Genotoxic Pyrrolizidine Alkaloids — Mechanisms Leading to DNA Adduct Formation and Tumorigenicity

1
National Center for Toxicological Research, Jefferson, AR 72079, USA
2
Department of Pharmacology, The Chinese University of Hong Kong, Shatin, Hong Kong
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2002, 3(9), 948-964; https://doi.org/10.3390/i3090948
Submission received: 7 June 2002
/
Accepted: 15 August 2002
/
Published: 30 September 2002
(This article belongs to the Special Issue Recent Advances in Environmental Health Research: Health Disparities, Toxicology and Carcinogenesis. Part I)
Abstract
:Plants that contain pyrrolizidine alkaloids are widely distributed in the world. Although pyrrolizidine alkaloids have been shown to be genotoxic and tumorigenic in experimental animals, the mechanisms of actions have not been fully understood. The results of our recent mechanistic studies suggest that pyrrolizidine alkaloids induce tumors via a genotoxic mechanism mediated by 6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine (DHP)-derived DNA adduct formation. This mechanism may be general to most carcinogenic pyrrolizidine alkaloids, including the retronecine-, heliotridine-, and otonecine-type pyrrolizidine alkaloids. It is hypothesized that these DHP-derived DNA adducts are potential biomarkers of pyrrolizidine alkaloid tumorigenicity. The mechanisms that involve the formation of DNA cross-linking and endogenous DNA adducts are also discussed.
Introduction
Pyrrolizidine alkaloid-containing plants are widely distributed in many geographical regions in the world [1,2,3]. Many pyrrolizidine alkaloids are highly toxic. Livestock are poisoned by grazing on plants containing pyrrolizidine alkaloids, causing livestock loss due to liver and pulmonary lesions [4,5,6,7,8]. It is now well recognized that a large variety of animal species are susceptible to pyrrolizidine alkaloid toxicity [1,2,3]. Pyrrolizidine alkaloids have also been found to contaminate human food sources, such as wheat, milk, honey, herbal medicines, and herbal teas, and this may potentially cause worldwide human health problems [9,10,11,12,13,14].
Although it has been known for several decades that pyrrolozidine alkaloids are genotoxic, particularly tumor induction, the mechanisms of action are not clear. We have recently determined that riddelliine, a representative carcinogenic pyrrolozidine alkaloid, induced liver tumors in rats through a genotoxic mechanism mediated by exogenous DNA adduct formation [15]. It is highly likely that this mechanism can be general to a large number of tumorigenic pyrrolizidine alkaloids. In this review we update the information concerning this mechanism. The role of the other two mechanisms, formation of DNA cross-linking and the possible formation of endogenous DNA adducts, is also discussed.
Pyrrolizidine alkaloids, widespread plant genotoxicants
It has been reported that about 3% of the world flowering plants contain toxic pyrrolozidine alkaloids [8]. Several hundred pyrrolizidine alkaloids have been isolated from plants and their structures have been well characterized [3]. The majority of these compounds are found in several genera of the plant families, particularly the three plant families, Boraginaceae, Compositae (Asteraceae), and Legumionsae (Fabaceae). The compounds most found are in the genus Senecio of the Compositae family.
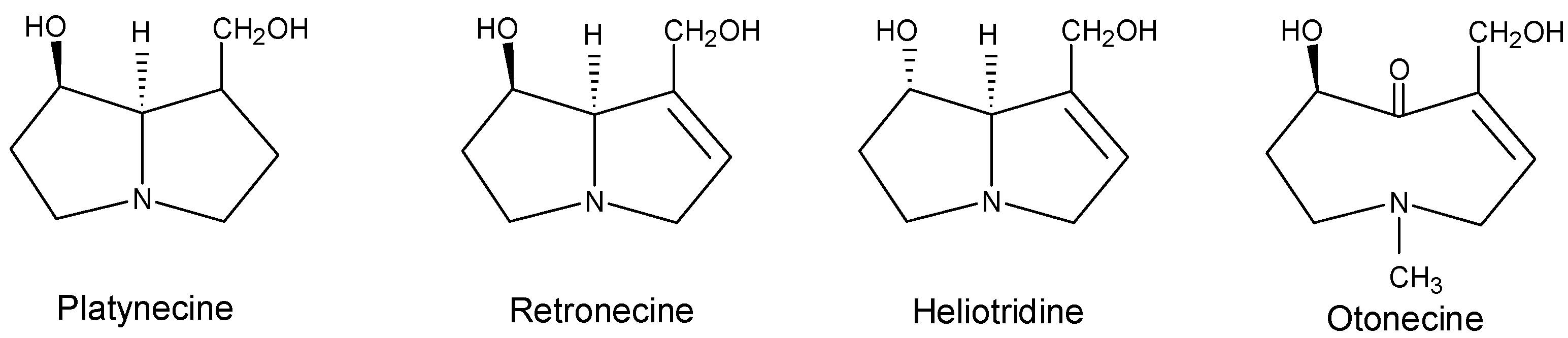
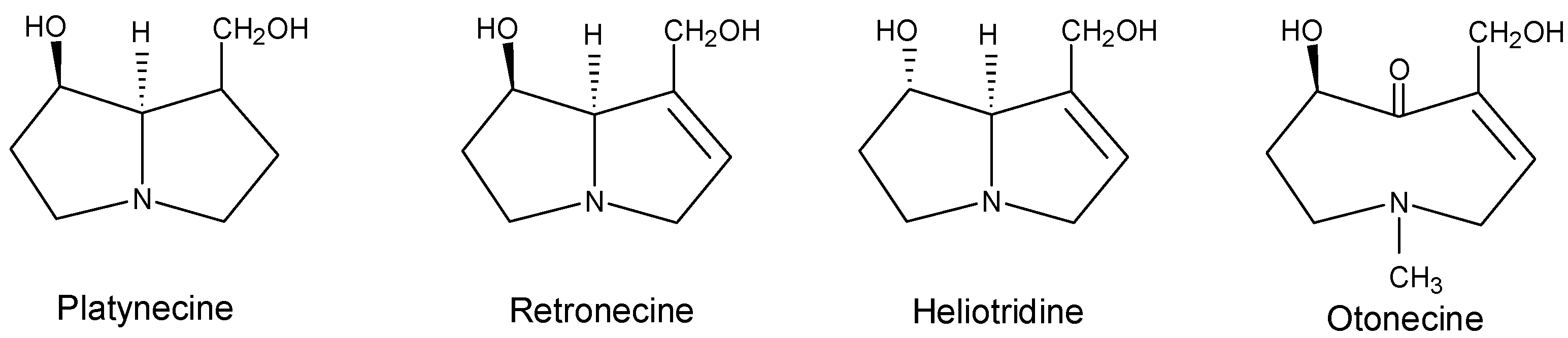
Hydrolysis products of a pyrrolizidine alkaloid are a necine base and a necic acid. The necic acids are four to six carbon-containing mono- or di-carboxylic acids, and are mostly branched-chained, unsaturated, hydroxylated, or epoxidized. Most of the pyrrolizidine alkaloids derived from esters of basic alcohols, the necine bases, have been found to exhibit toxic effects. The structures and numbering system of the four types of representative necine bases, platynecine, retronecine, heliotridine, and otonecine are shown in Figure 1. The platynecine type pyrrolizidine alkaloids do not contain a double bond in the base, and retronecine and heliotridine are enantiomers. Because of their abundance and toxicities, including hepatotoxicity and carcinogenicity, the retronecine- and heliotridine-derived pyrrolizidine alkaloids have received the most attention.
Pyrrolizidine alkaloids, particularly those from plants such as Senecio, Crotalaria, Heliotropium, and Amsinckia, have been found to exhibit acute toxicity, chronic toxicity, and genotoxicity. Acute poisoning causes massive hepatotoxicity with hemorrhagic necrosis. Chronic poisoning takes place mainly in liver, lungs, and blood vessels, and in some instances kidneys, pancreas, gastrointestinal tract, bone marrow, and brain [3]. Exposure over a longer period of time causes cell enlargement (megalocytosis), veno-occlusion in liver and lungs, fatty degeneration, nuclei enlargement with increasing nuclear chromatin, loss of metabolic function, inhibition of mitosis, fatty degeneration, proliferation of biliary tract epithelium, liver cirrhosis, nodular hyperplasia, and adenomas or carcinomas [3].
The genotoxicity of pyrrolizidine alkaloids includes DNA binding [15,16,17,18,19], DNA cross-linking [20,21,22,23,24], DNA-protein cross-linking [25,26], mutagenicity [15,27], and carcinogenicity [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42].
![Ijms 03 00948 g001]() Mutagenicity of plant extracts and pure pyrrolizidine alkaloids has been extensively studied [27]. Pyrrolizidine alkaloids, including clivorine, heliotrine, lasiocarpine, senkirkine, retrorsine, seneciphylline, and riddelliine, were found to be mutagenic in Salmonella typhimurium TA100 in the presence of the S9 activation enzyme system [3,19,27]. Pyrrolizidine alkaloids induce sister chromatid exchange and chromosomal aberrations in Chinese hamster ovary cells [3]. Pyrrolizidine alkaloids, such as heliotrine, are also teratogenic [1,43].
