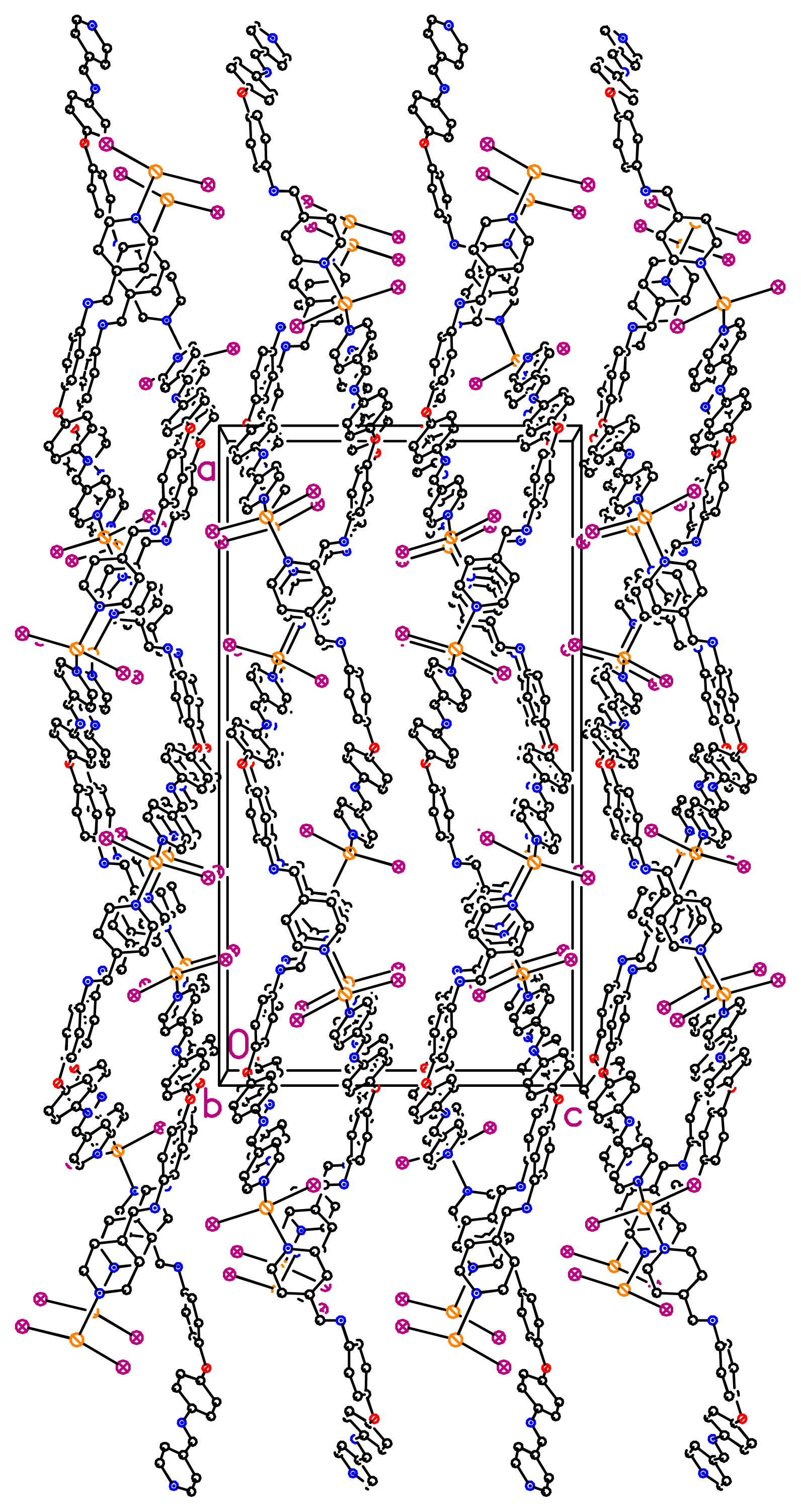
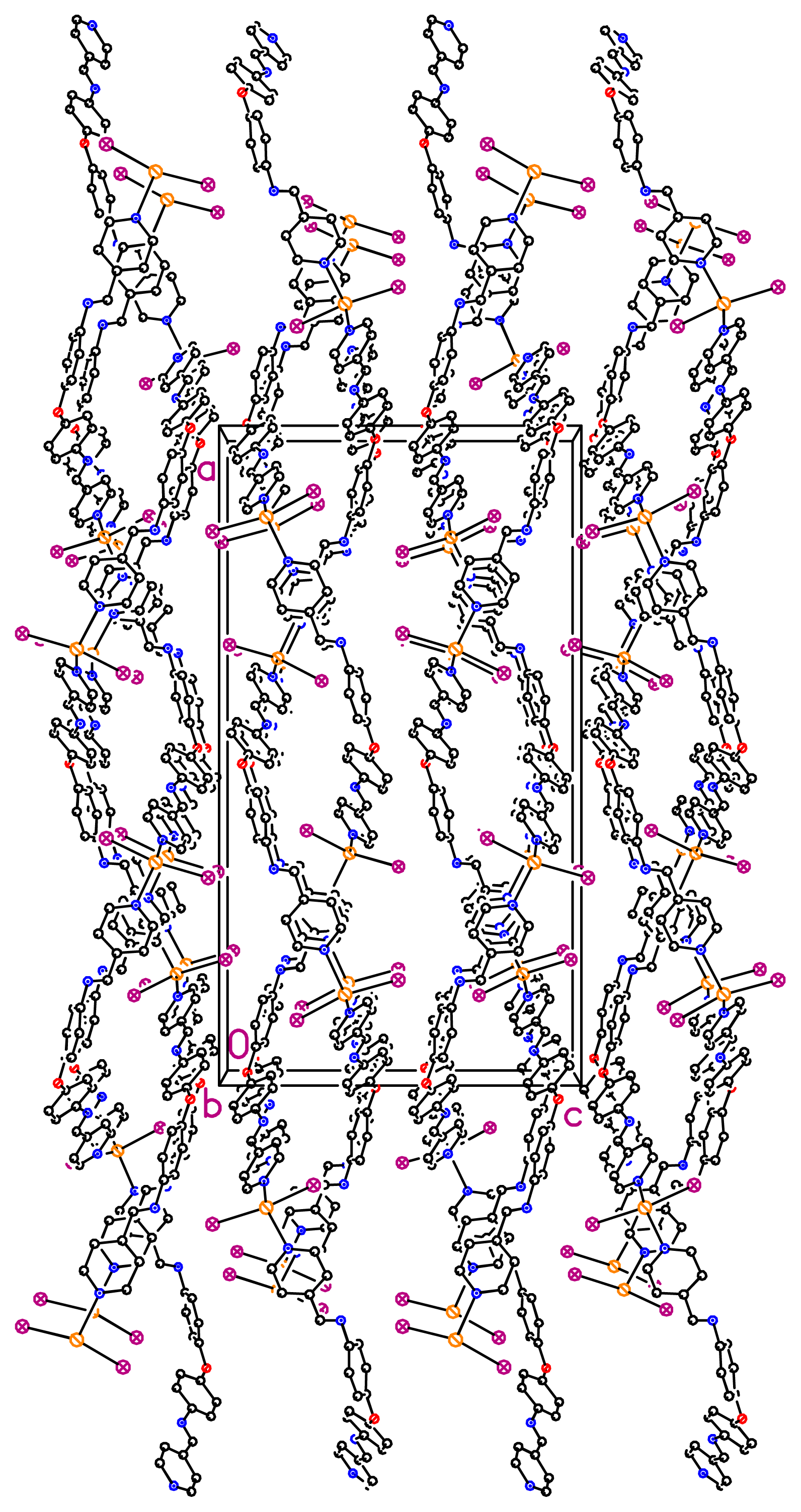
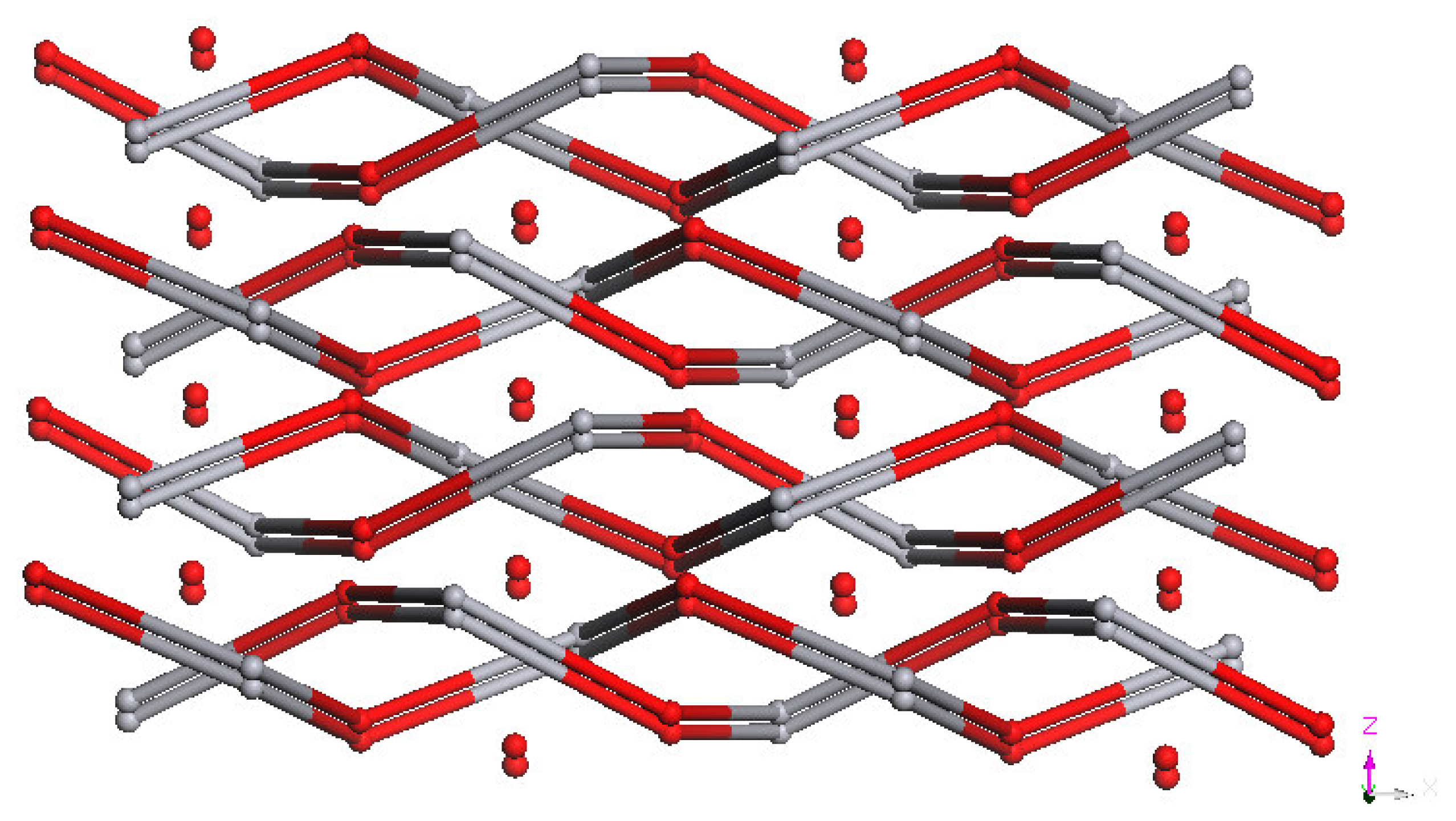
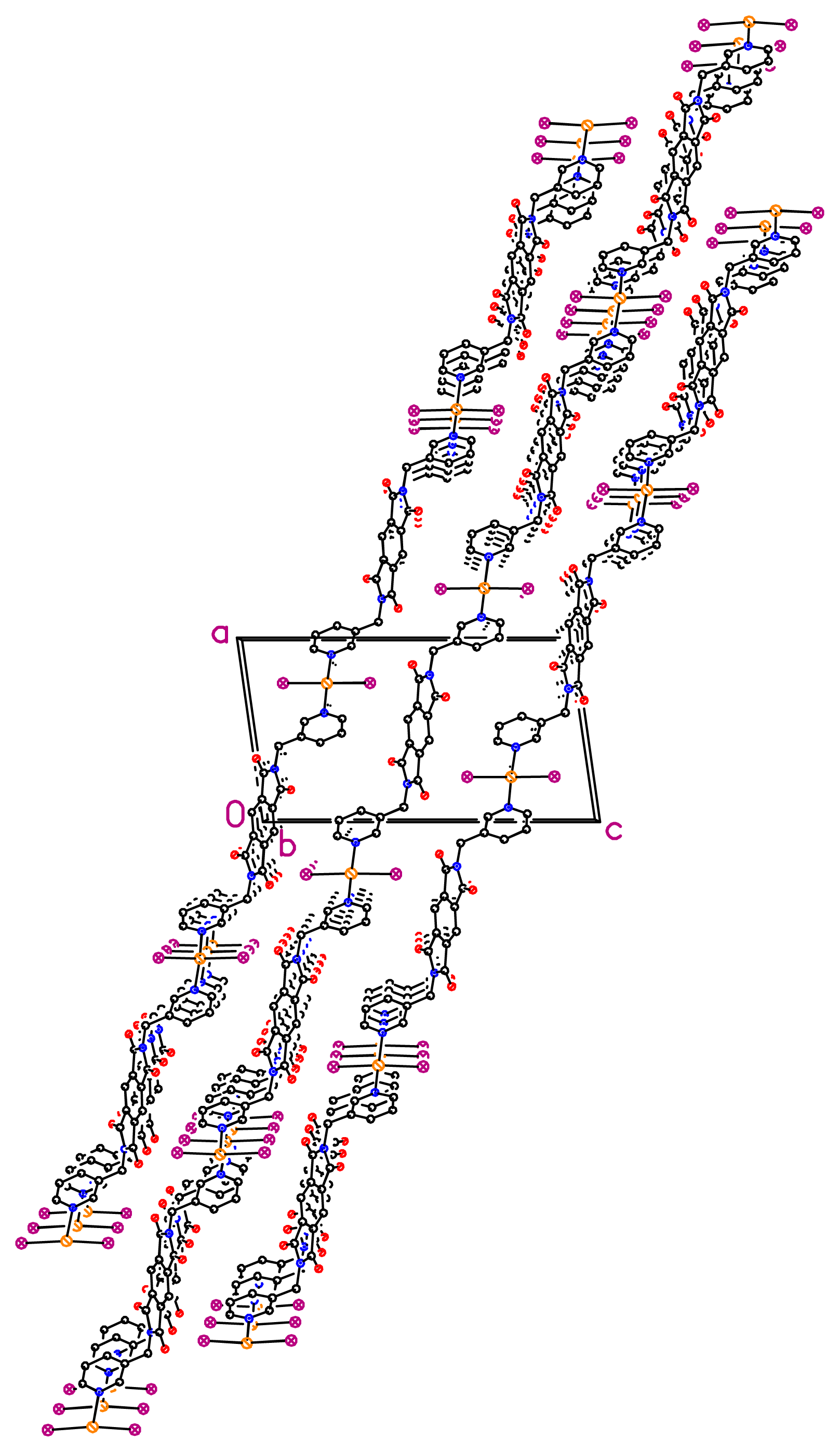
dHg–N = 2.41 Å) in a tetrahedral geometry (IHgN angle varying between 98.96° and 105.84°, IHgI angle of 