Introduction
The electric dipole moment
pe of a molecule is a quantity of fundamental importance in structural chemistry. When a molecule is subject to an external electric field
E, the molecular charge density may rearrange and hence the dipole moment may change [
1]. This change can be described by the tensor equation (1):
Here
α is a second rank tensor property called the dipole polarizability, and
β is the first of an infinite series of dipole hyperpolarizabilities and
pe,0, the permanent electric dipole moment, is the electric dipole moment in the absence of a field. Because the electric dipole moment may change when an external field is applied, the molecular potential energy U may also change according to the tensor equation (2):
Hyperpolarizabilities are known to be small in magnitude, and their effect is minimal for weak electric fields. They are however important quantities when the electric field is large, and molecules that exhibit large β values are of current interest because of their applications in electro optical devices [
2,
3] and in the general field of non-linear optics (NLO). Many such molecules can be represented by the idealized structure shown in
Figure 1.
Figure 1.
Schematic NLO conjugated molecule
Figure 1.
Schematic NLO conjugated molecule
It is thought that better materials can be made by modifying the strength of the activating group, the size and complexity of the conjugated system and the strength of the deactivating group. It is also thought that the presence of an activating group together with a deactivating group in a suitable ring position can enhance some electronic properties. The well-known phrase ‘push-pull mechanism’ is used to describe such phenomena.
A large number of such push-pull systems have been studied in recent years. Azoarene and stilbene derivatives are known to exhibit large nonlinear optical properties, which imply that they have large hyperpolarizabilities. There have been a number of theoretical studies of these quantities [
4,
5,
6]. The aim of this paper is to report a theoretical study of the dipole polarizabilities for the three series of molecules shown in
Figure 2. X and Y are a selection of simple activating and deactivating groups, as discussed below.
Figure 2.
Molecules studied.
Figure 2.
Molecules studied.
Choice of Ab Initio level of theory.
The experimental determination of a molecular polarizability is far from straightforward, especially if the molecule has little or no symmetry. The principal routes are studies of refractive index and relative permittivities, through Rayleigh and Raman scattering and through the quadratic Stark effect. These have been well reviewed elsewhere [
7], but a few words of background are in order.
For a molecule with symmetry, the principal axes of the polarizability tensor correspond to the symmetry axes; for molecules such as those discussed here workers often refer to the principal axes as
L (long),
M (medium) and
N (normal to the molecular plane) and so the principal values of the tensor are written α
LL, α
MM and α
NN . The mean value
(which is invariant to rotation of coordinate axes) can be determined from the refractive index
n of a gas of non-interacting particles according to the equation
where p is the pressure, k
B the Boltzmann constant, T the thermodynamic temperature and ∈
0 the permittivity of free space [
1].
In a condensed phase, the problem is more complicated because the separation between molecules is of the order of molecular dimensions and their interactions can no longer be ignored. As a result both the external field and the field due to the surrounding molecules polarize each molecule. The Lorenz-Lorentz equation:
applies to non-polar molecules in condensed phases and it can be derived from a detailed consideration of these ideas [
1]. Here, N is the number of molecules in volume V. In the case of molecules with a permanent dipole moment, it is necessary to take account of the orientation polarization. The Debye equation:
permits polarizabilities and dipole moments to be determined from measurements of the relative permittivity ∈
r and the density ρ as a function of temperature.
The anisotropy κ, usually defined as:
gives a measure of deviations from spherical symmetry since it would be zero for a spherically symmetric charge distribution.
An alternative route to polarizability is direct calculation, and there is a large literature on this topic [
8]. The general idea is to find a level of theory that gives highly accurate values for the polarizability tensor, but naturally this aim has to moderated with the cost of such calculations.
The principles involved in
Ab Initio polarizability calculations are well understood and can be illustrated by our results for azoarene (Series III with X = Y = H), shown in
Table 1.
Table 1.
Ab Initio calculations on azoarene.
Table 1.