Mutagenicity of plant extracts and pure pyrrolizidine alkaloids has been extensively studied [27]. Pyrrolizidine alkaloids, including clivorine, heliotrine, lasiocarpine, senkirkine, retrorsine, seneciphylline, and riddelliine, were found to be mutagenic in Salmonella typhimurium TA100 in the presence of the S9 activation enzyme system [3,19,27]. Pyrrolizidine alkaloids induce sister chromatid exchange and chromosomal aberrations in Chinese hamster ovary cells [3]. Pyrrolizidine alkaloids, such as heliotrine, are also teratogenic [1,43].
Figure 1.
Common necine bases of pyrrolizidine alkaloids.
Metabolism and metabolic activation of pyrrolizidine alkaloids
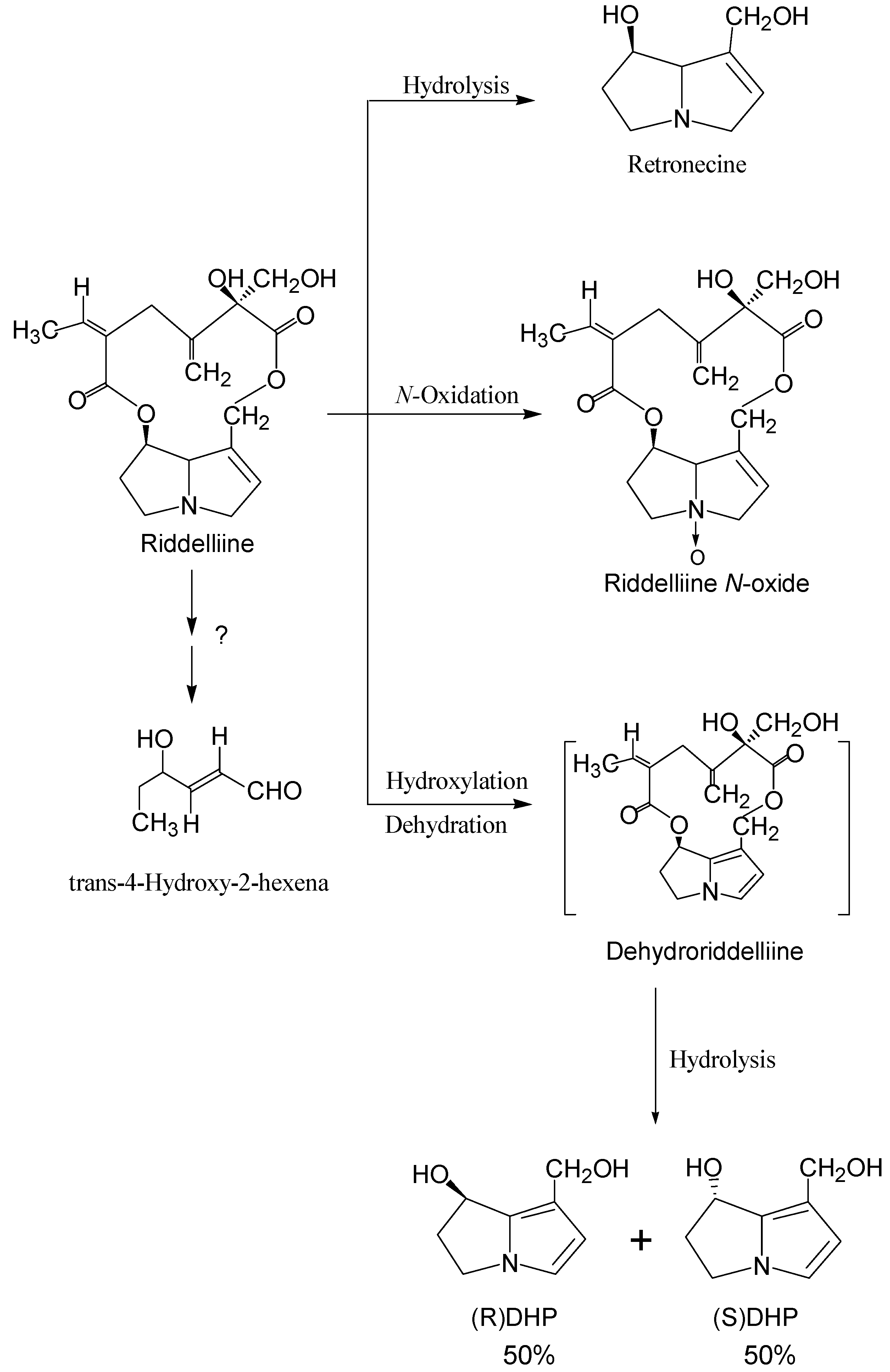
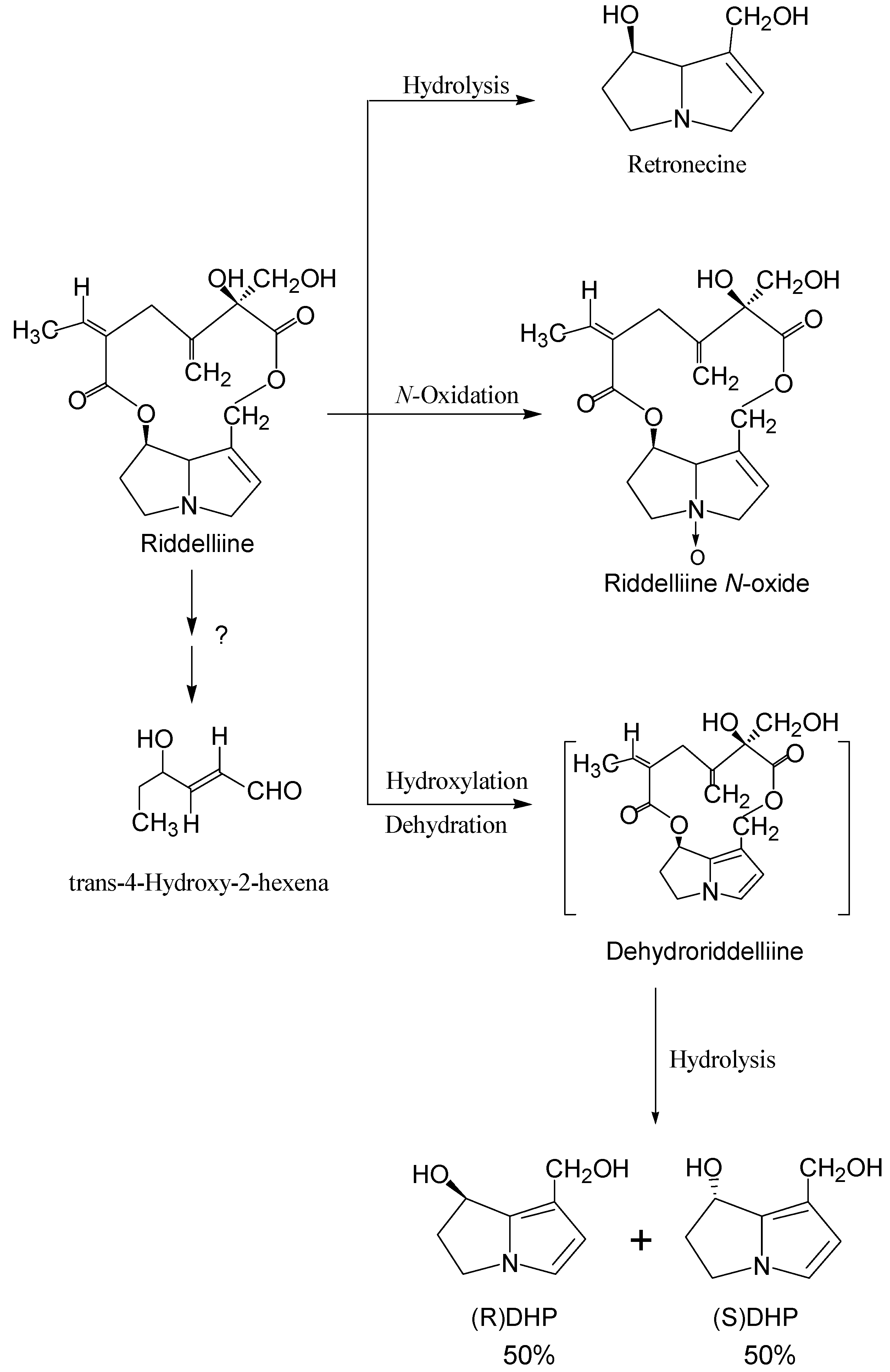
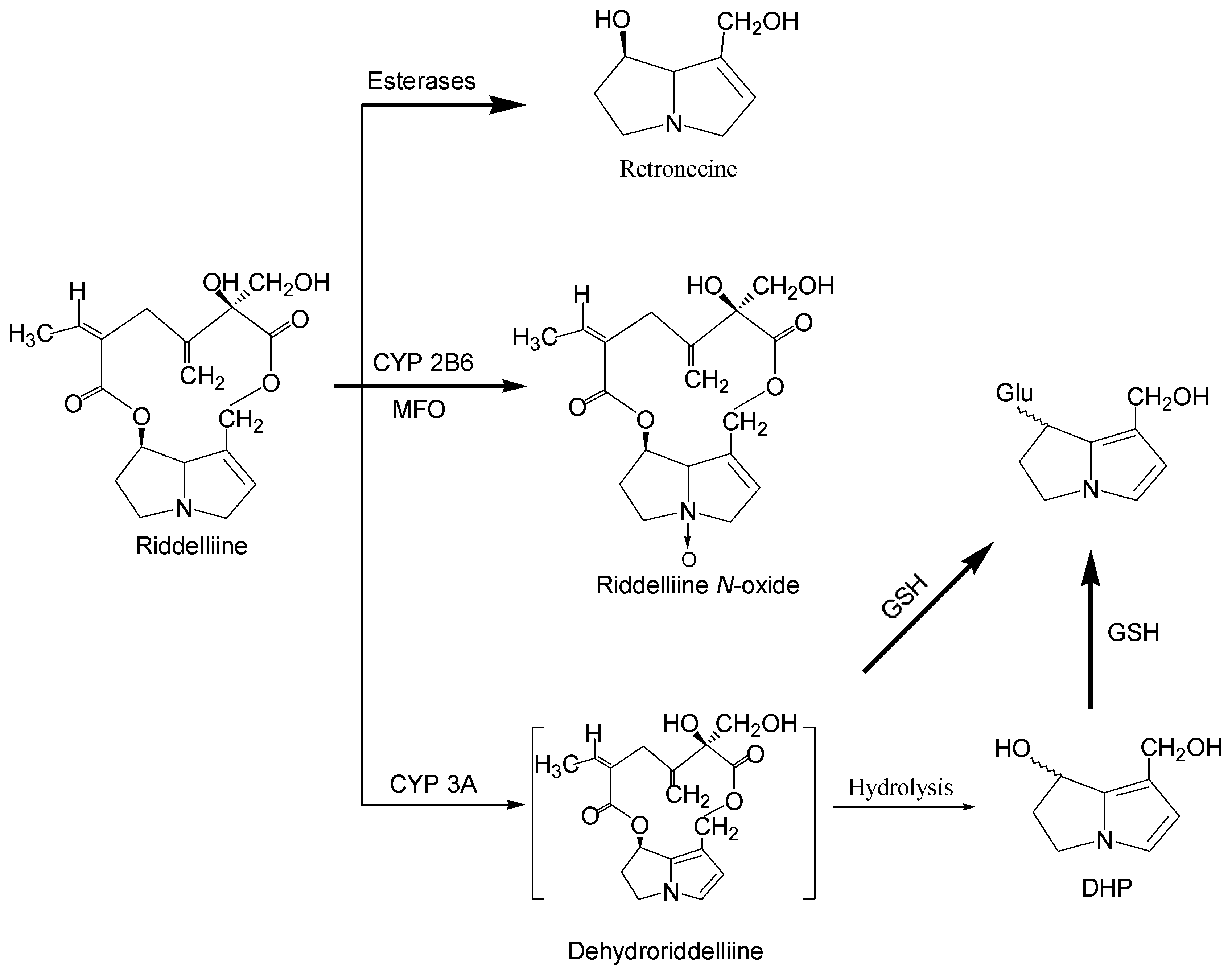
Like most of the genotoxic compounds, pyrrolizidine alkaloids require metabolic activation in order to exert genotoxicities [2,3,19,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64]. During the last several decades, metabolism of pyrrolizidine alkaloids has been extensively studied both in vitro and in vivo [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64]. There are three principal metabolic pathways; (i) hydrolysis of the ester functional group to form the necine bases (including heliotridine type, retronecine type, and otonecine type); (ii) oxidation of the necine bases to the corresponding necine N-oxides (heliotridine type and retronecine type); and (iii) hydroxylation at the C-3 or C-8 position of the necine base to form 3- or 8-hydroxynecine derivatives followed by dehydration, to form the corresponding dehydropyrrolizidine (pyrrolic) derivatives. The third pathway is generally considered to be the metabolic activation responsible for pyrrolizidine alkaloid intoxication. Using riddelliine as an example, the metabolic pathways are shown in Figure 2. It was reported in our laboratories that rat and mouse liver microsomal metabolism of riddelliine produced riddelliine N-oxide and 6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine (DHP) as major metabolites and retronecine as a minor metabolite [15,19,40]. Dehydroriddelliine was presumably formed which upon hydrolysis produced DHP. Although it was reported that trans-4-hydroxy-2-hexenal was formed by metabolism of senecionine [58], this metabolite was not found from metabolism of riddelliine [15] (Figure 2).
As expected, metabolism of pyrrolyzidine alkaloids occurs mainly in the liver. Metabolism of pyrrolizidine alkaloids to dehydropyrrolizidines is mainly catalyzed by cytochromes P-450, specifically both the CYP3A and CPY2B6 isozymes [46,47,48,49,50,51,52,53]. Metabolism of pyrrolizidine alkaloids to the corresponding N-oxides is catalyzed by both cytochrome P-450 and flavin-containing monooxygenase [48,52,65,66]. It has been established that hydrolysis of pyrrolizidine alkaloids of the
![Ijms 03 00948 g002]() ester group to form the necine base is a detoxification pathway and this pathway is catalyzed by liver microsomal and cytosolic carboxyesterase [3,51,54].
ester group to form the necine base is a detoxification pathway and this pathway is catalyzed by liver microsomal and cytosolic carboxyesterase [3,51,54].
Figure 2.
Metabolism pathways of riddelliine
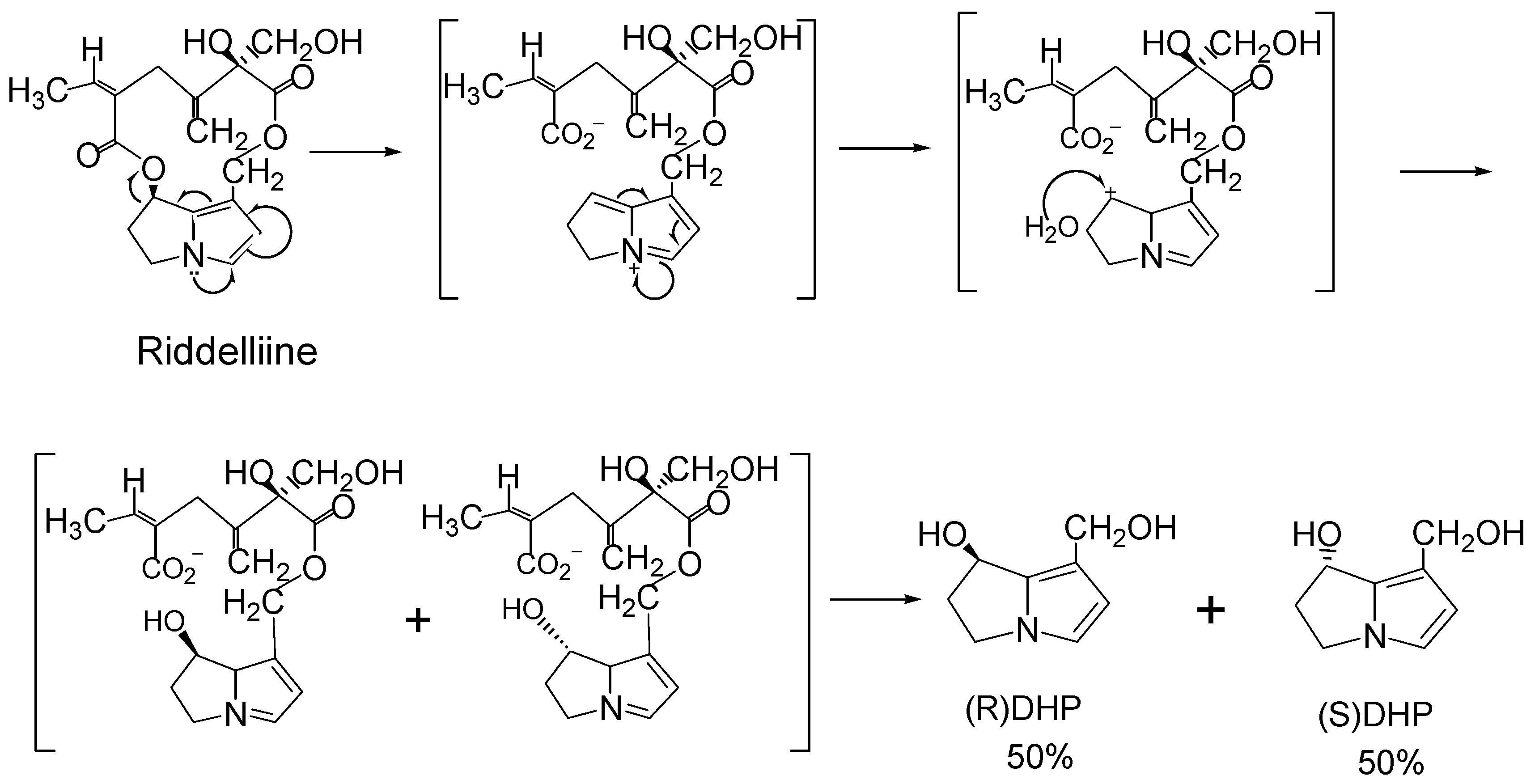
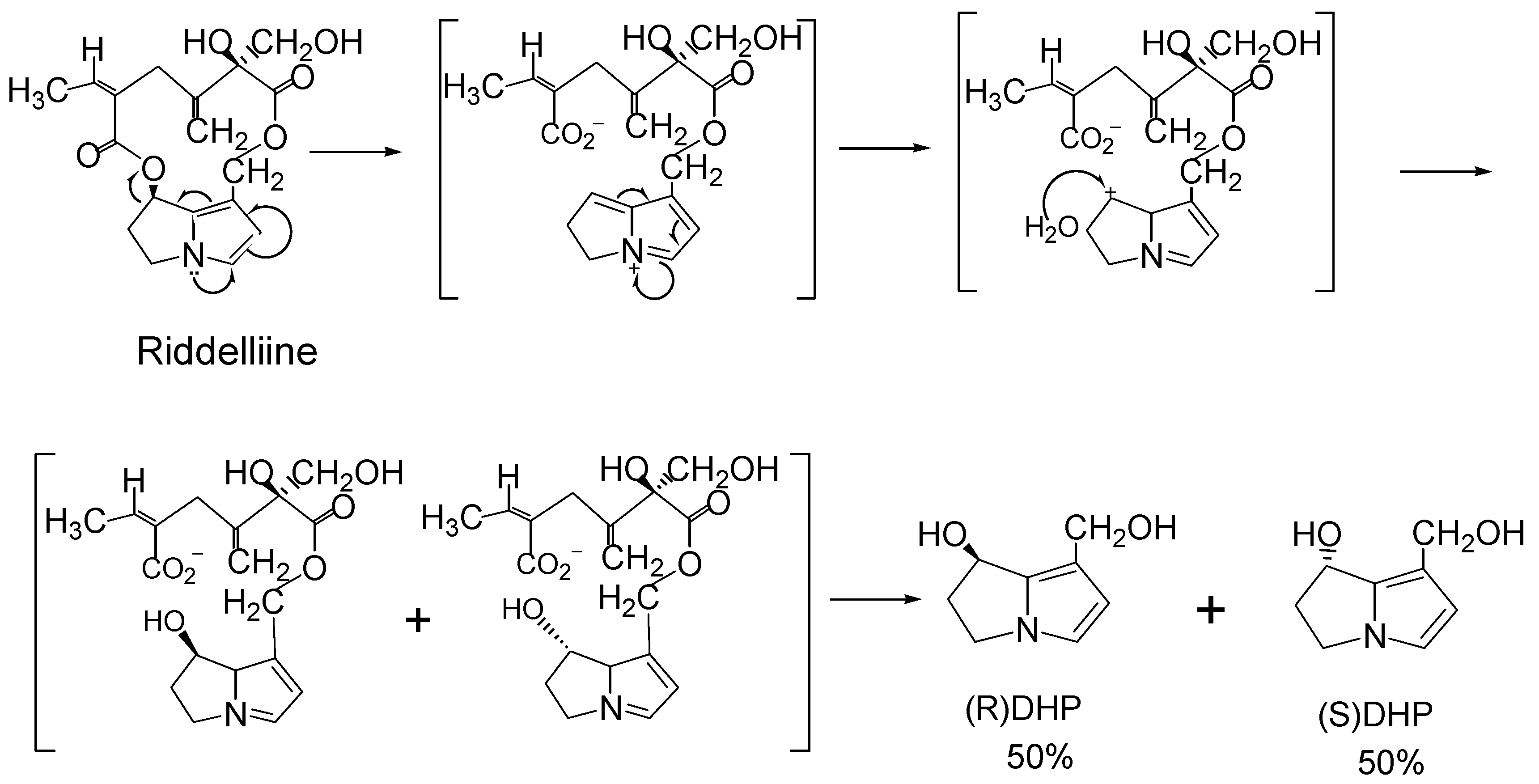