144.044° and NHgN angle of 94.6°). The nearest Hg···Hg separation in each helical chain is 23.775 Å. Unexpectedly, the X-ray diffraction study revealed the formation of a double stranded helical arrangement with a period of 33.405 Å (
Figures 2 and
3) resulting from a weak hydrogen bonding (
dC(8)–H(8)···I(2) = 3.115 Å and
θC(8)–H(8)···I(2) = 164.66°) between I atom and H atom of the phenyl unit, which is in the expected range (H···I < 3.35 Å and Y···I > 130°) [
34–
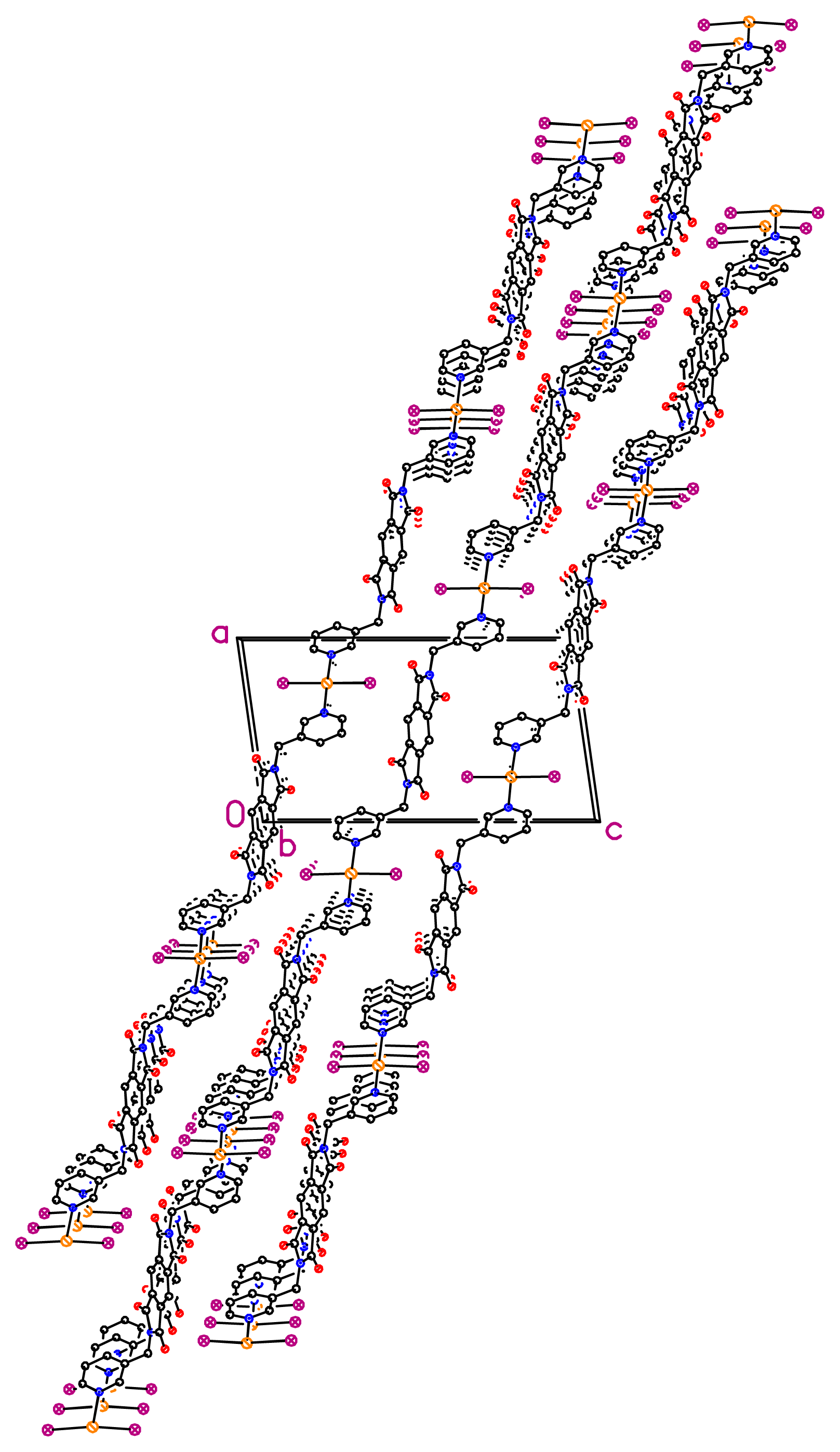
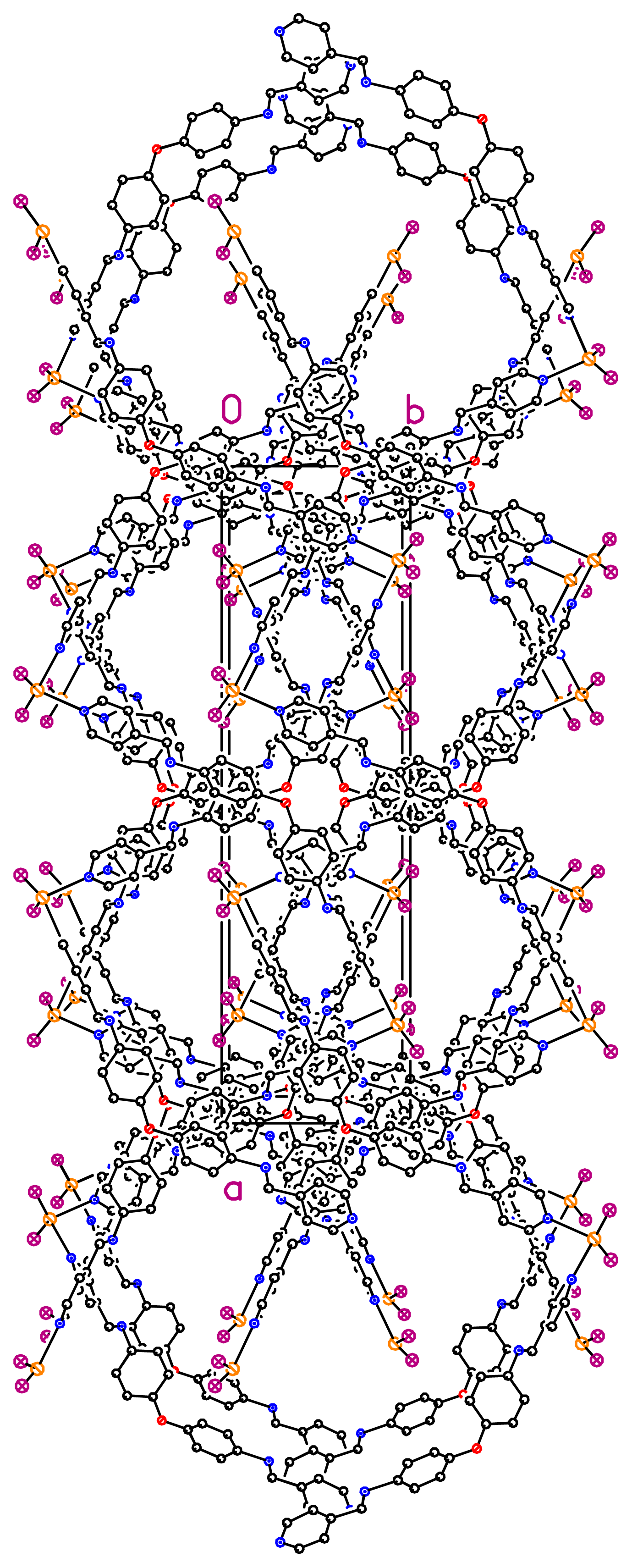
36]. When taking into account the lateral interconnection between double helices, the overall structure may be described as a 3-D coordination network (
Figure 4). Finally, it should be pointed out that related bis-monodentate Schiff bases L
3 and L
4 have been used to prepare interesting triple or double helices [
37,
38].
The ditopic ligand L2, contains a long rigid spacer of three fused rings and two freely-rotating pyridyl arms. It may take on either a cis-conformation to act as a ‘U’ type ligand or a transconformation to act as a ‘Z’ type ligand. Reaction of L2 with HgI2 in a DMF–MeOH mixture resulted in the complex 2, [HgI2(L2)·0.4CH3OH]∞, where single crystals suitable for X-ray diffraction grew on the wall of the tube containing the reaction mixture of L2 and HgI2.