Ab Initio calculations on azoarene.
| Level of Theory | αLL/au | αMM/au | αNN/au | <α>/au | κ |
|---|
| HF/6-311G(2d,1p) | 244.52 | 140.57 | 62.25 | 149.31 | 0.124 |
| HF/6-311++G(2d,1p) | 252.42 | 147.57 | 83.89 | 161.29 | 0.093 |
| B3LYP/6-311G(2d,1p) | 300.78 | 148.22 | 62.85 | 170.62 | 0.166 |
| B3LYP/6-311++G(2d,1p) | 311.19 | 156.10 | 84.35 | 183.88 | 0.133 |
| B3LYP/6-311++G(3d,2p) | 314.44 | 158.00 | 86.75 | 186.40 | 0.130 |
| B3LYP/6-311++G(2df,pd) | 311.02 | 155.95 | 84.39 | 183.79 | 0.132 |
| ‘Standard’ | 311.25 | 155.93 | 84.29 | 183.82 | 0.133 |
The atomic unit (au) of dipole polarizability is e
2 a
02 E
h-1 so that 1 atomic unit = 1.6488 × 10
-41 C
2 m
2 J
-1. The calculations here refer to an isolated gas-phase molecule at 0 K. Naturally, such a molecule has zero-point energy and there is a small but finite contribution to the polarizability from this effect [
8]. No attempt was made to estimate the vibrational contributions in this paper.
All calculations were done using Gaussian 03W [
9], with standard basis sets, integration points, cutoffs, etc. With the exception of the final row labelled ‘Standard’, all calculations were begun by optimizing the molecular geometry at the level of theory specified; polarizabilities were then calculated at the same level of theory using the standard Gaussian 03W keyword ‘Polar’. This keyword means that the polarizabilities were obtained analytically rather than by numerical differentiation.
The true experimental value for an isolated molecule of azoarene in the gas phase is unknown, but the
Table 1 entries suggest values of about 186 au for <α>, and 0.13 for the anisotropy. The first
Table 1 entry shows why HF-LCAO calculations with standard basis sets are no longer thought adequate for accurate polarizability calculations; the mean value of 149.31 is poor and the perpendicular component is particularly badly represented. Addition of diffuse functions (second entry) gives a pleasing increase in the perpendicular component, but the mean value is still poor.
The third entry is the KS-LCAO equivalent to the first entry, and illustrates the importance of electron correlation for these calculations. The fourth entry shows once again the importance of diffuse functions. The fifth and sixth entries suggest that there is no real advantage to be gained by trying to improve the polarization functions beyond the usual (2d,1p) set.
The rows of
Table 1 are arranged in order of computer resource (measured as time taken to run the calculation using Gaussian03 W on a standard AMD Athlon XP2600+ PC). On detailed comparison, it turns out that the geometries predicted by the very sophisticated basis sets including ++ and extended polarization functions are almost identical to those given by B3LYP/6-311G(2d,1p), so a compromise was chosen in order to save computer resources.
Ab Initio geometries reported from now on are at the B3LYP/6-311G(2d,1p) optimized level of theory, whilst the dipole polarizabilities are at the B3LYP/6-311++G(2d,1p) level of theory but with the same geometry. This is the meaning of the term ‘Standard’ in
Table 1. In a commonly used notation [
9], the polarizabilities are therefore calculated at the B3LYP/6-311G(2d,1p)// B3LYP/6-311++G(2d,1p) level of theory. The anisotropy κ does not appear to be a very sensitive descriptor, and will not be mentioned again.
Calculations on monosubstituted benzenes
The first step is to consider the monosubstituted benzenes, which correspond to Series I with X = H. We will report only the mean polarizabilities for this series, but draw attention to any interesting behaviour in the tensor components.
Table 2 shows various data for the molecules studied.
Table 2.
Various quantities for Series I with X = H
Table 2.