With respect to the C-O bond at the C7 position, retronecine-type and heliotridine-type pyrrolizidine alkaloids are enantiomers and are optically active. However, the studies by Buhler and co-workers [53,56,57] and by our laboratories [19; unpublished data] found that metabolism of riddelliine, monocrotaline, retrorsine, jacobine and lasiocarpine by rat liver microsomes did not produce the optically active necine bases dehydroretronecine (DHR; R-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine; R-DHP) or dehydroheliotridine (DHH; S-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine; S-DHP) [3]. However, all these metabolism produced the racemic (+/-)6,7-dihydro-7-
![Ijms 03 00948 g003]() hydroxy-1-hydroxymethyl-5H-pyrrolizine (R/S-DHP; DHP), instead of the optically pure DHR or DHH enantiomers. Taking metabolism of riddelliine as an example (Figure 3), formation of the racemic DHP metabolite involves: (i) metabolism of riddelliine (or other retronecine-type and heliotridine-type pyrrolizidine alkaloids) produces dehydroriddelliine (or the corresponding pyrrolic metabolites); (ii) these pyrrolic metabolites (intermediates) are converted to the carbonium ions located at the C7 of the necine base; and (iii) reaction of the carbonium ion with water, by attacking at both sides of the necine plane, to form the racemic DHP.
hydroxy-1-hydroxymethyl-5H-pyrrolizine (R/S-DHP; DHP), instead of the optically pure DHR or DHH enantiomers. Taking metabolism of riddelliine as an example (Figure 3), formation of the racemic DHP metabolite involves: (i) metabolism of riddelliine (or other retronecine-type and heliotridine-type pyrrolizidine alkaloids) produces dehydroriddelliine (or the corresponding pyrrolic metabolites); (ii) these pyrrolic metabolites (intermediates) are converted to the carbonium ions located at the C7 of the necine base; and (iii) reaction of the carbonium ion with water, by attacking at both sides of the necine plane, to form the racemic DHP.
Figure 3.
Scheme of metabolism of riddelliine leading to (±)DHP.
It has been well studied that the pyrrolic metabolic metabolites are capable of binding with glutathione to form glutathione conjugates and this enzymatic reaction, catalyzed by glutathione S-transferase (GST), may be the major detoxification pathway for toxicity induced by pyrrolizidine alkaloids [60,61,62,63,64].
There is a lack of reports on the investigation of metabolic activation of otonecine-type pyrrolizidine alkaloid and the induced hepatotoxicity [3]. Recently Lin and co-workers studied rat liver microsomal metabolism of clivorine, an otonecine-type pyrrolizidine alkaloid, and observed several new findings [60,64]. From the study, a dehydroretronecine (DHR) metabolite was identified as a metabolite, which clearly confirmed that an otonecine-type pyrrolizidine alkaloid (clivorine) can be enzymatically converted to a metabolite (DHR) that was formed from metabolism of retronecine-type pyrrolizidine alkaloids [3]. The other finding is that a pair of glutathione conjugates with the glutathionyl group located at the C7 of the necine base (7-GSH-DHR), were identified from metabolism in the presence of glutathione [60,64]. Based on this finding, the formation of carbonium ion located at the C7 of the necine base was proposed [64].
