Introduction
MDR or multidrug resistance is responsible for many forms of resistance in bacteria, fungi and human tumours [
1]. This resistance functions by the presence of membrane bound efflux pumps, which actively export therapeutics from the cell resulting in a low intracellular ineffective concentration of the drug [
2]. These pumps recognize a wide variety of structurally unrelated compounds [
3] and it is believed that MDR inhibitors bind directly to the hydrophobic region of the efflux pump thus preventing the drug transport [
4].
There has been much research conducted to find inhibitors of these proteins, particular in human tumour resistance as reviewed by Stouch and Gudmundsson [
5]. Some progress has been reported on the structure-activity relationships (SARs) for inhibitors of bacterial efflux pumps [
6], but further work is necessary to fully explain the mechanism of MDR efflux inhibition, since most potent inhibitors of the NorA MDR pump of
Staphylococcus aureus come from totally different chemical classes. This is unusual since it seems that there is no common pharmacophore that causes inhibition of MDR.
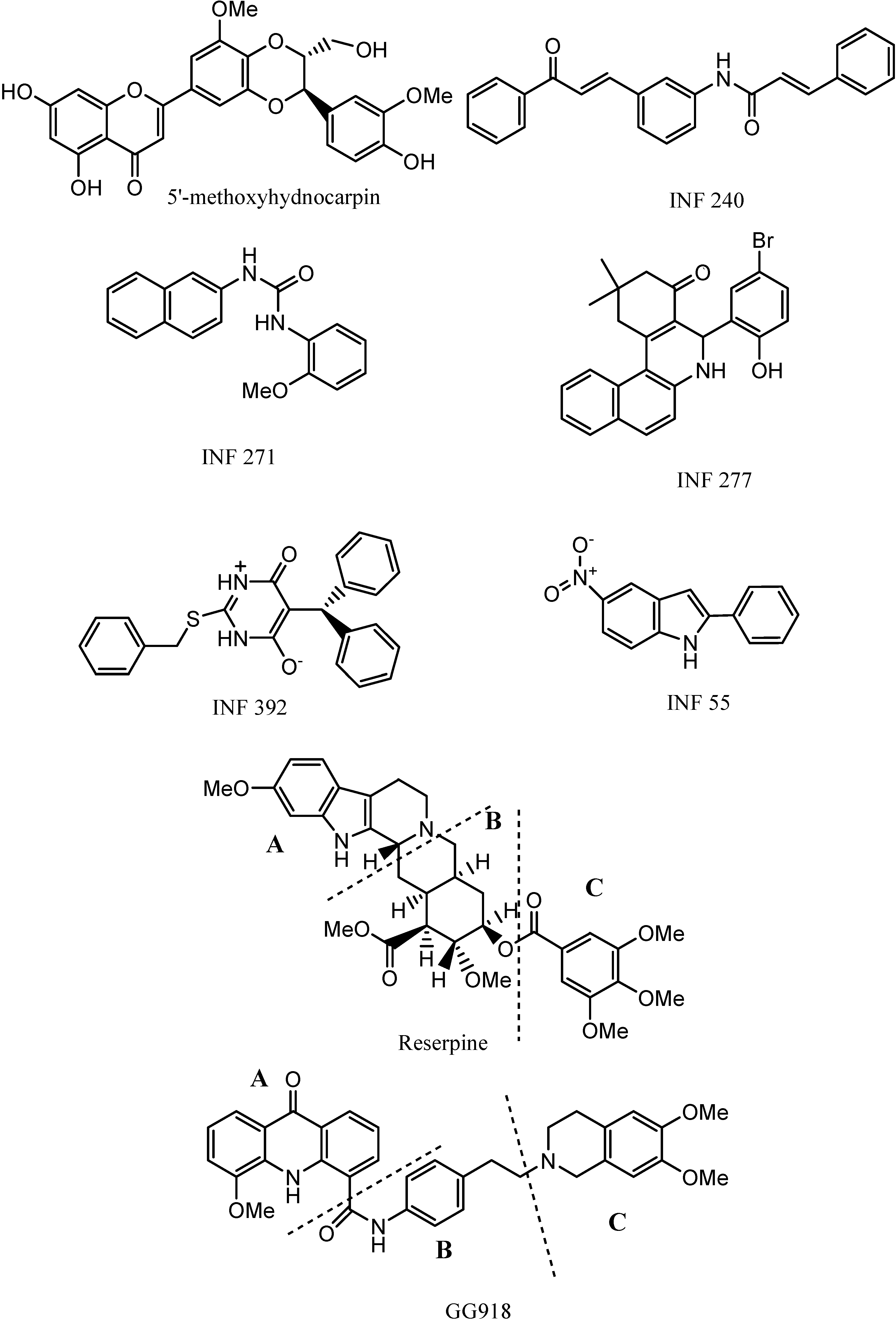
We have modelled and explored biomolecular similarities of a series of representative MDR inhibitors of the NorA pump from different classes (
Scheme 1) on the basis of molecular interaction potentials and report here some structural features required for MDR inhibition.
Results and Discussion
The efflux pump NorA plays an important role in resistance to fluoroquinolone antibiotics of the major human pathogen
Staphylococcus aureus, which is highly problematic in the clinical environment [
14]. The restoration of antibiotic efficacy could be achieved by using inhibitors, molecules that potentiate the activity of standard antibiotics against MDR cells. Efflux inhibitors are from a wide range of structural classes and representative molecules for different classes were studied here. The experimental results of MDR modulation for chosen inhibitors are shown in
Table 1. These results are taken from the literature [
15] or from one of the authors [
16,
17]. Compounds with no potential for MDR inhibition are shown in
Scheme 2. It is believed that inhibitors of these transport processes act by directly binding to hydrophobic regions of MDR proteins causing inhibition of antibiotic removal [
4]. Since a wide range of MDR inhibitors have been discovered and with no apparent pharmacophore detected, we have assumed that interactions important for MDR inhibition must be non-specific. The molecular electrostatic potential could be very important for the formation of a potential hydrogen bond network and other interactions between MDR inhibitor and efflux pump, and it could also play a significant role in molecular recognition.
In the absence of an explanation for the MDR inhibition mechanism, we have decided to evaluate the importance of molecular electrostatic potential in these processes. The
ab initio optimised geometries of selected inhibitors of the NorA efflux pump were compared using atom-centred point-charge distribution and results are presented in
Table 2 in the form of a similarity matrix. Reserpine and GG918 had to be split into three portions, and those are defined in the
Scheme 1.
Table 1.
MDR modulation results for NorA inhibitors (5’-MHC depicted 5’-methoxyhydnocarpin).
Table 1.
MDR modulation results for NorA inhibitors (5’-MHC depicted 5’-methoxyhydnocarpin).
| Inhibitor | Drug | Inhibition | Reference |
| INF 240 | Ciprofloxacin | 0.12* | Markham et al., 1999. [15] |
| INF 271 | Ciprofloxacin | 0.12* | Markham et al., 1999. [15] |
| INF 277 | Ciprofloxacin | 0.15* | Markham et al., 1999. [15] |
| INF 392 | Ciprofloxacin | 0.28* | Markham et al., 1999. [15] |
| INF 55 | Ciprofloxacin | 0.25* | Markham et al., 1999. [15] |
| 5’-MHC | Norfloxacin | 4** | Stermitz et al., 2000. [18] |
| Reserpine | Norfloxacin | 4** | Gibbons et al., 2003. [16] |
| GG918 | Norfloxacin | 8** | Gibbons et al., 2003a. [16] |
| Diterpene from Lycopus europaeus | Norfloxacin | ND | Gibbons et al., 2003b. [16] |
| Epigallocatechin-3-O-gallate | Norfloxacin | ND | Gibbons, unpublished data [2003] |
The similarity index for all pairs of inhibitors was between 0.644 and 0.932, depending on the size of the compared molecules. The results for the parts of reserpine and GG918 are to be taken with caution due to the relatively small sizes of examined moieties. Direct correlation between similarity and fold of modulation cannot be fully examined due to differences in the representation of experimental data, however, the following could be emphasised:
- a)
INF 271 and INF 277 have a similar potency of MDR inhibition and similarity index is also high – 0.817;
- b)
5'-methoxyhydnocarpin (5’-MHC) and INF 240 are potent MDR inhibitors and have a high similarity index.