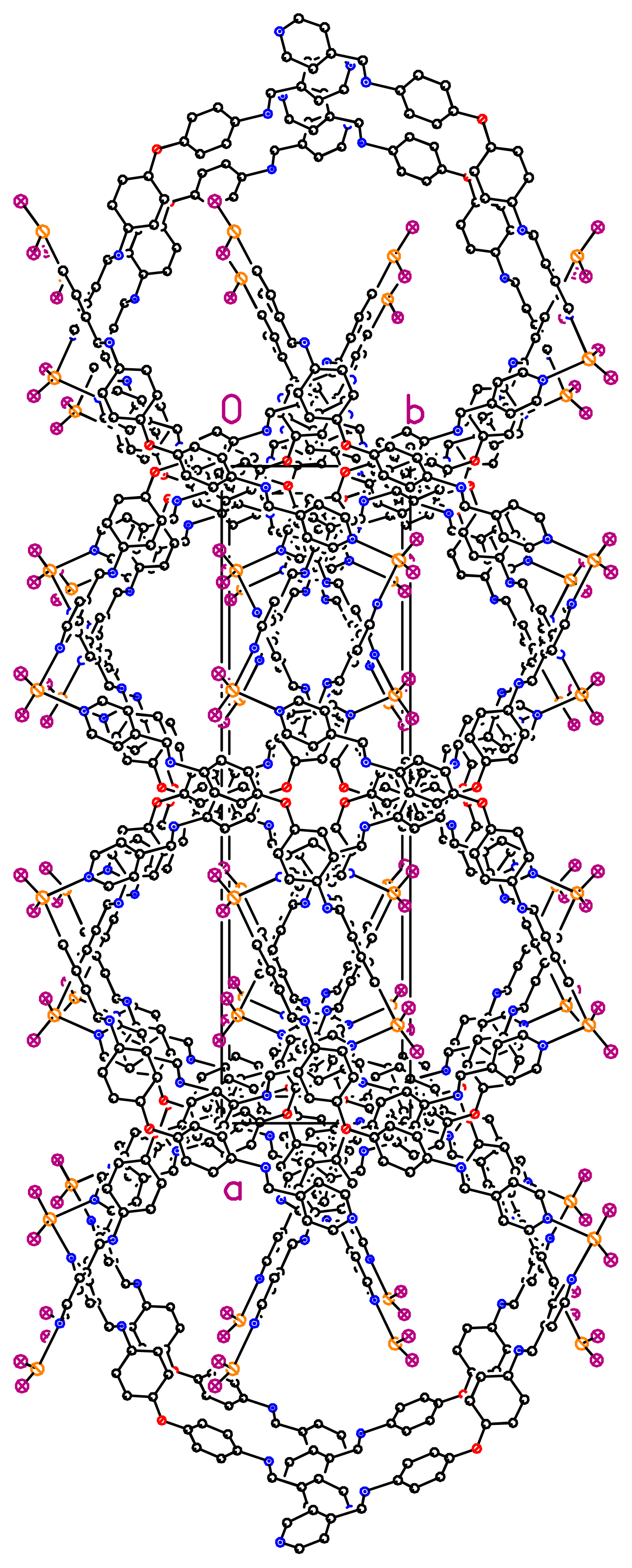
As shown in
Figure 5, each ligand coordinates to two Hg(II) ions and each Hg(II) ion is coordinated by two L
2 to generate the 1D zigzag chains, which are parallel with each other. All the 1D chains are stacked parallel along the
a axis (
Figure 6), and they are separated by 6.226 Å along the
b axis and 6.542 Å along the
c axis; these values are equal to the unit cell parameter
b and one third of the length of the unit cell parameter
c, respectively. The nearest inter-chain Hg···Hg separation, 10.951 Å, is almost the same as the unit cell parameter
a, whereas the nearest Hg···Hg separation in each zigzag chain is 18.013 Å. The Hg(II) is in a distorted tetrahedral geometry with its coordination sphere completed by two I– anions. The two Hg–I distances (
dHg–I = 2.640 Å) are virtually equivalent, as are the two Hg– N bond distances (
dHg–N = 2.426 Å). The N–Hg–N angle, which is 89.27°, is dramatically smaller than the I–Hg–I angle, which is 142.64°, and IHgN angle varies between 98.29° and 108.16°. The two ligands coordinated to the same Hg(II) ion are crystallographically independent, and each possesses a crystallographic inversion center that is situated in the middle of the central benzene ring. As a result, both adopt a distorted ‘Z’ conformation, where the two pyridyl units of L
2 are parallel with each other.
Unlike related semirigid ditopic ‘Z’ type ligand L
5, which can self-assemble with HgI
2 and afford the zigzag chains that interweave into an interesting clothlike 2D network in a ‘two-over/two-under’ (2O/2U) fashion [
39], L
2 can self-assemble with HgI
2 and generate the 1D zigzag chains. The result may be related to their symmetry and flexibility: L
2 has lower symmetry and more rigidity than L
5.