Significant species differences in susceptibility to pyrrolizidine alkaloid intoxication have been reported in livestock and laboratory animals. In general, cattle, horses, rats, and mice more resemble humans and are highly susceptible to pyrrolizidine alkaloid intoxication, whereas sheep, rabbits, and guinea pigs are resistant to pyrrolizidine alkaloid toxicity [3,67,68]. The mechanism of hepatotoxicity induced by pyrrolizidine alkaloid has been extensively investigated, and it is well established that metabolic activation of pyrrolizidine alkaloid in the liver to form the reactive pyrrolic metabolites plays a key role in causing hepatotoxicity [3]. It has been reported that the species difference in susceptibility to the pyrrolizidine alkaloid intoxication is mainly due to the variations in the balance between the formation of the toxic pyrrolic metabolites and the detoxification pathways to generate non-toxic N-oxides and/or hydrolyzed metabolites [3,69,70]. The species difference in susceptibility to the otonecine-type pyrrolizidine alkaloid induced hepatotoxicity was studied by Lin et al. [unpublished data]. The in vitro metabolic activation of clivorine in both male rat and human was similar but was different from that in guinea pig. This may explain the species difference in susceptibility to this toxic compound.
We recently conducted metabolism of riddelliine N-oxide by liver microsomes of F344 female rats and found that both riddelliine and dehydroretronecine (DHR) are major metabolites [unpublished data]. Since riddelliine is a tumorigen and DHR is the reactive metabolite of riddelliine, these results suggest that riddelliine N-oxide is also biologically active. Thus, although formation of pyrrolizidine alkaloid N-oxides from metabolism of pyrrolizidine alkaloids is generally considered a detoxification pathway [3], our results provide evidence that metabolism of pyrrolizidine alkaloids to the corresponding N-oxide metabolites may not necessarily be a detoxification process.
Structure-activity relationship concerning genotoxicity
The platynecine-type pyrrolizidine alkaloids, with the structure shown in Figure 1, do not contain a double bond at the base and have been found that pyrrolizidine alkaloids of this type are not genotoxic. The pyrrole metabolites (dehydropyrrolizidines) formed from metabolism of the other three types of pyrrolizidine alkaloids (Figure 1) have been found to be the active species and are proposed to be responsible for most of the genotoxicity of the parent pyrrolizidine alkaloids. The hydrolysis metabolites are much less toxic than the substrates and thus are considered detoxified products. Therefore, the relative ease of dehydropyrrolizidine formation compared to hydrolysis product formation is crucial in determining a pyrrolizidine alkaloid’s toxicity. Structural features, particularly steric hindrance, have been found to be important factors in both hydroxylation (leading to dehydrogenation) and hydrolysis reactions [3,71,72]. Steric hindrance around the ester functional groups of the molecule can inhibit the enzymatic hydrolysis process [3,72]. In general, the allylic ester group in some diester compounds (e.g., senecionine) is more easily hydrolyzed than the ester group at the other side of the molecule. This is because the allylic ester is less sterically hindered, and the double bond enhances hydrolysis electronically.