Due to the difference in the size of molecules, a more detailed analysis was carried out by visually comparing inhibitors.
Table 2.
Final similarity matrix for MDR inhibitors of the NorA pump calculated by MIPSIM using point charges. Three parts of reserpine were denoted as Reserpine A, Reserpine B and Reserpine C, and three parts of GG918 were denoted as GG918 A, GG918 B, and GG918 C. Similarity between parts that belong to the same molecule were not considered.
Table 2.
Final similarity matrix for MDR inhibitors of the NorA pump calculated by MIPSIM using point charges. Three parts of reserpine were denoted as Reserpine A, Reserpine B and Reserpine C, and three parts of GG918 were denoted as GG918 A, GG918 B, and GG918 C. Similarity between parts that belong to the same molecule were not considered.
| | 5’-MHC | INF 240 | INF 271 | INF 277 | INF 392 | INF 55 |
| 5’-MHC | 1.000 | | | | | |
| INF 240 | 0.840 | 1.000 | | | | |
| INF 271 | 0.735 | 0.733 | 1.000 | | | |
| INF 277 | 0.704 | 0.673 | 0.817 | 1.000 | | |
| INF 392 | 0.740 | 0.751 | 0.781 | 0.722 | 1.000 | |
| INF 55 | 0.729 | 0.707 | 0.866 | 0.803 | 0.725 | 1.000 |
| Reserpine A | 0.690 | 0.735 | 0.870 | 0.754 | 0.719 | 0.932 |
| Reserpine B | 0.691 | 0.616 | 0.817 | 0.813 | 0.739 | 0.808 |
| Reserpine C | 0.699 | 0.649 | 0.717 | 0.695 | 0.677 | 0.737 |
| GG918 A | 0.669 | 0.713 | 0.818 | 0.755 | 0.707 | 0.915 |
| GG918 B | 0.644 | 0.703 | 0.759 | 0.699 | 0.670 | 0.839 |
| GG918 C | 0.716 | 0.692 | 0.795 | 0.722 | 0.646 | 0.824 |
| Diterpene from Lycopus europaeus | 0.681 | 0.566 | 0.763 | 0.807 | 0.722 | 0.720 |
| Epigallocatechin-3-O-gallate | 0.682 | 0.670 | 0.825 | 0.868 | 0.733 | 0.750 |
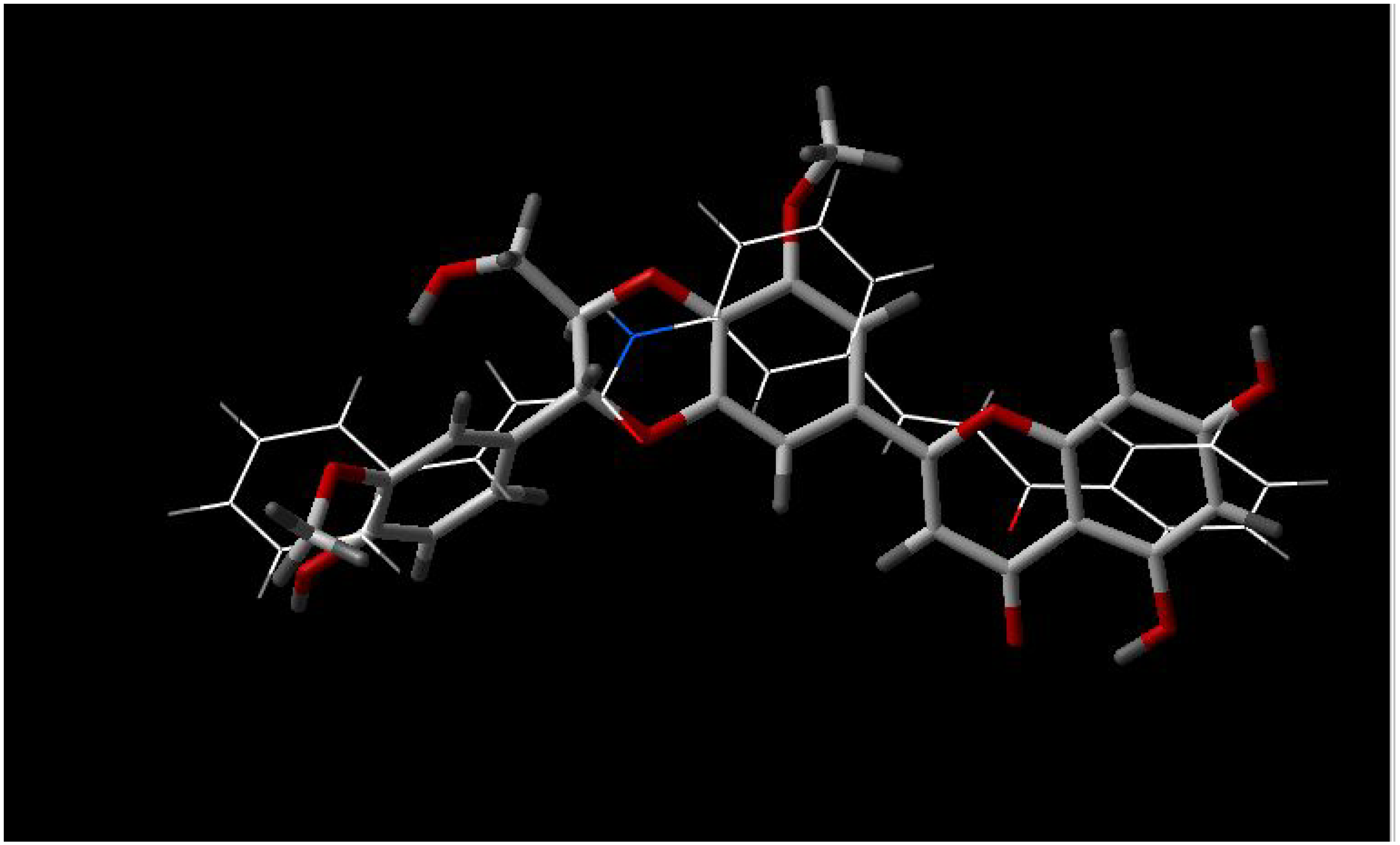
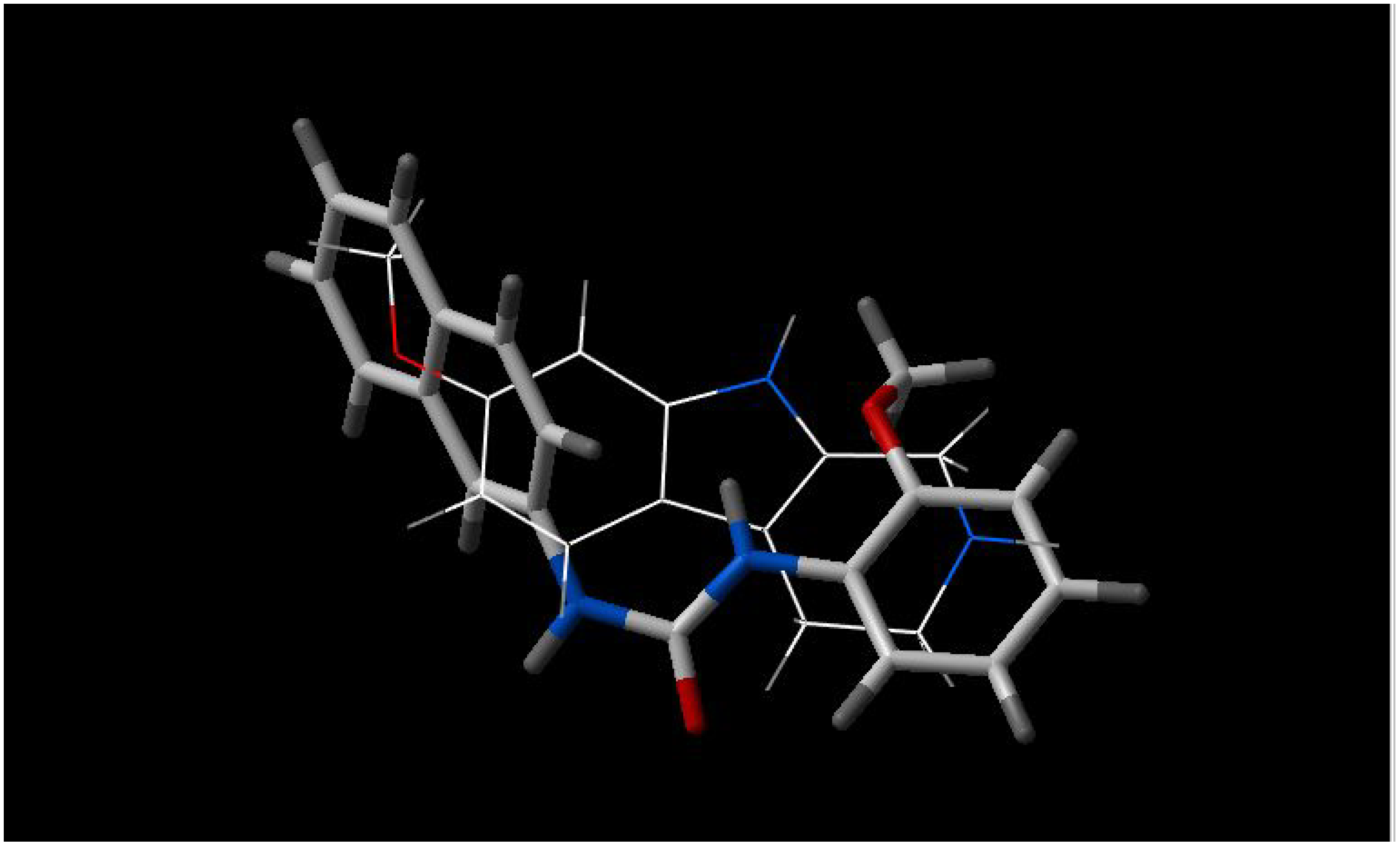
From the figures of the best fit between pairs of inhibitors, some observations are apparent:
- a)
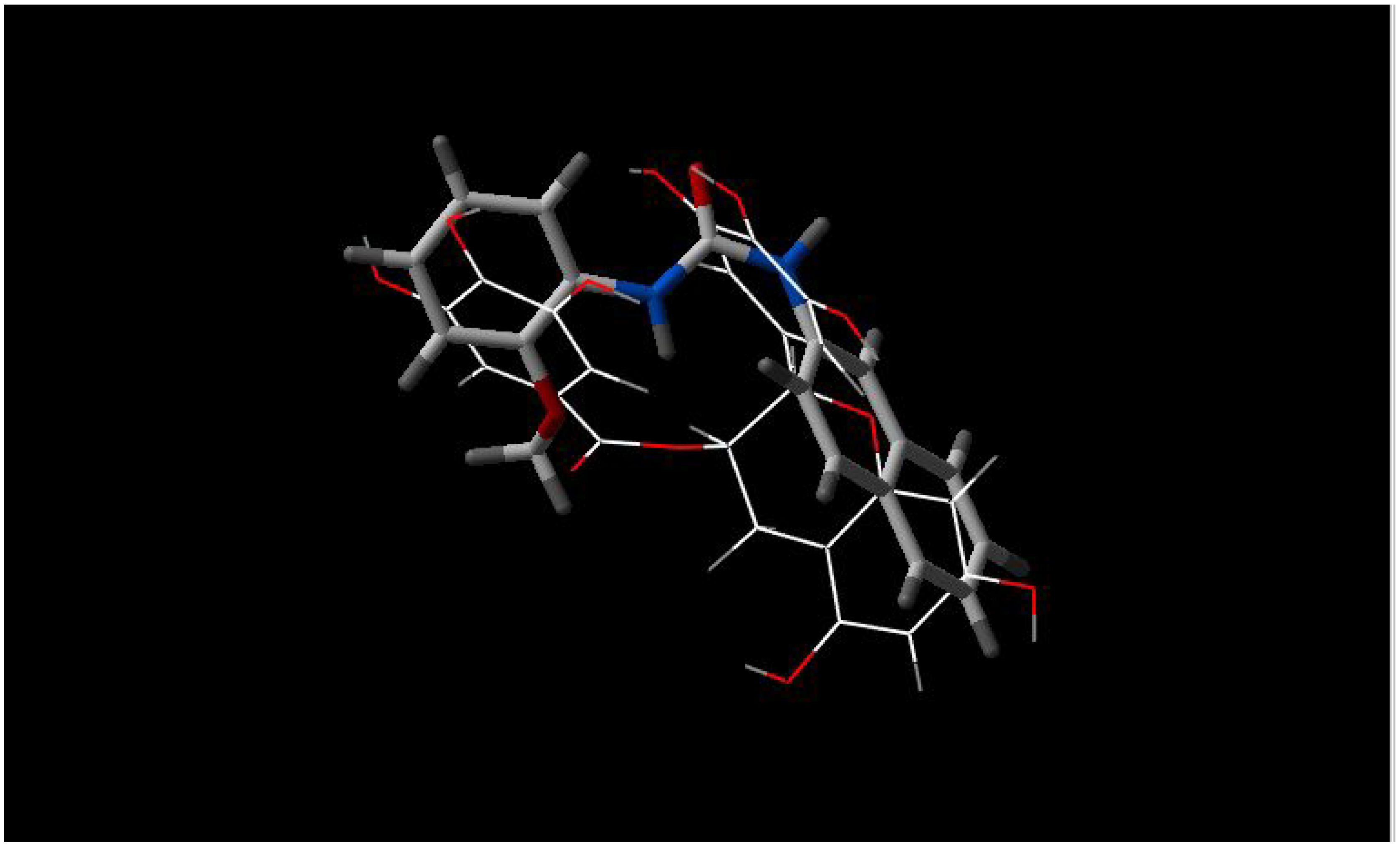
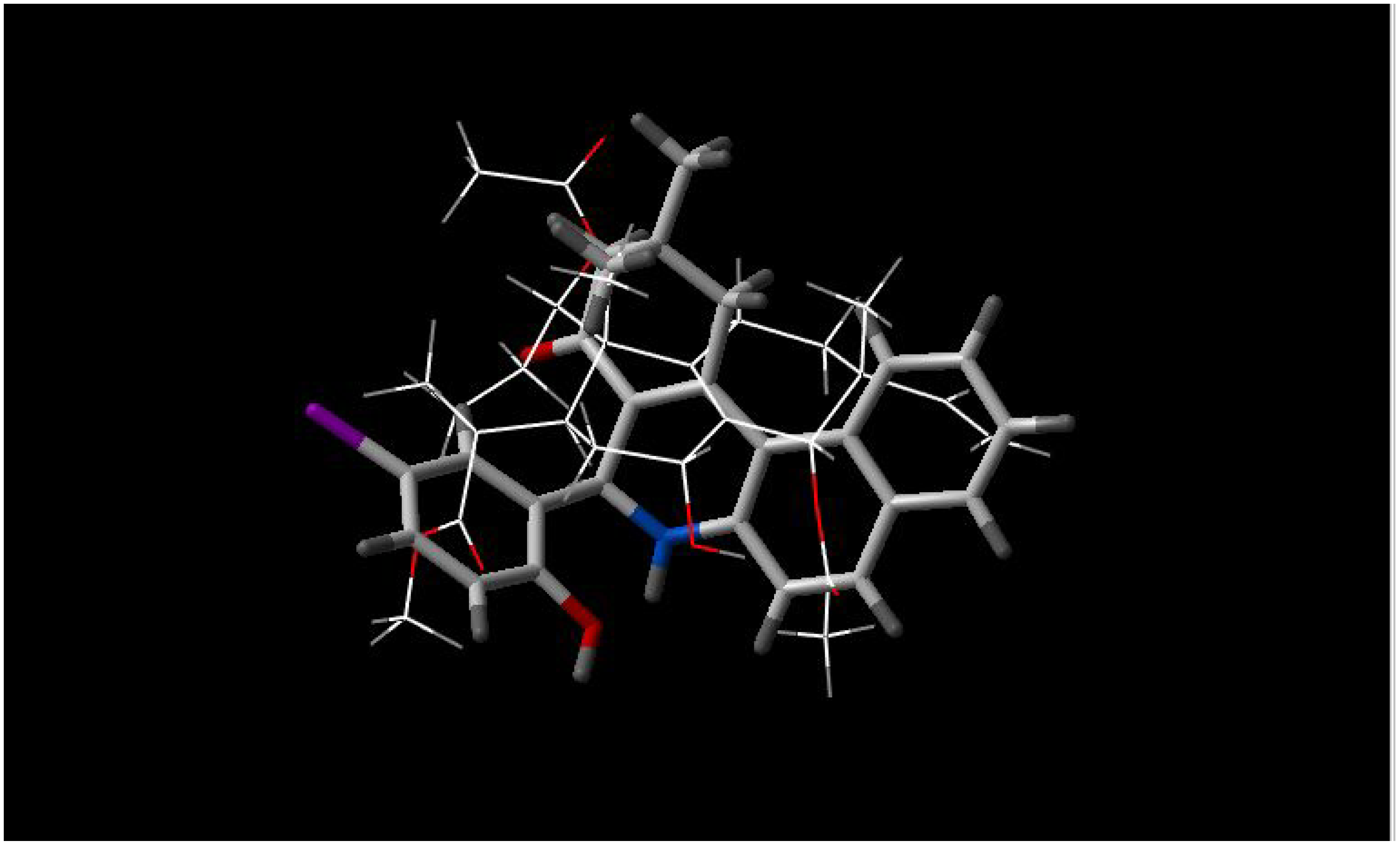