Various quantities for Series I with X = H
| Y | Ab Initio <α>/ au | Increment/ au | Volume/ Å3 | Miller/ Å3 | AM1 <α> /au |
|---|
| H | 67.307 | 0.00 | 332.58 | 10.43 | 48.83 |
| OH | 72.800 | 5.49 | 353.40 | 11.07 | 53.27 |
| NH2 | 79.425 | 12.12 | 367.69 | 11.78 | 61.99 |
| CHO | 84.772 | 17.47 | 386.41 | 12.36 | 63.43 |
| CN | 84.923 | 17.62 | 389.57 | 12.29 | 64.31 |
| OCH3 | 85.665 | 18.36 | 407.96 | 12.91 | 63.80 |
| NO2 | 85.676 | 18.37 | 393.30 | 12.15 | 66.05 |
The second column of
Table 2 shows the mean polarizability, and the third column is the polarizability difference on substitution of Y into benzene. These values give us an ’increment’ for use when discussing possible additivities in the polarizability mean values for later series of molecules. The table values are arranged in order of increasing increment. The largest increment is seen to be due to NO
2, and the two smallest increments are due to OH and NH
2.
According to a recent undergraduate organic chemistry textbook [
10], the order of our chosen groups from highest activating to highest deactivating is as follows: NH
2 > OH > OCH
3 > (zero) -C(=O)H > CN > NO
2, with the point on the scale where groups are neither activating or deactivating denoted ‘zero’. There is no particular reason why our increments should correlate exactly with Loudon’s scheme; the latter is to do with the chemical reactivity of a given substituted molecule C
6H
5X compared to benzene, ours to do with the response of the ground state molecule C
6H
5X to an applied electric field. We will see later that the scheme gives a good indication of increments for disubstituted species.
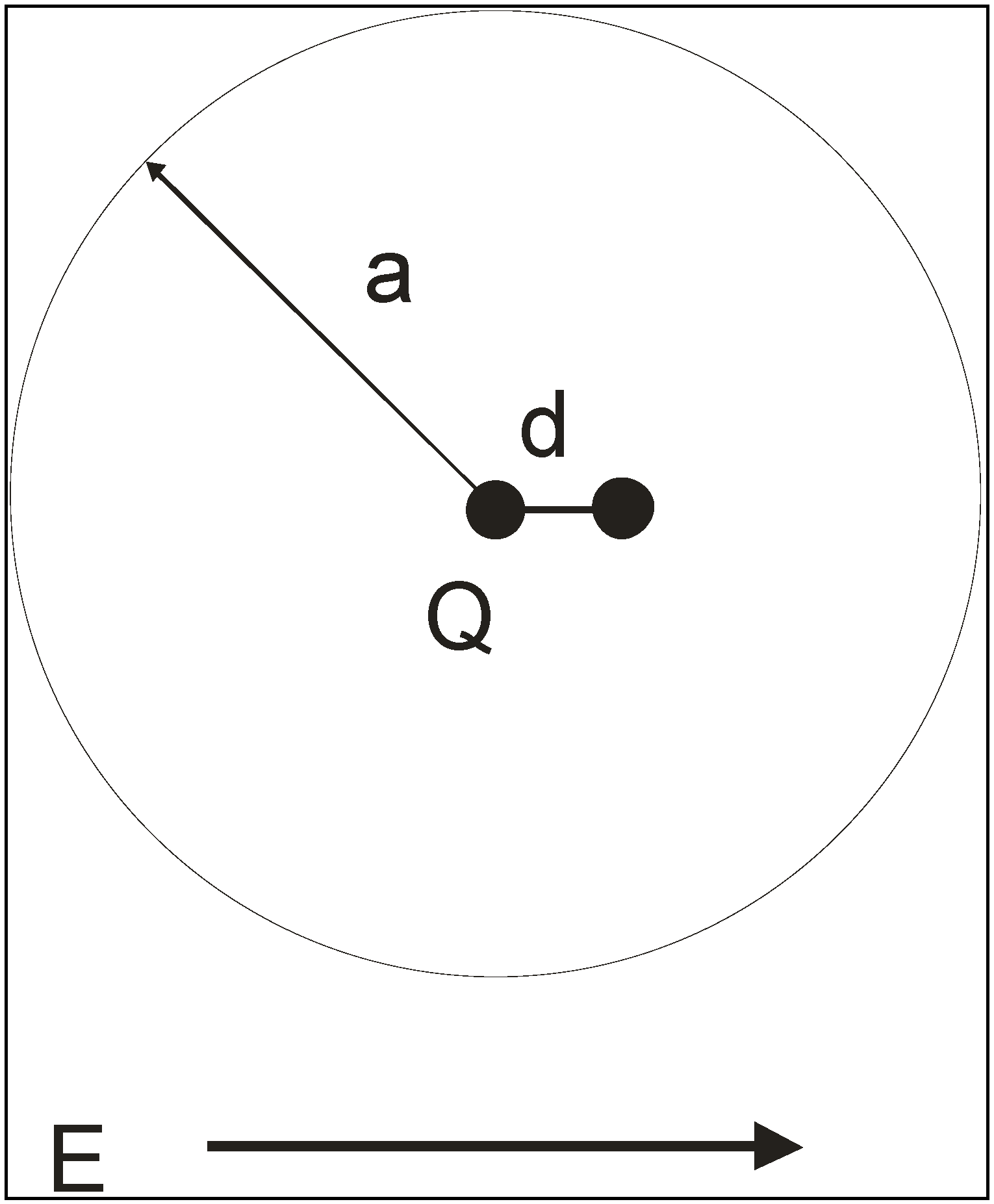
Figure 3.
Atom in an external electric field
Figure 3.
Atom in an external electric field
Dipole polarizabilities are often used in QSAR studies, where the aim is to give a reliable but quick estimate of <α>, as part of the process of high-throughput screening.
Ab Initio polarizability calculations are prohibitively expensive in a QSAR context, even for such simple molecules. One therefore looks to less rigorous but reliable procedures. Consider therefore a typical neutral atom modelled as the sphere of charge shown in
Figure 3. The radius is
a and the nuclear charge is
Q. We switch on the electric field
E which displaces the nucleus by a relative distance
d from the original atomic centre. At this point there is a force on the nucleus
QE due to the applied field and one due to the electron density. According to Gauss’ electrostatic theorem [
1], the latter force is
At equilibrium these two forces must be equal and so the displacement