Tumorigenicity of pyrrolizidine alkaloids
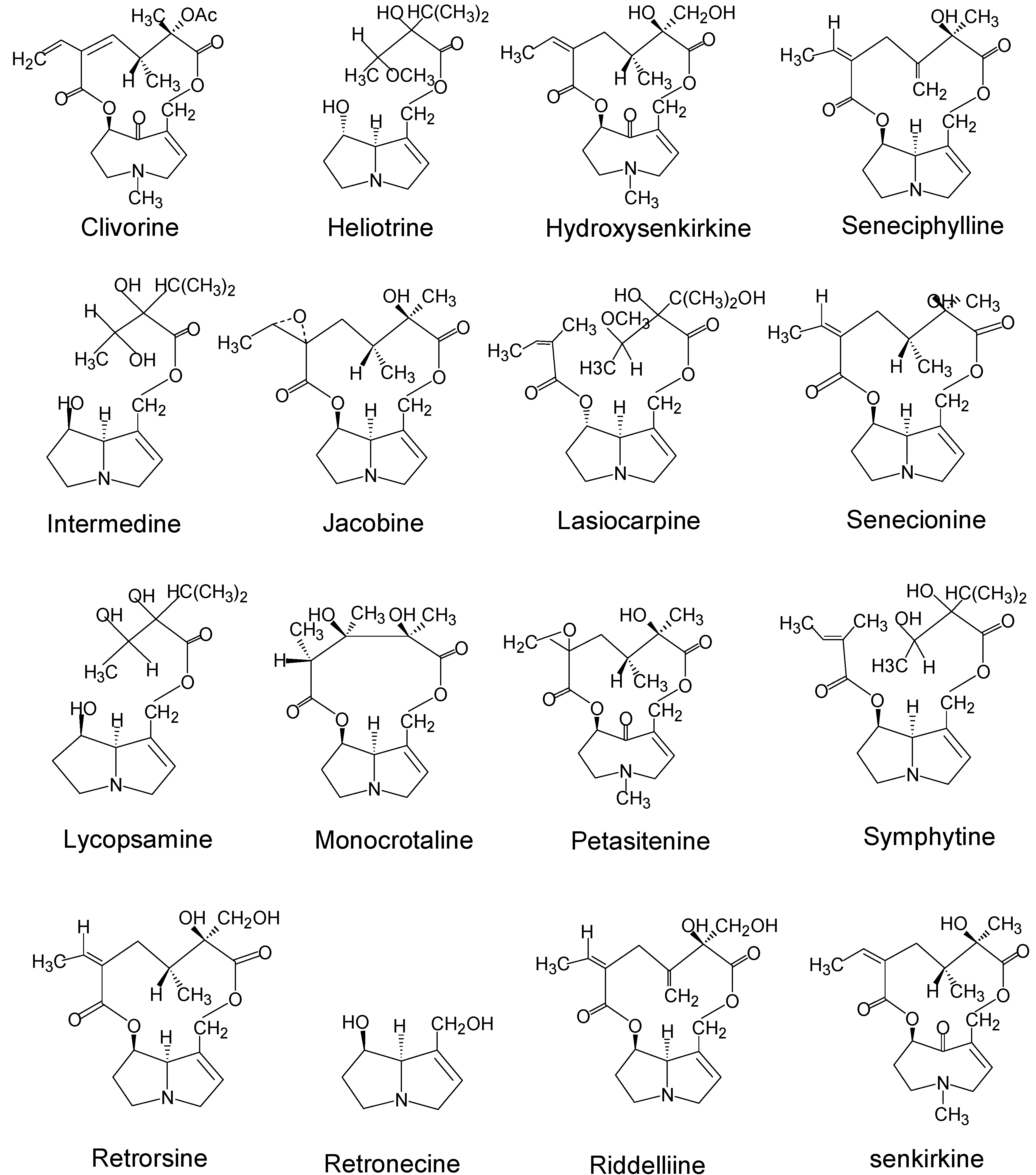
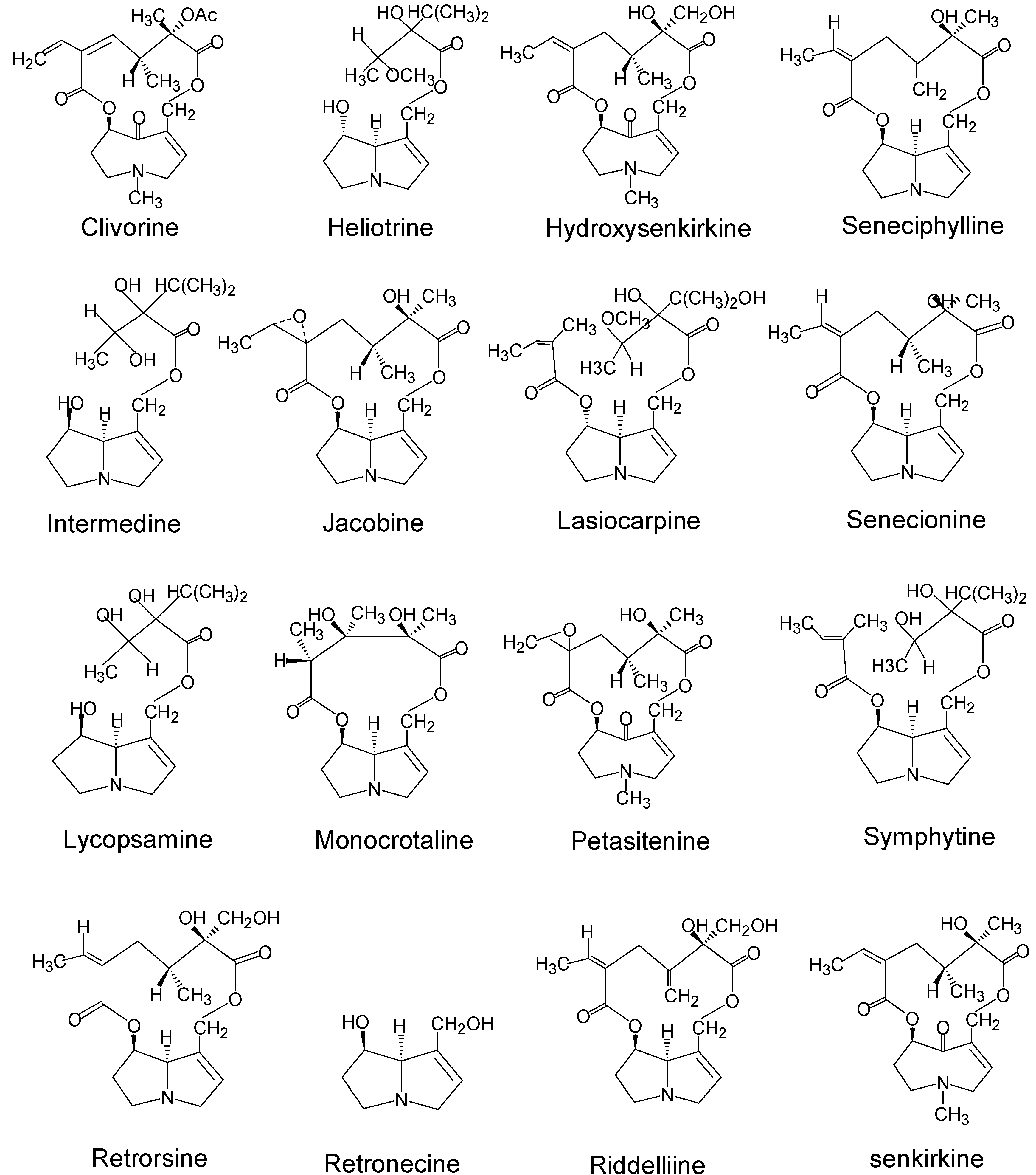
Pyrrolizidine alkaloids were among the first naturally occurring carcinogens identified in plants [3]. Sixteen pyrrolizidine alkaloids and one pyrrolizidine alkaloid N-oxide (isatidine) from the three plant families, Boraginaceae, Compositae (Asteraceae), and Legumionsae (Fabaceae), have been shown to induce tumors in experimental animals (Table 1). The names and structures of the tumorigenic pyrrolizidine alkaloids are shown in Figure 4. Among the tumorigenic pyrrolizidine alkaloids so far identified, two (heliotrine and lasiocarpine) are heliotridine-type pyrrolizidine alkaloids, three (clivorine, senkirkine, and hydroxysenkirkine) are otonecine-type pyrrolizidine alkaloids, and the majority listed in Table 1 belong to retronecine-type pyrrolizidine alkaloids. This confirms that the retronecine-type pyrrolizidine alkaloids are the most abundant and most genotoxic (tumorigenic) pyrrolizidine alkaloids.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyrrolizidine alkaloids | Plant species (Family)1 | References |
| Clivorine | Ligularia dentata Hara (Compositae) | 28 |
| Heliotrine | Heliotropium (Boraginaceae) | 29 |
| Hydroxy-senkirkine | Senecio (Compositae) | 30,31 |
| Intermedine | Amsinckia (Boraginaceae) | 32,33 |
| Jacobine | Senecio L. (Compositae) | 34,35 |
| Lasiocarpine | Heliotropium (Boraginaceae) | 28,36,37 |
| Lycopasamine | Amsinckia (Boraginaceae) | 32,33 |
| Monocrotaline | Crotalaria (Leguminosae) | 29 |
| Patasitenine | Senecio (Compositae) | 38 |
| Retrorsine | Senecio (Compositae) | 33,34,39 |
| Retronecine | Crotalaria (Leguminosae) | 30 |
| Riddelliine | Senecio (Compositae); Crotalaria (Leguminosae) | 39 |
| Seneciphylline | Senecio (Compositae) | 38,39 |
| Symphytine | Symphytum officinale L (Boraginaceae) | 41,42 |
| Senkirkine | Senecio (Compositae); Petasites (Compositae) | 11,38,41 |
| Senecionine2 | Senecio (Compositae) | 11,34 |
| Isatidine (Retrorsine N-oxide) | Senecio (Compositae), Crotalaria (Leguminosae) | 33,34 |
1Represents one of the main sources2Not based on testing of the pure compound, but based on testing of the Senecio plants (such as Senecio jacobaea L.) that contain senecionine.