5'-methoxyhydnocarpin and INF 240 have the same shape and some polar groups in a similar position (
Figure 1). Note the absence of nitrogen atom in 5’-MHC;
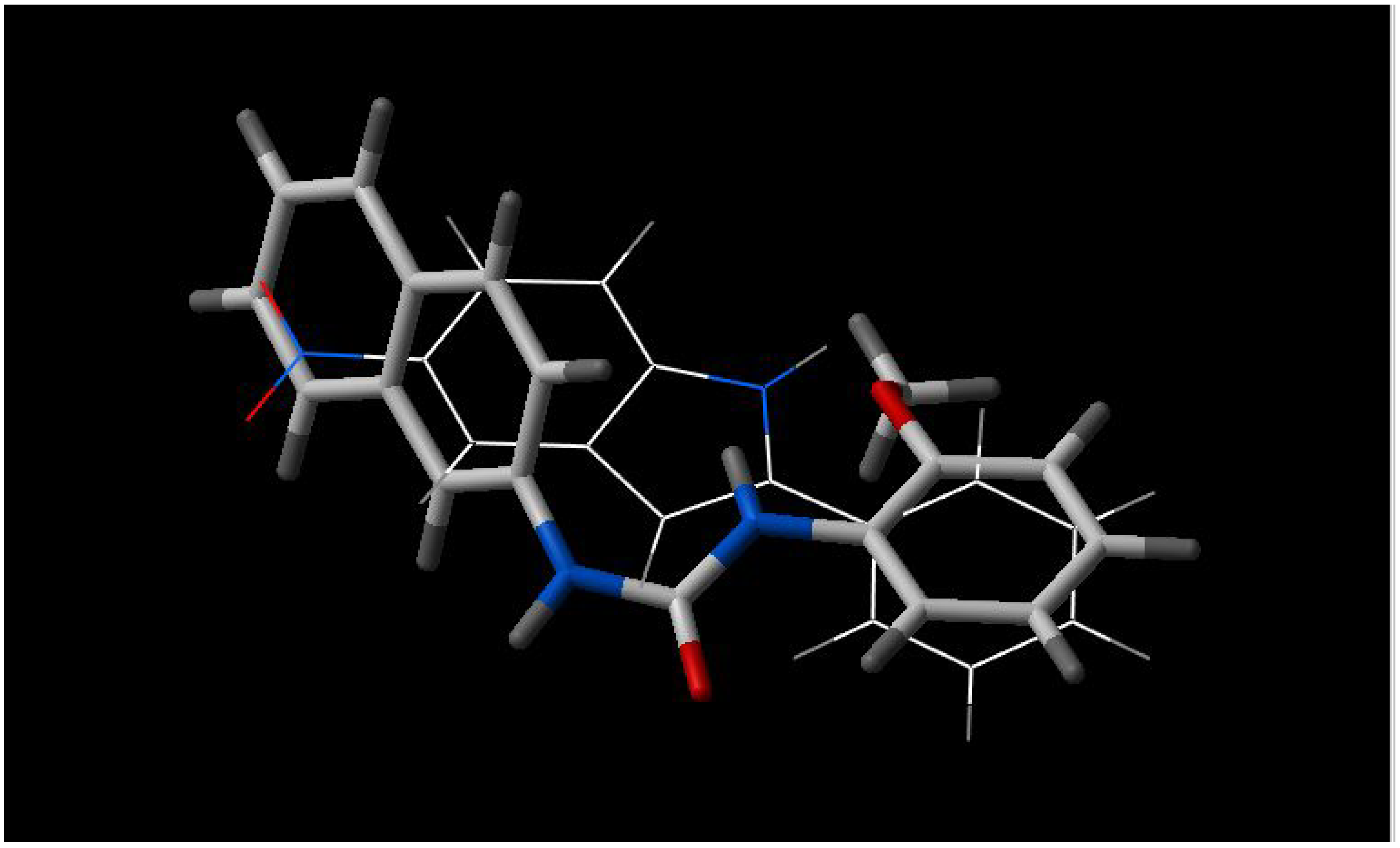
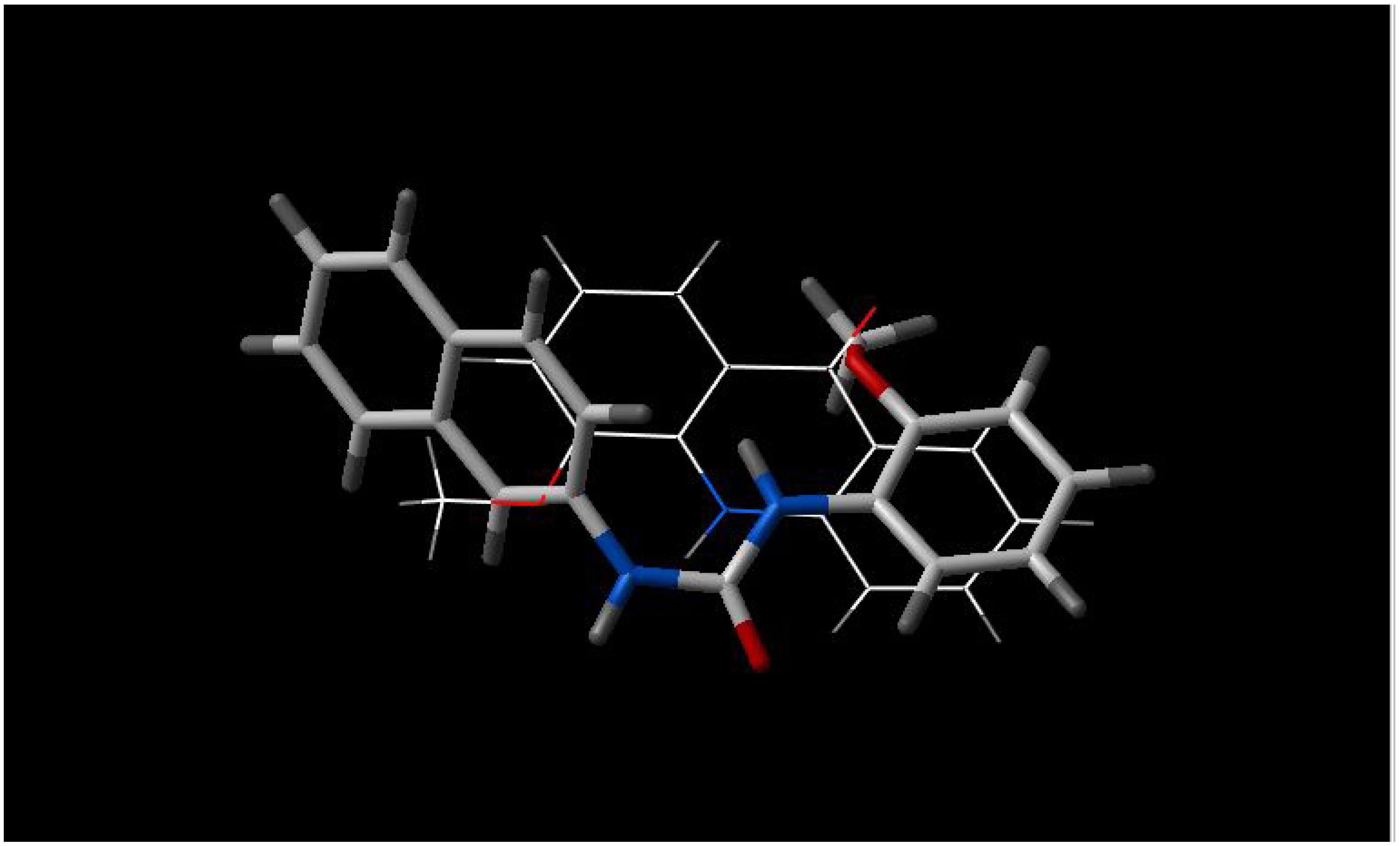
- b)
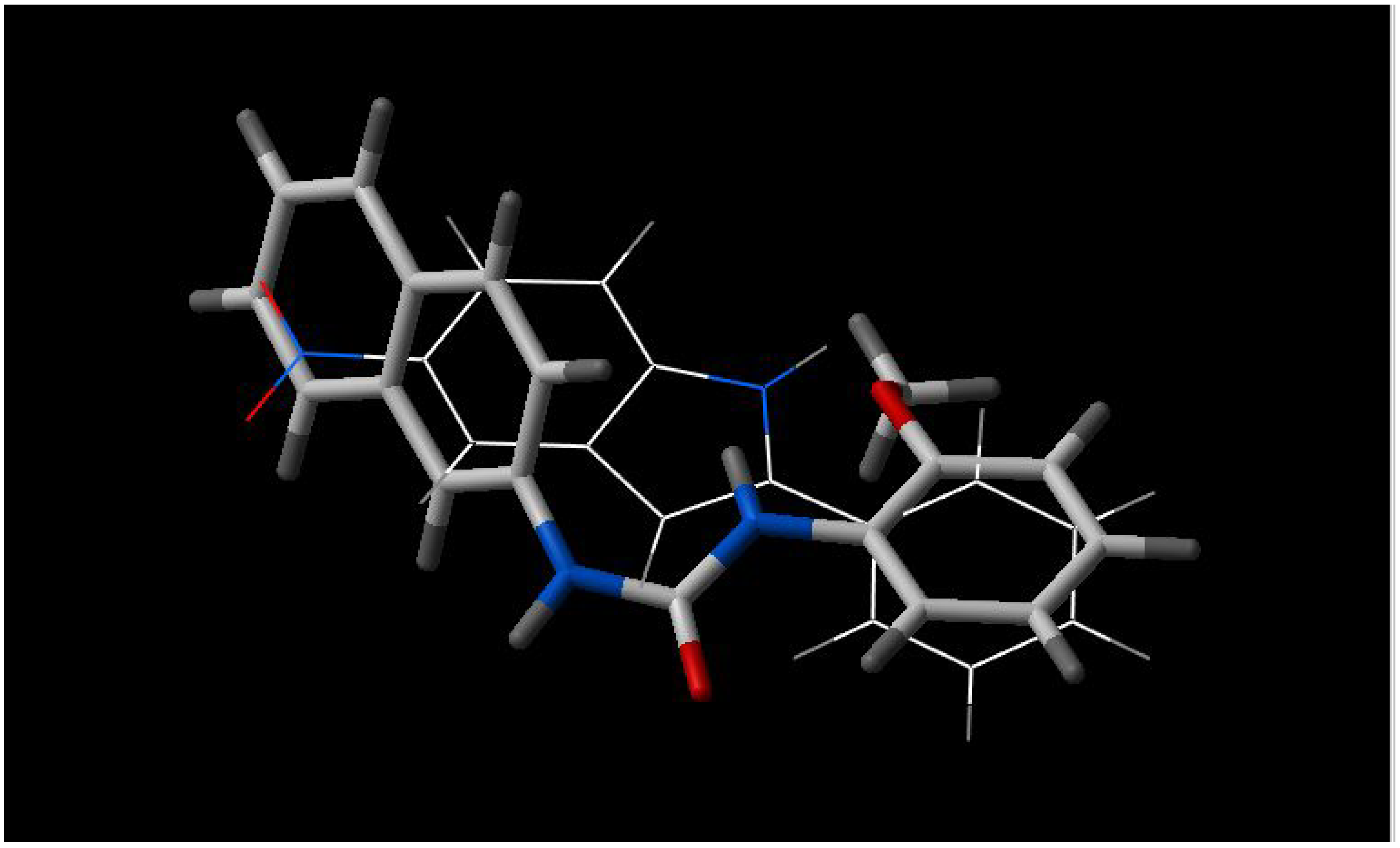
INF 271 and INF 277 have a similar shape and both have a nitrogen atom in the middle of the molecule (
Figure 2);
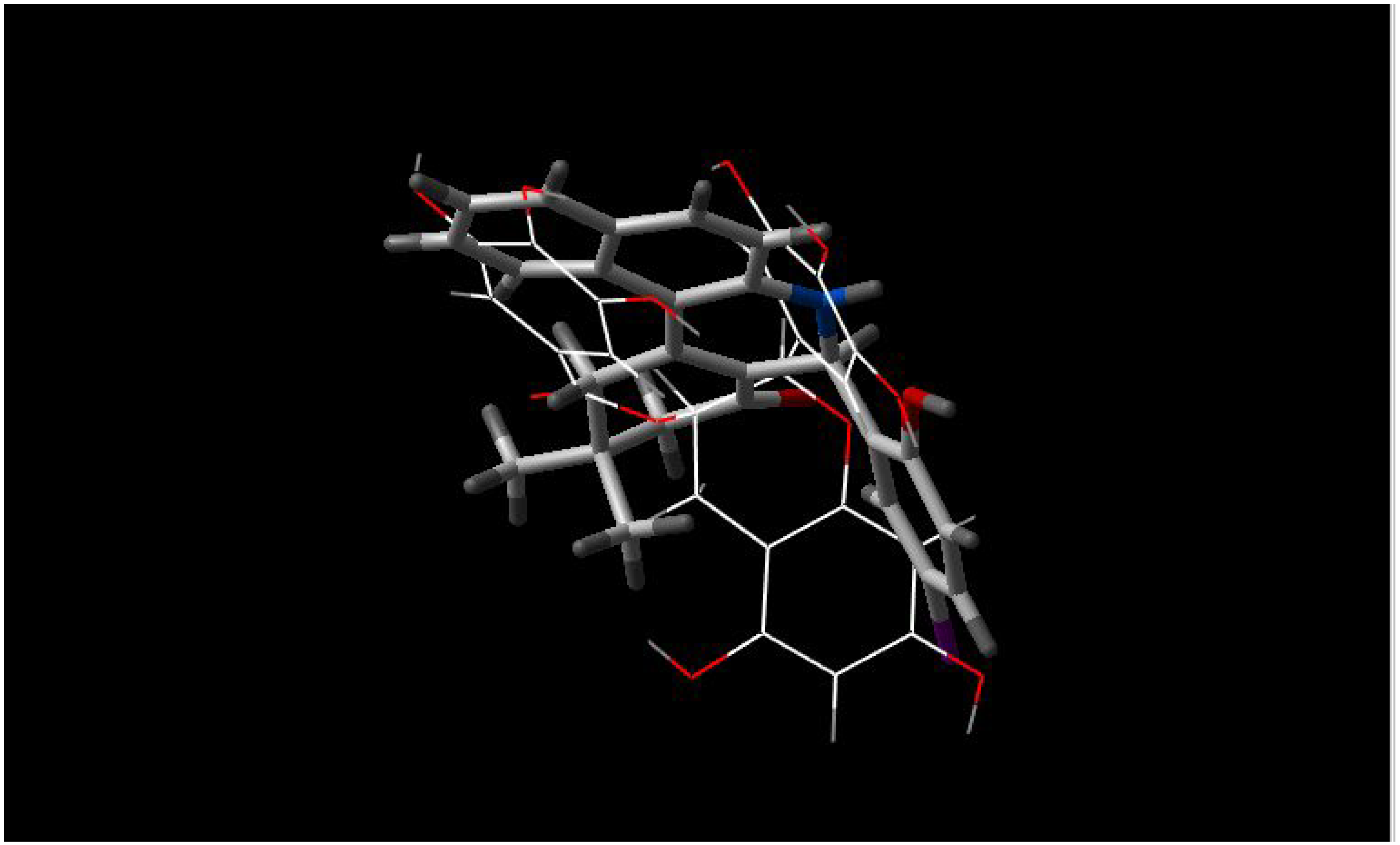
- c)
INF 55 is planar and different in shape compared to INF 271, however both molecules have a nitrogen atom in the middle of molecule and aromatic rings from both molecules are almost parallel to each other (
Figure 3);
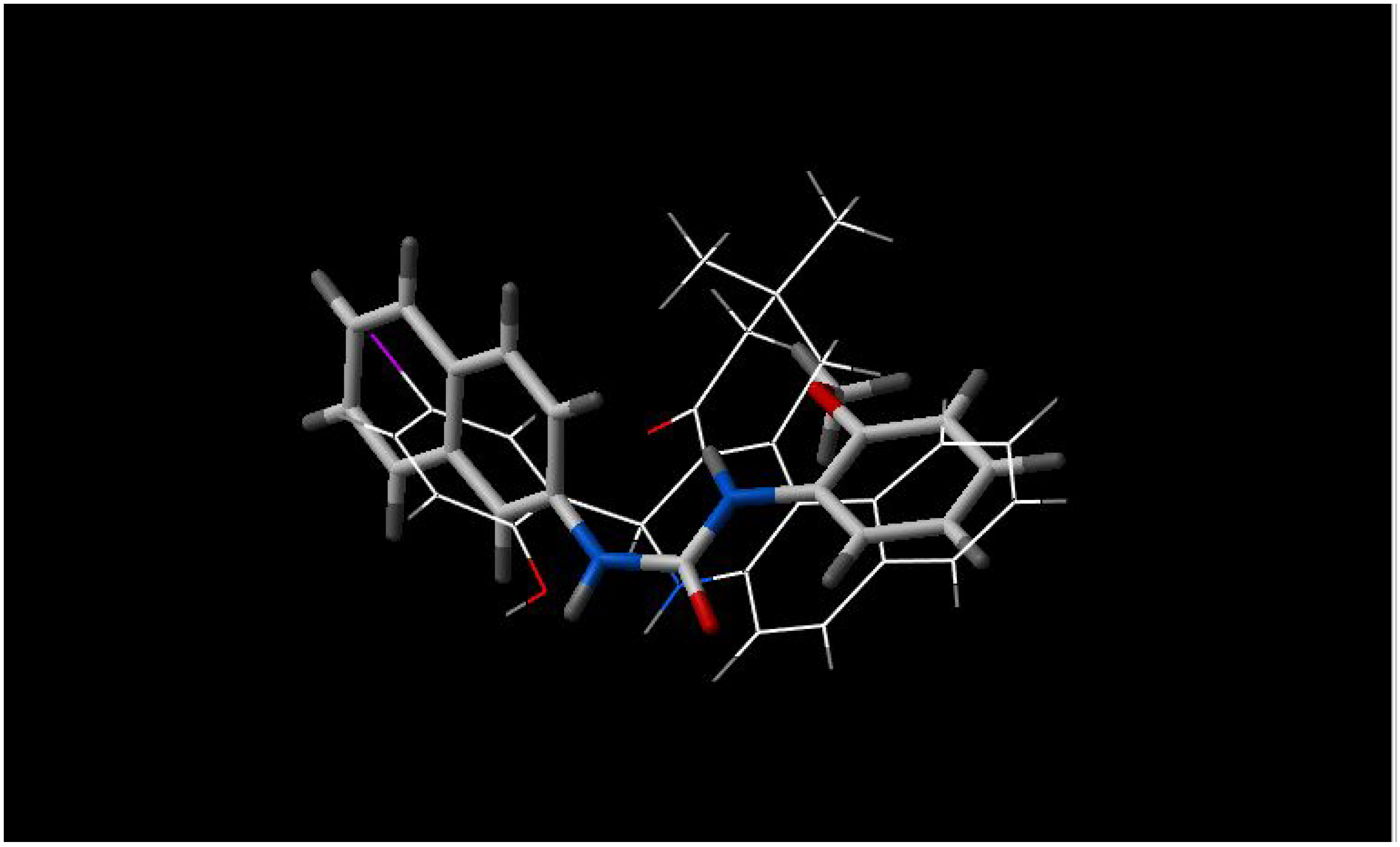
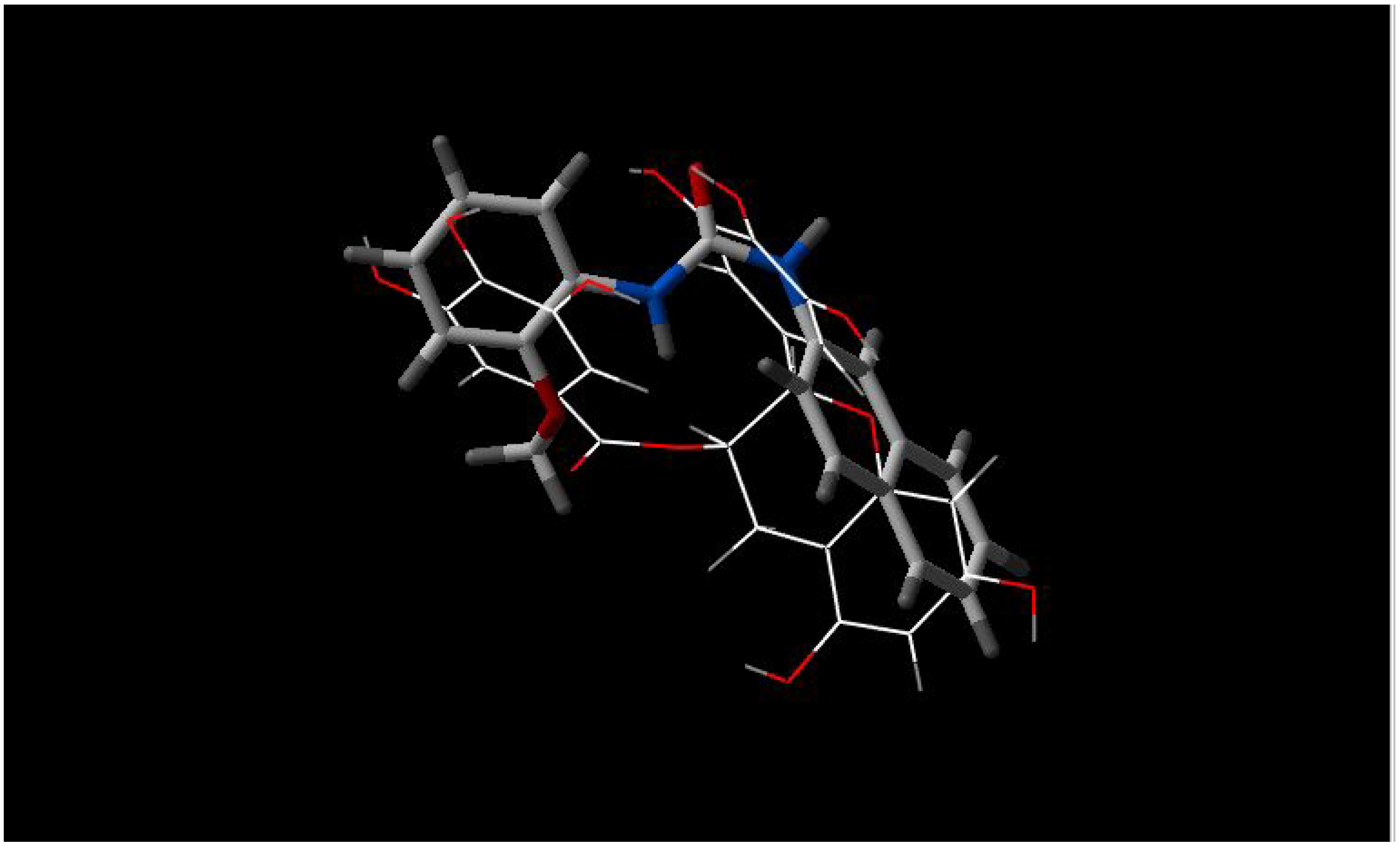
- d)
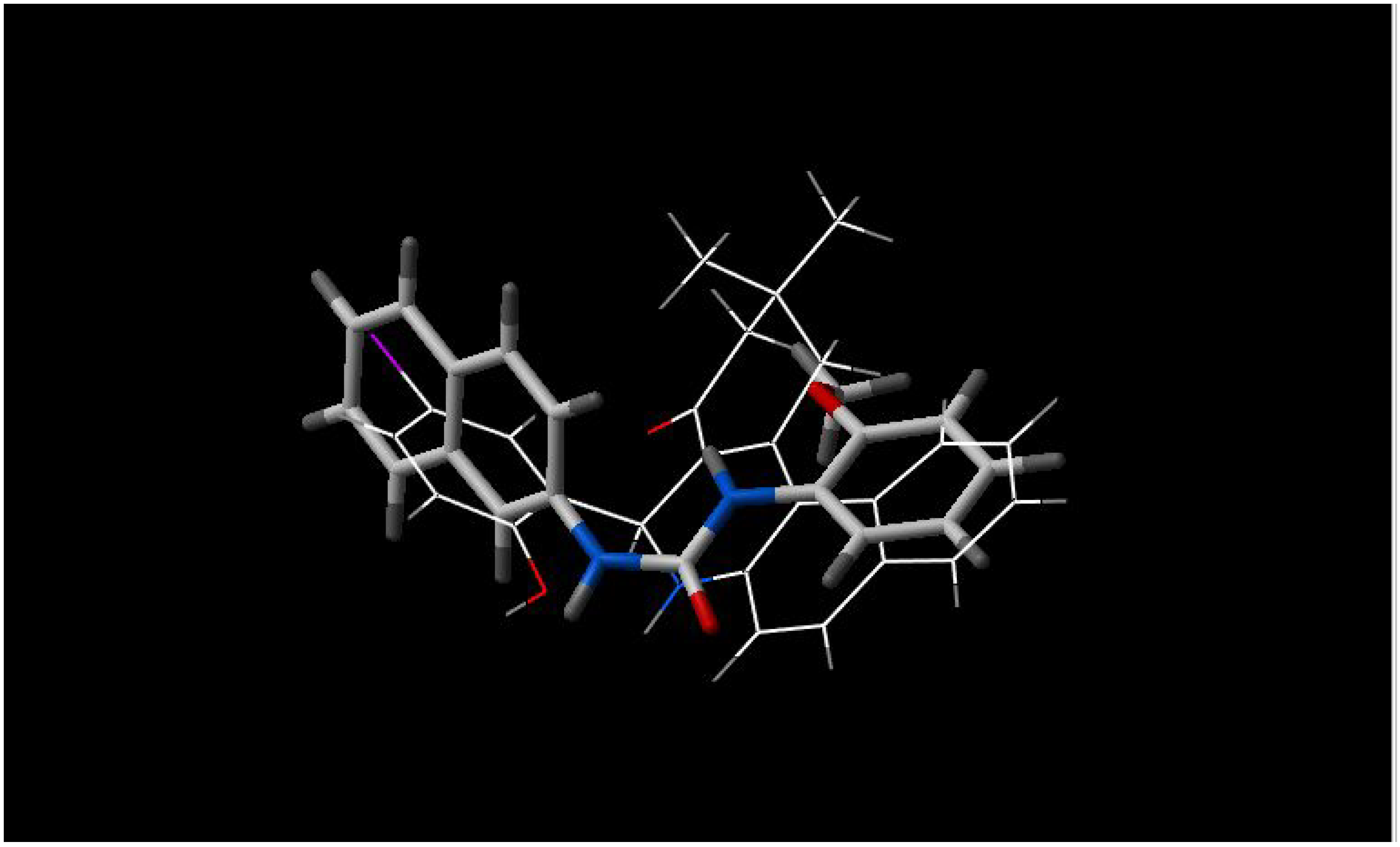
there is high similarity between INF 271 and parts of Reserpine and GG918 (
Figure 4 and
Figure 5, respectively), again with a nitrogen atom in the middle of the molecule;
- e)
all potent MDR inhibitors have aromatic rings in the areas that contain high similarity.