d satisfies
The induced electric dipole moment is Qd and the polarizability is Qd / E hence
The derivation can be easily extended to a closed volume of arbitrary shape, not necessarily a sphere, and apart from the factor 4πε0 the polarizability of an atom is determined in this model by its volume. For this reason, workers in the field speak about ‘polarizability volumes’ and quote their results in volume units.
Molecular volumes are routinely determined in QSAR studies, and typical values are shown in column 4. The numerical conversion factor between Å
3 and atomic units of polarizability is
and so the prediction from this simple model would be <α> = 6.748 / × 332.58 au = 2244 au for benzene. Whilst the quantitative agreement with experiment is clearly nonexistent, our molecular volumes do give a good least square fit to the
Ab Initio mean polarizability values, with a correlation coefficient of 0.94.
Rewriting equation (5) in terms of molar quantities defines the molar refractivity
Here M is the molar mass, N
A the Avogadro constant and ρ the density. It is an experimental fact that molar refractivities are additive properties at the molecular level, and a view has long prevailed that the molar refractivity of a molecule is a sum of the molar refractivities of the constituent parts (atoms/ groups). Extensive tables of additive atom and group molar refractivities are available [
11]. These tables have been extended to molecular polarizabilities with the compilations of Denbigh [
12] and others. Such values are thought to be unreliable for molecular polarizability calculations.
The definitive reference in this field appears to be that due to K. J. Miller [
13]. Miller pointed out the need to take account of the atomic environment in molecular calculations, and this is usually done by assigning parameters in which each atom is characterized by its state of atomic hybridization. Miller and Savchik [
14] proposed a functional form
where τ
A is an atomic hybrid component for each atom
A in a given state of hybridization. N is the total number of electrons. In fact, Miller and Savchik omitted the factor 4πε
0 and so most computer packages quote the results as polarizability volumes (typically Å
3). These are shown in column 5 of
Table 2. The Miller method gives a mean polarizability of 70.38 au for benzene, in much better agreement with the
Ab Initio value than the crude molecular volume. It is clear that polarization volumes are not to be interpreted as molecular volumes. A linear regression between the Miller and the
Ab Initio <α> values as shown in
Table 2 gives a regression coefficient of 0.93.