Figure 4.
Names and structures of tumorigenic pyrrolizidine alkaloids.
Mechanisms leading to tumorigenicity
Although the carcinogenesis by pyrrolizidine alkaloids has long been studied, the mechanisms that these compounds induce tumors in experimental animals are not clear. We recently reported that a genotoxic mechanism mediated by exogenous DNA adduct formation was involved in the induction of liver tumors in rats and mice fed riddelliine [15]. The following presents this finding, and discuss the other probably activation pathways.
1. Formation of exogenous DNA adducts
Riddelliine is a genotoxic pyrrolizidine alkaloid isolated from plants of the genus Senecio, which grow in rangelands of the western United States [3,40]. In view of the human exposure to riddelliine, this compound was nominated by the U.S. Food and Drug Administration (FDA) for genotoxicity and carcinogenicity testing conducted by the National Toxicology Program (NTP) [40]. The NTP 2-year carcinogenicity bioassay showed that riddelliine induced liver hemangiosarcomas in male and female rats and male mice, hepatocellular adenomas and carcinomas in male and female rats, and lung alveolar adenomas in female mice [40].
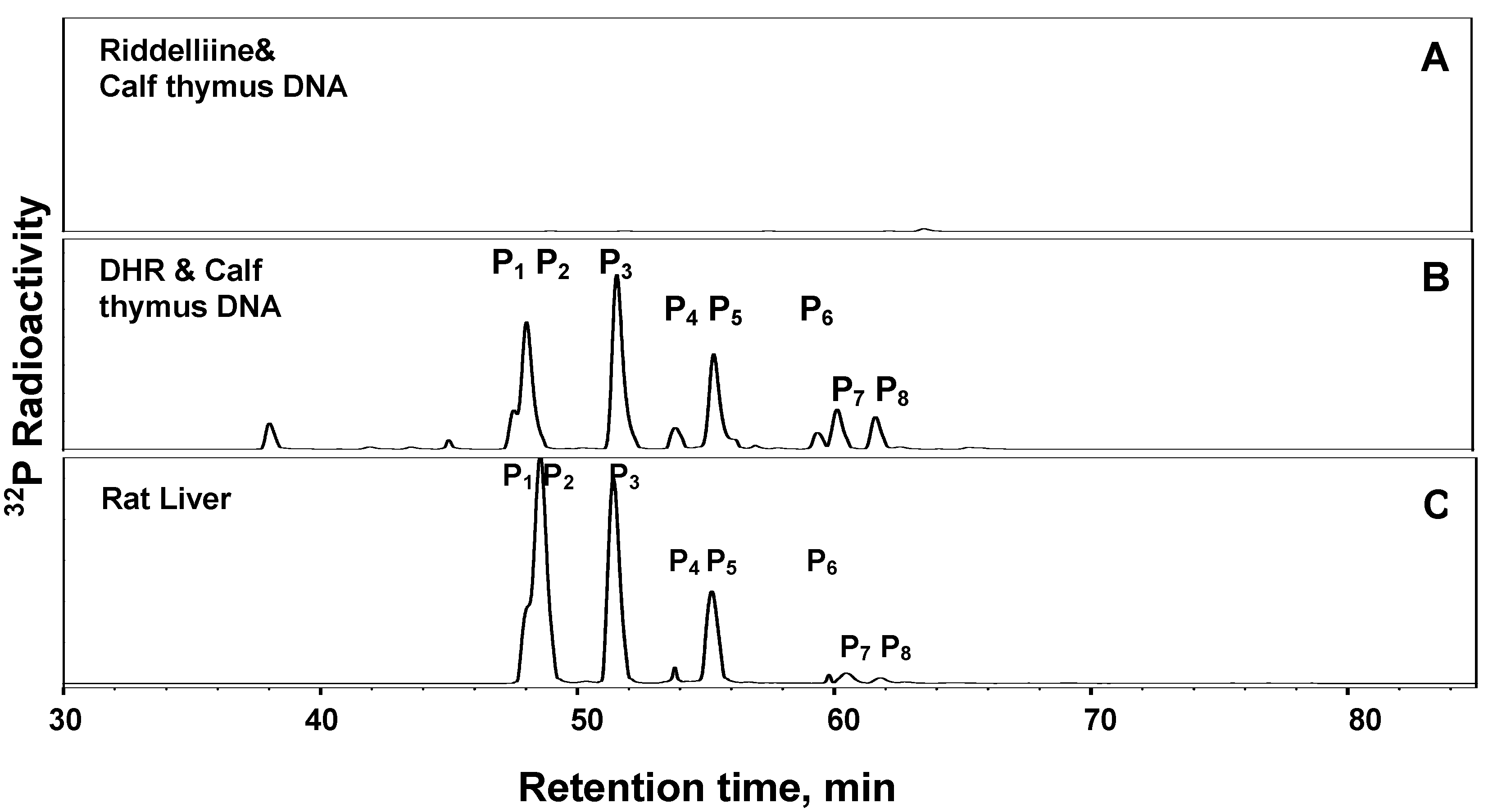
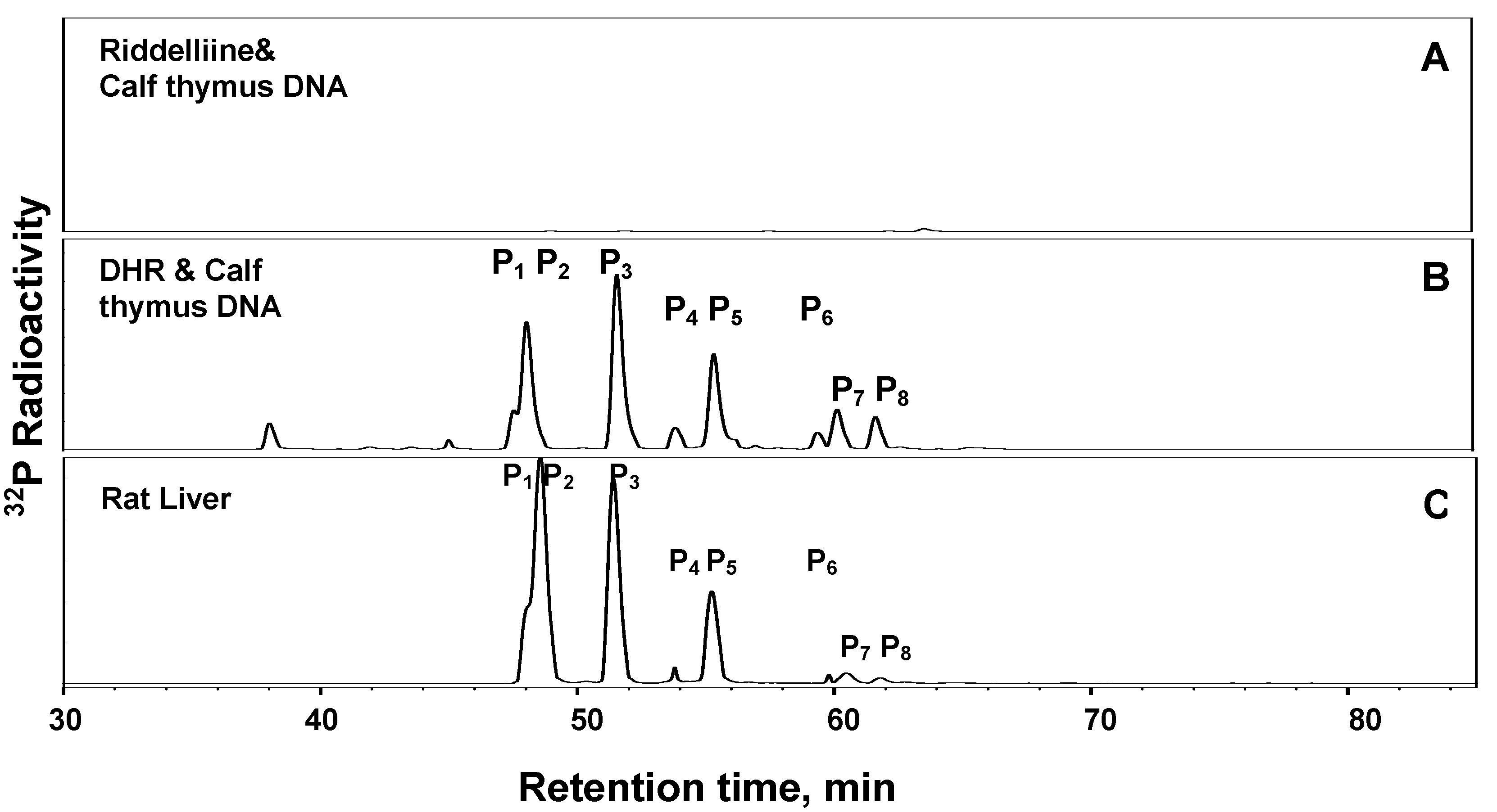
The mechanisms by which riddelliine induces liver tumors in female F344 rats were studied by Yang et al. [15]. As shown in Figure 2, metabolism of riddelliine by liver microsomes of female rats generated riddelliine-N-oxide and dehydroretronecine (DHR) as metabolites [15]. Subsequent metabolism studies indicated that the racemic (+)DHP was formed, instead of the optical active DHR enantiomer (unpublished data). Employing 32P-postlabeling/HPLC methodology [73], reaction of the synthetically prepared DHR with calf thymus DNA resulted in the formation of eight DNA adducts, which were not formed in the absence of DHR (Figure 5A and Figure 5B). Two of these adducts were identified as enantiomers of DHP-derived 7’-deoxyguanosin-N2-yl adducts (DHR-3’-dGMP) [15]. Six other DHP-derived DNA adducts were subsequently characterized as DHP-modified dinucleotides [74 and data to be submitted for publication]. It was then found that metabolism of riddelliine in the presence of calf thymus DNA resulted in the set of eight DNA adducts that were identical to those obtained from the reaction of DHR with calf thymus DNA (data not shown). Furthermore similar DNA adduct profile was detected in the livers of F344 female rats administered riddelliine (Figure 5C) [15]. A dose-response relationship was obtained between the dose administered to the rats and the level of the total (eight) DHP-derived adducts [15]. These results suggested that riddelliine induces liver tumors (hemangiosarcomas and hepatocellular adenomas and carcinomas) in rats through a genotoxic mechanism and that the eight DHP-derived DNA adducts are responsible in part, if not all, for the liver tumor development.