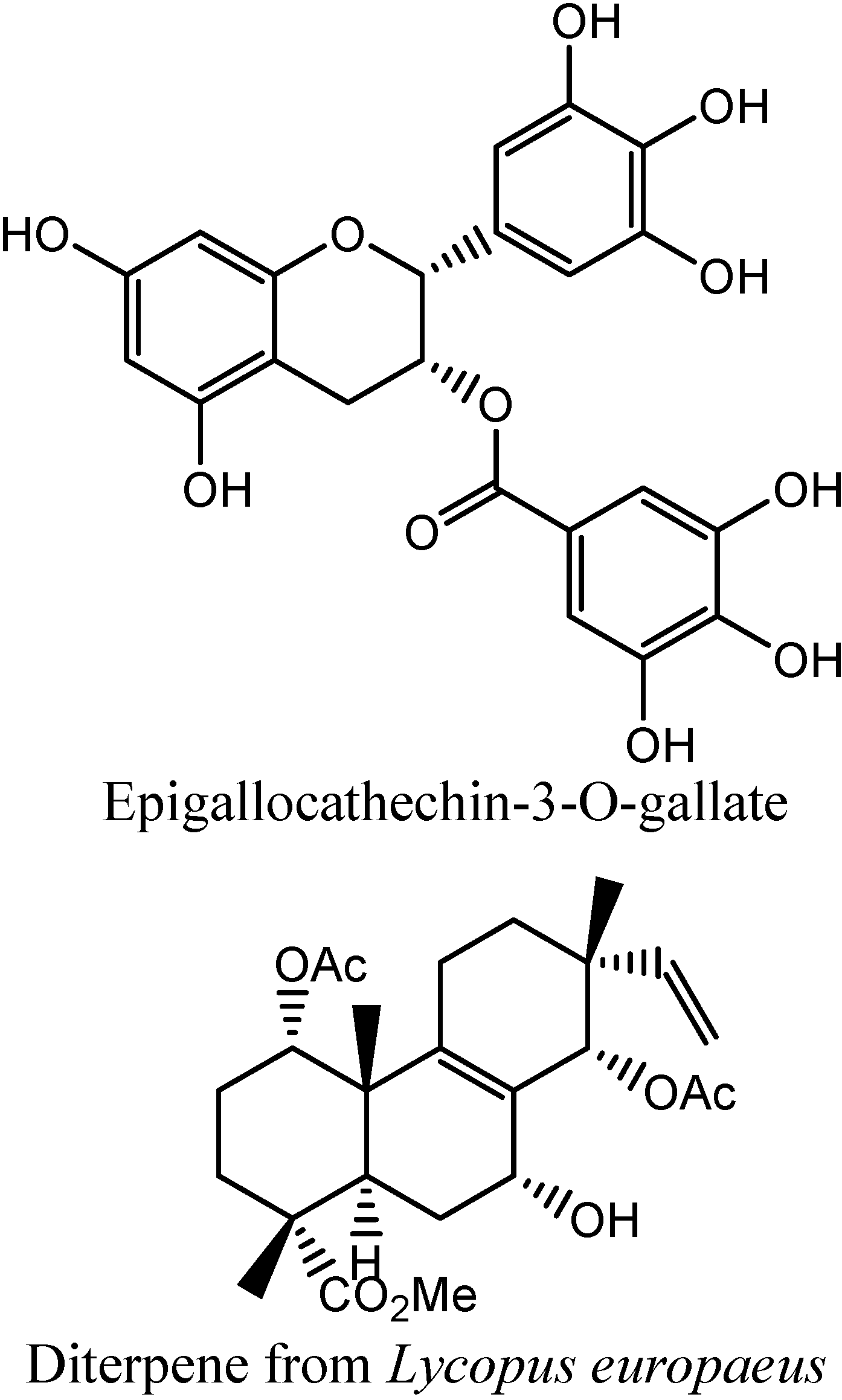
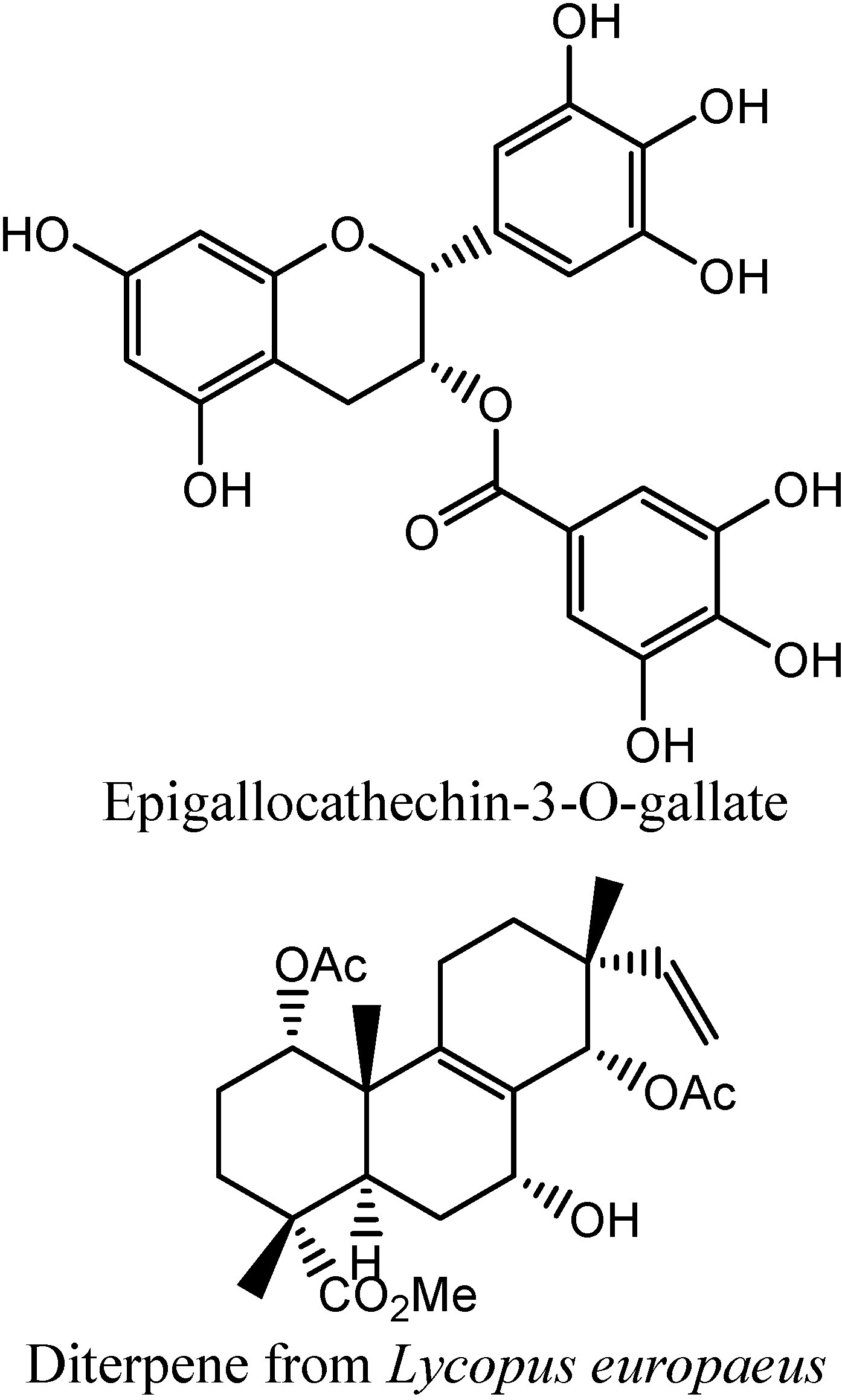
In
Figure 6 and
Figure 7, similarities between epigallocatechin-3-O-gallate (EGG) and two Influx compounds are depicted. However, EGG is not a potentiator of drugs in bacterial MDR processes, and possibly is therefore not an inhibitor of bacterial MDR efflux. This can possibly be explained by visually examining the best fit between EGG and the two Influx compounds (INF271 and INF277). The similarity index is very high for both of these combinations but it can be observed that there is a poor fit between EGG and the aromatic moieties of both of these compounds. Looking at the fit between INF277 and the diterpene from
Lycopus europaeus, there is a high calculated similarity but again this diterpene is not a potentiator of MDR drugs and this is probably due to the poor fit that this has with aromatic moieties of other MDR inhibitors e.g. INF277 (
Figure 8).
Figure 1.