Finally we should consider the use of semiempirical HF-LCAO techniques such as CNDO/2, MINDO3, PM3 and AM1 for calculations of dipole polarizabilities. The final column of
Table 2 shows AM1 calculations of <α>. All geometries were optimized before calculation of <α>. There is a good least squares correlation between the AM1 and the
Ab Initio results, with a correlation coefficient of 0.96, but semiempirical calculations of the tensor α (rather than the mean value) tend to give extremely poor values for the normal component α
NN. For benzene we find
Calculations on disubstituted benzenes (Series I)
Table 3 records values of
Ab Initio calculations on Series I C
6H
4X with a further X substituted in the
para position (giving X-C
4H
4-X).
Table 3.
Various quantities for the X-C6H4-X series
Table 3.
Various quantities for the X-C6H4-X series
| X | Ab Initio <α> /au | Increment /au | Additive increments from Table 2 /au |
|---|
| H | 67.31 | 0.00 | 0.00 |
| OH | 77.81 | 10.50 | 10.99 |
| NH2 | 90.87 | 23.56 | 24.24 |
| CHO | 103.86 | 36.56 | 34.93 |
| CN | 105.40 | 38.10 | 35.23 |
| OCH3 | 106.19 | 38.88 | 36.72 |
| NO2 | 104.60 | 37.29 | 37.74 |
The second column gives the mean polarizability, the third column the difference between the disubstituted molecule and benzene, whilst the final column shows the sum of increments that would be expected for a purely additive model (with results taken from
Table 2). The values in columns 3 and 4 are little different, but there is no obvious trend. We now consider the selection of
para-substituted X-C
6H
4-Y molecules shown in
Table 4, where X and Y are activating/deactivating groups.
Table 4.
Various quantities for the X-C6H4-Y series
Table 4.
Various quantities for the X-C6H4-Y series
| X, Y | Ab Initio <α> /au | Increment /au | Additive increment from
Table 2. /au | Volume / Å3 | Miller / Å3 |
|---|
| H,H | 67.31 | 0.00 | 0.00 | 332.58 | 10.43 |
| OH, CN | 91.72 | 24.41 | 23.11 | 410.18 | 12.92 |
| NH2, CN | 99.82 | 32.52 | 29.73 | 424.43 | 13.64 |
| OH, CHO | 92.24 | 24.93 | 22.96 | 408.53 | 12.99 |
| NH2, CHO | 101.20 | 33.89 | 29.58 | 421.21 | 13.71 |
| OCH3, NO2 | 108.86 | 41.55 | 36.73 | 470.44 | 14.75 |
| OCH3, CN | 106.44 | 39.13 | 35.97 | 465.75 | 14.76 |
| OCH3, CHO | 107.25 | 39.94 | 35.82 | 464.6 | 14.83 |
| NH2, NO2 | 104.93 | 37.62 | 30.49 | 428.06 | 13.63 |
| OH, NO2 | 93.83 | 26.52 | 23.86 | 413.44 | 12.91 |
Again, column 2 shows the
Ab Initio mean polarizabilities, column 3 the difference between the molecule and benzene and column 4 the value that this increment would have in a purely additive model using the values given in
Table 2. In every case, disubstitution produces a larger enhancement of <α> than we would have expected from pure additivity of the two functional groups. This behaviour is consistent with the push-pull mechanism. The largest push-pull enhancement is for the pair X = NH
2, Y = NO
2 (7.14 au), which is entirely consistent with the order of activating/ deactivating groups mentioned above. For clarity, the enhancements are shown in
Table 5, arranged in order of increasing value.
Table 5.
Push-pull enhancements to <α>
Table 5.