The rate of riddelliine metabolism was higher with PB-microsomes than with control-microsomes [15]. When metabolism was incorporated with the CYP3A4 enzyme inhibitor, triacetyleandomycin, the formation of DHP was 84% reduced [unpublished data]. These overall results suggest that CPY3A isozymes are the major metabolizing enzymes responsible for riddelliine metabolism. These results are consistent with those reported on the metabolism of senecionine to DHP by male and female human liver microsomes [48].
In order to examine the relationship between DNA adduct levels and the incidence of hemangiosarcomas, we have measured DHP-derived DNA adduct levels in purified rat and mouse liver endothelial cells, the cells of origin for the hemangiosarcomas [75,76]. F344 rats and B6C3F1 mice were treated by gavage five days per week for two weeks with riddelliine at 1.0 mg/kg for rats and 3.0 mg/kg for mice. One, three, seven, and 28 days after the last dose, the DHP-derived DNA
![Ijms 03 00948 g005]() adduct levels in the endothelial cells were significantly greater than in the parenchymal cells. The DNA adduct levels in rat endothelial cells were greater than in the mouse endothelial cells [75,76]. These results indicate that the riddelliine-induced cell-specific DNA adducts in liver cells correlate with the potency of riddelliine for the induction of liver hemangiosarcomas.
adduct levels in the endothelial cells were significantly greater than in the parenchymal cells. The DNA adduct levels in rat endothelial cells were greater than in the mouse endothelial cells [75,76]. These results indicate that the riddelliine-induced cell-specific DNA adducts in liver cells correlate with the potency of riddelliine for the induction of liver hemangiosarcomas.
Figure 5.
32P-Postlabeling/HPLC analysis of DHP-derived DNA adducts contained in (A) reaction of riddelliine with calf thymus DNA without microsomal activation; (B) modified DNA from reaction of DHP with calf thymus DNA; and (B) livers of rats treated orally gavaged with 1.0 mg/kg/day 5 days/week beginning at weaning and continuing until sacrifice at three months [15]. The eight chromatographic peaks eluted at 47.6, 48.3, 51.4, 53.9, 55.3, 60.1, 61.0, and 62.6 min are the identified DHP-derived DNA adducts designated as P1, P2, P3, P4, P5, P6, P7 and P8, respectively. P4 and P6 are DHP-3'-dGMP adducts, and the other six adducts are DHP-derived dinucleotides.
Figure 5.
32P-Postlabeling/HPLC analysis of DHP-derived DNA adducts contained in (A) reaction of riddelliine with calf thymus DNA without microsomal activation; (B) modified DNA from reaction of DHP with calf thymus DNA; and (B) livers of rats treated orally gavaged with 1.0 mg/kg/day 5 days/week beginning at weaning and continuing until sacrifice at three months [15]. The eight chromatographic peaks eluted at 47.6, 48.3, 51.4, 53.9, 55.3, 60.1, 61.0, and 62.6 min are the identified DHP-derived DNA adducts designated as P1, P2, P3, P4, P5, P6, P7 and P8, respectively. P4 and P6 are DHP-3'-dGMP adducts, and the other six adducts are DHP-derived dinucleotides.
To study the relevance of the mechanistic studies in experimental to humans, metabolism of riddelliine was conducted by male and female human liver microsomes, generating DHP and riddelliine N-oxide as major metabolites [77]. The levels of DHP and riddelliine N-oxide were in quantities comparable to those from rat liver microsomal metabolism. For DHP formation, the kinetic parameters, Vmax and Km, between human and rat liver microsomal metabolism are comparable [77]. When metabolism in the presence of calf thymus DNA, the same set of eight DHP-derived DNA adducts was formed. Both the metabolism pattern and DNA adduct profile are closely similar to those formed from rat liver microsomes. These results strongly indicate the use of rodents for studying the mechanisms of liver tumor induction by riddelliine is highly relevant to humans.
The formation of DHP-derived DNA adducts in blood DNA of rats fed riddelliine was also studied [78]. In the adduct formation and removal experiment, both male and female F344 rats were administered riddelliine by gavage at a single dose of 10.0 mg/kg body weight in 0.1 M phosphate buffer. It was found that during the 48 to 168 hour time-period, the adduct levels in female rat blood were 4-fold greater than those in male rats. In the dose responsive experiment, female rats were gavaged 0.1 and 1.0 mg/kg doses of riddelliine for three consecutive days. The levels of the DHP-derived DNA adducts in blood of rats received 0.1 and 1.0 mg/kg doses were 12.9 and 51.8 adducts/107 nucleotides [78]. These results suggest that: (i) leucocyte DNA can bind with DHP to form the same set of DHP-derived DNA adducts generated in liver; and (ii) DHP-derived DNA adducts in blood can serve as a potential non-invasive biomarkers for assessing the exposure to riddelliine.
2. Formation of DNA cross-linking and DNA-protein cross-linking
It has been shown that a variety of chemicals can bind to DNA in a cross-link fashion both in vivo and in cultured cells. Similarly, a number of bifunctional pyrrolizidine alkaloids and their pyrrolic derivatives have been found to to form DNA cross-linking and DNA-protein cross-links [20,21,22,23,24,25,26]. Nevertheless, rather than in vivo experiments, these studies were mainly conducted in cultured cells, such as cultured bovine kidney epithelial cells.
To study on a structure-activity basis with the mammalian Mardin Darby bovine kidney epithelial cell line, Hincks et al. [21] determined DNA cross-linking and DNA-protein cross-linking capability of three classes of pyrrolizidine alkaloids. These include: (i) macrocycles compounds - seneciphylline, riddelliine, retrorsine, senecionine, and monocrotaline; (ii) open diester compounds - heliosupine and latifoline; and (iii) retronecine. The relative potency in causing DNA cross-linking and DNA-protein linking of these compounds are: seneciphylline > riddelliine > retrorsine > senecionine > heliosupine > monocrotaline > latifoline > retronecine. In most cases, higher levels of DNA cross-linking than the DNA-protein cross linking were found among these compounds. Since cross-linking ability well correlated with their ability to inhibit colony formation and thus was suggested to be involved in the biological activity of these compounds [21].
DNA-protein cross-linking potency of the pyrrolizidine alkaloids tested has also been found to well correlate with known potency differences in animal toxicity [26]. Thus, Kim et al. [26] proposed that DNA-protein cross-linking activity is probably involved in pyrrolizidine alkaloid induced diseases. Since most of these tested compounds are tumorigens, formation of DNA cross-linking and DNA-protein cross-linking may lead to tumor formation. However, further investigation is warranted for confirmation of this possible mechanistic pathway.
3. Formation of endogenous DNA adducts
Chemical carcinogens may exert tumorigenicity through secondary mechanisms, such as oxidative stress, hypomethylation/hypermethylation, induction of lipid peroxidation and formation of endogenous DNA adducts, induction of peroxisome proliferation, and modulation of endocrine disruptors. It was reported that trans-4-hydroxy-2-hexenal was formed by metabolism of senecionine [58,79]. This metabolite may be formed either from the enzymatic cleavage of the senecionine molecule, or from senecionine-induced lipid peroxidation. It is known that trans-4-hydroxy-2-hexenal is highly toxic and other α,β-unsaturated aldehydes (e.g., malondialdehyde, crotonaldehyde etc.) are carcinogenic, mutagenic, and form DNA adducts. Consequently, formation of trans-4-hydroxy-2-hexenal from metabolism of senecionine has been considered an activation pathway [79]. Furthermore, Miranda et al. [59] reported that monocrotaline-induced toxicity can be inhibited by the antioxidant, butylated hydroxyanisole (BHA). As such, these findings suggest that induction of lipid peroxidation by pyrrolizidine alkaloids may be involved in pyrrolizidine alkaloid-induced toxicity, including tumorigenicity. This warrants for further investigation.