Best fit of optimized 5'-methoxyhydnocarpin (sticks) and INF 240 (wireframe) structures.
Figure 1.
Best fit of optimized 5'-methoxyhydnocarpin (sticks) and INF 240 (wireframe) structures.
Figure 2.
Best fit of optimised INF 271 (sticks) and INF 277 (wireframe) structures.
Figure 2.
Best fit of optimised INF 271 (sticks) and INF 277 (wireframe) structures.
Figure 3.
Best fit of optimised INF 271 (sticks) and INF 55 (wireframe) structures.
Figure 3.
Best fit of optimised INF 271 (sticks) and INF 55 (wireframe) structures.
Figure 4.
Best fit of optimised INF 271 (sticks) and Reserpine A (wireframe) structures.
Figure 4.
Best fit of optimised INF 271 (sticks) and Reserpine A (wireframe) structures.
Figure 5.
Best fit of optimised INF 271 (sticks) and GG918 A (wireframe) structures.
Figure 5.
Best fit of optimised INF 271 (sticks) and GG918 A (wireframe) structures.
Figure 6.
Best fit of optimised INF277 (sticks) and Epigallocatechin-3-O-gallate (wireframe) structures.
Figure 6.
Best fit of optimised INF277 (sticks) and Epigallocatechin-3-O-gallate (wireframe) structures.
Figure 7.
Best fit of optimised INF271 (sticks) and Epigallocatechin-3-O-gallate (wireframe) structures.
Figure 7.
Best fit of optimised INF271 (sticks) and Epigallocatechin-3-O-gallate (wireframe) structures.
Figure 8.
Best fit of optimised INF277 (sticks) and diterpene from Lycopus europaeus (wireframe) structures.
Figure 8.
Best fit of optimised INF277 (sticks) and diterpene from Lycopus europaeus (wireframe) structures.
We have also studied some theoretical parameters i.e. clogP and logP, (
Table 3), however there is no obvious correlation with MDR modulation results. However clogP is generally lower for non-MDR inhibitors, the exception to this is INF55, which is a small molecule and one of the poorest MDR inhibitor of the INF series.
Table 3.
Theoretically calculated values of clogP, logP, surface area and volume of all studied MDR inhibitors and non potentiators by different methods.
Table 3.