In L
1, the central ether oxygen atom can introduce enhanced flexibility into the ligand backbone, as suggested by Hannon
et al.[
37], and this enhanced flexibility permits the ligand to support helical chain arrays, thus
1 forms an interestingly infinite cross-linked double helical structure, whereas in L
2, the central benzene ring may increase the rigidity, thus
2 forms the one-dimensional zigzag chains.
In conclusion, by using bis-monodentate ligands, the formation of two coordination polymers was demonstrated. The one forms an interestingly infinite cross-linked double helical structure through weak hydrogen bonding (C–H···I), whereas another forms the one-dimensional zigzag chains, which are parallel with each other. The results may be related to the flexibility of ligands.
3. Experimental Section
3.1. Synthesis of the Complex
Preparation of [HgI2(L1)·0.5H2O]∞(1): A solution of L1 (12 mg) in 3 mL DMF was slowly mixed with a solution of HgI2 (12 mg) in 10 mL MeOH. The resulting mixture was left standing for several days to give a colorless crystalline product. Yield: 60%. 1H NMR (ppm, DMSO-d6): 8.731 (s, CH=N), 7.865(d, Hpy), 7.432 (d, Hph), 7.120 (d, Hph). IR (cm−1, KBr): 3423w, 3034w, 2887w, 1624w, 1605m, 1581m, 1492s, 1417m, 1367w, 1321w, 1282w, 1243s, 1207w, 1157m, 1058m, 1008m, 872w, 837m, 817w, 714w, 544m. Preparation of [HgI2(L2)·0.4CH3OH]∞(2): A solution of L2 (12 mg) in 3 mL DMF was slowly mixed with a solution of HgI2 (12 mg) in 10 mL MeOH. The resulting mixture was left standing for several days to give a colorless crystalline product. Yield: 60%. 1H NMR (ppm, DMSOd6): 8.617 (s, Hpy), 8.502 (d, Hpy), 8.259 (s, Hph), 7.956 (s, Hph), 7.791 (d, Hpy), 7.402 (m, Hpy), 4.878 (s, CH2). IR (cm−1, KBr): 3468w, 3033w, 1771m, 1714s, 1672m, 1391s, 1347m, 1313m, 1194w, 1157w, 1093m, 931w, 796w, 733m, 704w, 558w.
3.2. X-ray Crystallographic Analysis
Crystal data for [HgI
2(L
1)·0.5H
2O]
∞(1): HgI
2C
24H
19N
4O
1.5,
M = 841.81, orthorhombic,
a = 32.096(6),
b = 9.2588(19),
c = 17.656(4) Å,
U = 5246.8(18) Å
3,
T = 293(2) K, space group
Pcca,
Z = 8,
Dc = 2.126 gcm
−3,
μ (Mo Kα) = 8.245 mm
−1, crystal size 0.51 × 0.27 × 0.23 mm. Final refinement statistics:
R1 = 0.0269,
wR2 = 0.0627, and GOF = 1.003 for 3938 reflections with
I > 2σ (I). Crystal data for [HgI
2(L
2)·0.4CH
3OH]
∞(2): HgI
2C
22.4H
15.6N
4O
4.4,
M = 865.58, Monoclinic,
a = 10.812(2),
b = 6.2260(12),
c = 19.627(4) Å, β = 98.11(3)°,
U = 1308.1(4) Å
3,
T = 293(2) K, space group
P2/n,
Z = 2,
Dc = 2.198 gcm
−3,
μ (Mo Kα) = 8.279 mm
−1, crystal size 0.31 × 0.15 × 0.10 mm. Final refinement statistics:
R1 = 0.0428,
wR2 = 0.1071, and GOF = 1.037 for 2998 reflections with
I > 2σ(I).The diffraction data were collected on a Rigaku RAXISRAPID automated diffractometer at room temperature using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structure was solved by direct methods and successive difference maps (SHELXS 97) [
40] and refined by full-matrix least squares on
F2 using all uniqe data (SHELXL 97) [
41]. CCDC-270022 – CCDC-270023 the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK; Fax: (+44) 1223-336033; or
[email protected].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}