Push-pull enhancements to <α>
| X,Y | Enhancement / au |
|---|
| H,H | 0 |
| OH, CN | 1.30 |
| OH, CHO | 1.97 |
| OH, NO2 | 2.66 |
| NH2, CN | 2.78 |
| OCH3, CN | 3.16 |
| OCH3, CHO | 4.12 |
| NH2, CHO | 4.30 |
| OCH3, NO2 | 4.83 |
| NH2, NO2 | 7.14 |
A linear regression between the molecular volumes and the Ab Initio mean polarizabilities gives a correlation coefficient of 0.93, with a corresponding correlation coefficient of 0.95. Although not shown here, AM1 calculations of the mean polarizability give values that correlate well with the Ab Initio values (with a correlation coefficient of 0.99), but the AM1 αNN absolute values are as usual extremely low.
Calculations on disubstituted stilbene and azoarene (Series II and III)
In
Table 6, we record the polarizability principal components for the 10 disubstituted stilbenes.
Table 6.
Principal values of α for the X-C6H4-CH=CH-C6H4-Y series
Table 6.
Principal values of α for the X-C6H4-CH=CH-C6H4-Y series
| X, Y | αLL /au | αMM /au | αNN. /au |
|---|
| H,H | 324.33 | 162.65 | 89.41 |
| OH, CN | 439.91 | 170.63 | 96.22 |
| NH2, CN | 487.65 | 173.53 | 100.14 |
| OH, CHO | 439.67 | 178.54 | 95.66 |
| NH2, CHO | 480.89 | 181.99 | 99.84 |
| OCH3, NO2 | 488.91 | 191.94 | 105.34 |
| OCH3, CN | 473.32 | 181.61 | 106.52 |
| OCH3, CHO | 474.03 | 189.23 | 106.00 |
| NH2, NO2 | 502.82 | 184.35 | 99.27 |
| OH, NO2 | 452.91 | 180.76 | 95.08 |
These are given primarily for reference, given the compute-intensive nature of the calculations and the sensitivity of the normal component to level of theory. In
Table 7, we record a number of quantities for discussion.
Table 7.
Various quantities for the X-C6H4-CH=CH-C6H4-Y series
Table 7.
Various quantities for the X-C6H4-CH=CH-C6H4-Y series
| X, Y | Ab Initio <α> /au | Increment /au | Additive increment from Table 2. /au | Volume / Å3 | Miller / Å3 |
|---|
| H,H | 192.13 | 0.00 | 0.00 | 633.02 | 23.57 |
| OH, CN | 235.59 | 43.46 | 23.11 | 710.36 | 26.06 |
| NH2, CN | 253.78 | 61.65 | 29.73 | 724.06 | 26.77 |
| OH, CHO | 237.95 | 45.83 | 22.96 | 710.34 | 26.13 |
| NH2, CHO | 254.24 | 62.11 | 29.58 | 721.72 | 26.84 |
| OCH3, NO2 | 262.06 | 69.93 | 36.73 | 770.34 | 27.88 |
| OCH3, CN | 253.82 | 61.69 | 35.97 | 767.00 | 27.90 |
| OCH3, CHO | 256.42 | 64.29 | 35.82 | 764.80 | 27.96 |
| NH2, NO2 | 262.15 | 70.02 | 30.49 | 726.86 | 26.76 |
| OH, NO2 | 242.92 | 50.79 | 23.86 | 713.67 | 26.05 |
Table 8 and
Table 9 are the corresponding Tables for the disubstituted azobenzenes.
Table 8.
Principal values of α for the X-C6H4-N=N-C6H4-Y series
Table 8.
Principal values of α for the X-C6H4-N=N-C6H4-Y series
| X, Y | αLL /au | αMM /au | αNN /au |
|---|
| H,H | 311.25 | 155.93 | 84.29 |
| OH, CN | 429.06 | 163.58 | 91.29 |
| NH2, CN | 481.94 | 166.36 | 95.07 |
| OH, CHO | 422.02 | 171.40 | 90.78 |
| NH2, CHO | 469.15 | 174.80 | 94.84 |
| OCH3, NO2 | 464.36 | 184.83 | 100.40 |
| OCH3, CN | 464.67 | 174.40 | 101.57 |
| OCH3, CHO | 458.10 | 181.92 | 101.06 |
| NH2, NO2 | 497.59 | 177.74 | 94.17 |
| OH, NO2 | 427.04 | 173.85 | 90.16 |
Table 9.