Theoretically calculated values of clogP, logP, surface area and volume of all studied MDR inhibitors and non potentiators by different methods.
| Inhibitor | clogP (Chem3D) | logP (Chem3D) | logP (SciLogP) | logP (Vega) | lipole (Vega) | Surface area (A2) | Volume (A3) |
| 5’-MHC | 3.47 | 2.33 | 6.23 | 5.43 | 2.92 | 482.9 | 410.5 |
| INF 240 | 5.14 | 4.73 | 6.04 | 3.03 | 1.43 | 396.5 | 328.5 |
| INF 271 | 4.28 | 3.38 | 5.67 | 3.76 | 2.77 | 316.9 | 263.9 |
| INF 277 | 6.02 | 5.12 | 6.02 | 6.73 | 2.29 | 408.0 | 361.2 |
| INF 392 | 4.25 | 5.60 | 6.15 | 4.48 | 1.29 | 419.8 | 354.0 |
| INF 55 | 0.93 | 3.68 | 5.09 | 4.92 | 5.12 | 248.7 | 207.2 |
| Reserpine | 3.85 | 2.69 | 6.08 | 4.38 | 1.10 | 656.0 | 546.6 |
| GG918 | 4.21 | 5.03 | 6.12 | 5.60 | 0.89 | 629.9 | 536.0 |
| Diterpene from Lycopus europaeus | 2.45 | 3.65 | 6.23 | 3.34 | 0.73 | 497.84 | 425.59 |
| Epigallocatechin-3-O-gallate | 1.49 | 2.07 | 6.21 | 6.62 | 2.61 | 429.3 | 363.6 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}