Various quantities for the X-C6H4-N=N-C6H4-Y series
Table 9.
Various quantities for the X-C6H4-N=N-C6H4-Y series
| X, Y | Ab Initio <α> /au | Increment /au | Additive increment from Table 2. /au | Volume / Å3 | Miller / Å3 |
|---|
| H,H | 183.82 | 0.00 | 0.00 | 618.29 | 22.15 |
| OH, CN | 227.98 | 44.16 | 23.11 | 698.05 | 24.64 |
| NH2, CN | 247.79 | 63.97 | 29.73 | 711.11 | 25.36 |
| OH, CHO | 228.07 | 44.24 | 22.96 | 694.77 | 24.71 |
| NH2, CHO | 246.26 | 62.44 | 29.58 | 708.53 | 25.43 |
| OCH3, NO2 | 249.86 | 66.04 | 36.73 | 757.75 | 26.47 |
| OCH3, CN | 246.88 | 63.06 | 35.97 | 754.4 | 26.48 |
| OCH3, CHO | 247.03 | 63.20 | 35.82 | 752.9 | 26.55 |
| NH2, NO2 | 256.50 | 72.68 | 30.49 | 714.32 | 25.35 |
| OH, NO2 | 230.35 | 46.53 | 23.86 | 702.9 | 24.63 |
Once again, we focus on the push-pull contribution to the mean polarizability, defined as the difference between column 2 and column 3, i.e., the differences between the substituted and unsubstituted molecules. For clarity, these are recorded in
Table 10, in order of increasing magnitude for the two series.
Table 10.
Push-pull enhancements to the mean polarizability.
Table 10.
Push-pull enhancements to the mean polarizability.
| Stilbene series | Azoarene series |
|---|
| Groups X, Y | Enhancement / au | Groups | Enhancement / au |
|---|
| H,H | 0 | H,H | 0 |
| OH,CN | 20.35 | OH,CN | 21.05 |
| OH,CHO | 22.87 | OH,CHO | 21.28 |
| OCH3,CN | 25.71 | OH,NO2 | 22.66 |
| OH,NO2 | 26.93 | OCH3,CN | 27.08 |
| OCH3,CHO | 28.47 | OCH3, CHO | 27.38 |
| NH2,CN | 31.91 | OCH3, NO2 | 29.31 |
| NH2,CHO | 32.53 | NH2,CHO | 32.86 |
| OCH3,NO2 | 33.21 | NH2,CN | 34.23 |
| NH2,NO2 | 39.53 | NH2,NO2 | 42.19 |
For both series, the smallest enhancement is due to the pair OH/ CN, and the largest enhancement due to the pair NH2/ NO2. The overall combinations vary between the two series, but the enhancements are consistent with the standard activating/ deactivating order mentioned above.
Finally we consider the likely reliability of various easily-computed indices such as the molecular volume, the Miller empirical volume polarizabilities and AM1 polarizabilities discussed above. Linear regressions were done for each of these quantities against the B3LYP mean polarizabilities <α>, and the regression coefficients
r are given in
Table 11.
Table 11.
Linear regression coefficients r for the Stilbene/ azoarene series II and III.
Table 11.
Linear regression coefficients r for the Stilbene/ azoarene series II and III.
| Correlation of <α>B3LYP with | Stilbene | Azoarene |
|---|
| Molecular volume | 0.80 | 0.77 |
| <α>Miller | 0.85 | 0.83 |
| <α>AM1 | 0.85 | 0.88 |
The correlation coefficients are all well below 0.95, which value if often taken to justify a straight line relationship. It therefore seems that none of the three simpler procedures gives a reliable estimate of <α> for these two latter series of molecules.




{kind=link}
{kind=link}
{kind=link}