Perspectives
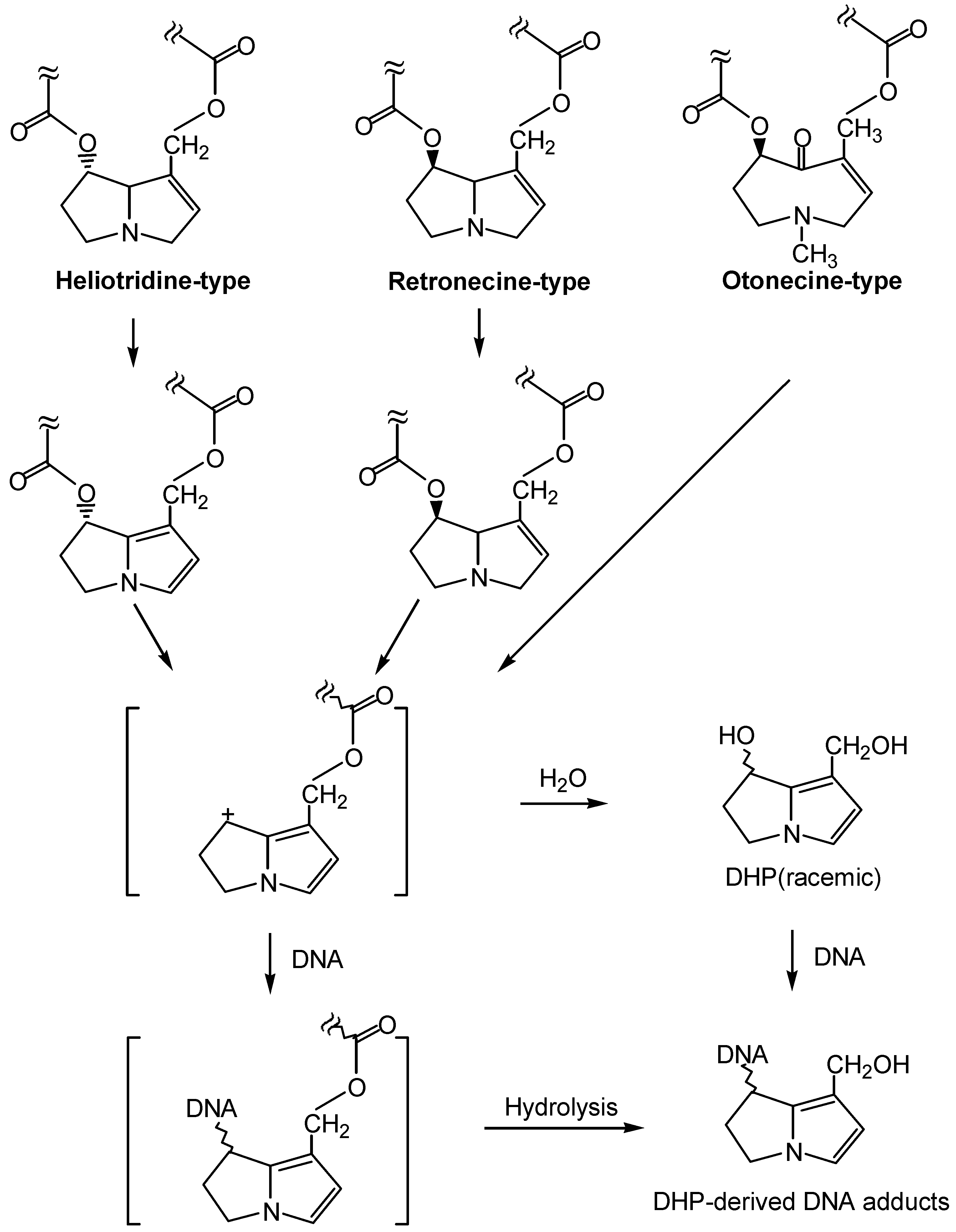
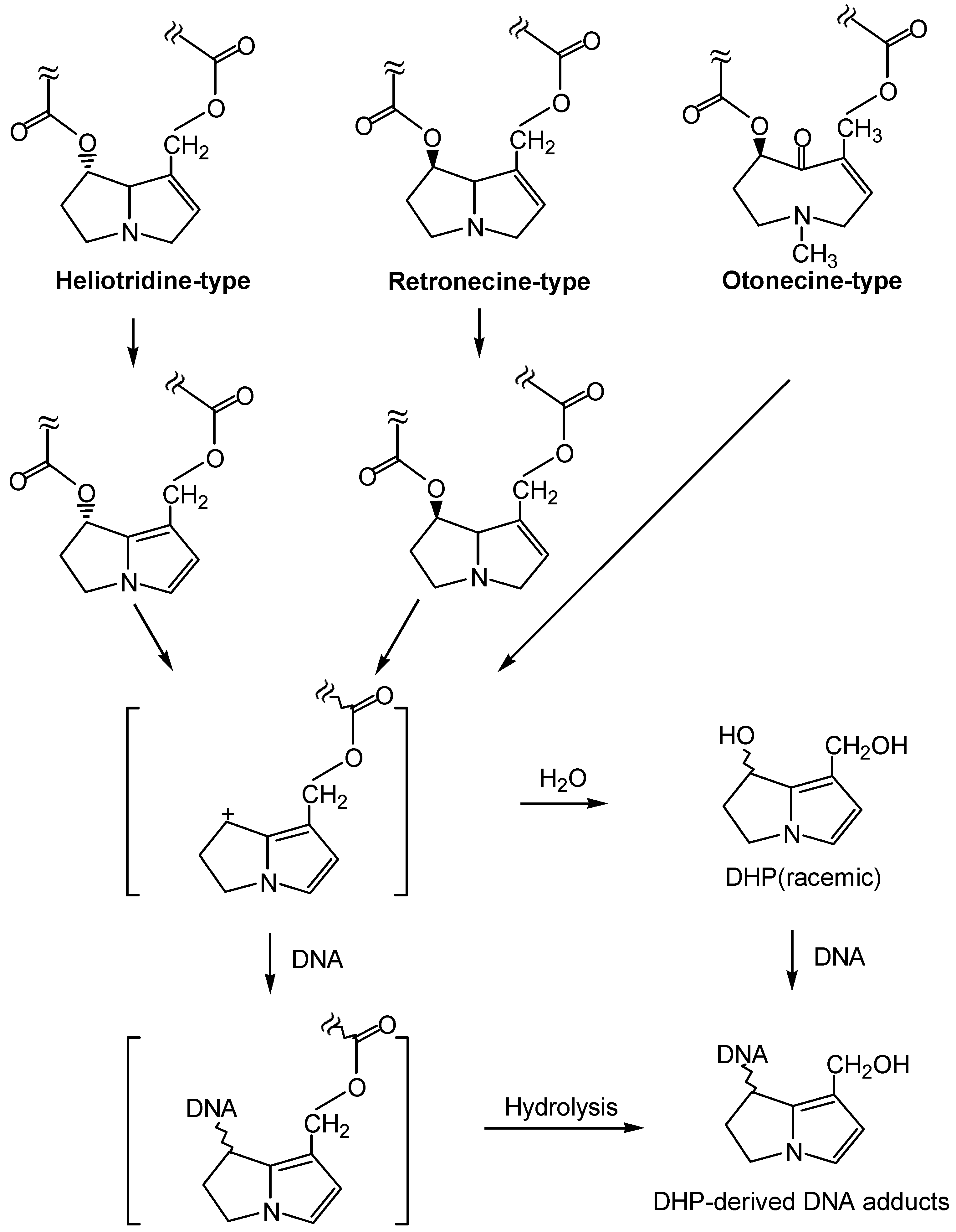
We have recently determined that riddelliine induced liver tumors through a genotoxic mechanism mediated with DHP-derived DNA adduct formation. These eight DHP-derived DNA adducts exhibited dose response for tumor formation. These eight DHP-derived DNA adducts were also formed in liver of rats fed DHR, which itself is a tumorigen (unpublished data). It has been reported that DHP (e.g., DHR) is a common metabolite formed from metabolism of many retronecine-type, heliotridine-type, and otonecine-type pyrrolizidine alkaloids. Based on our findings and the information described above, it is highly possible that this genotoxic mechanism mediated with DHP-derived DNA adduct formation is a general mechanism of induction of tumors by pyrrolizidine alkaloids. The proposed general mechanism is shown in Figure 6. As such, we hypothesize that these DHP-derived DNA adducts are potential biomarkers of pyrrolizidine alkaloid tumorigenicity as well as pyrrolizidine alkaloid exposure. We also hypothesize that these DNA adducts may also be responsible for the other genotoxicities, including mutagenicity and teratogenicity, of pyrrolizidine alkaloids. These hypotheses warrant further investigation.
If this genotoxic mechanism is indeed general in pyrrolizidine alkaloid-induced tumorigenicity and other genotoxicity, inhibition of DNA adduct formation should be one of the most effective intervention strategies for prevention of human health risk posed by pyrrolizidine alkaloids. Based on metabolism point of view, decrease of the formation of pyrrole and DHP formation would inhibit DNA adduct formation. It can be achieved by increase of the processes of hydrolysis and N-oxidation of pyrrolizidine alkaloids, since the resulting necine base and N-oxides are biologically inactive or much less active (Figure 7). Thus, enhancement of the enzyme levels of esterase, mixed function oxygenase, cytochrome 2B6 may play an effective intervention role. Another strategy on intervention is that once the pyrrolic and DHP metabolites are formed, they can be detoxified by reacting with glutathione catalyzed by glutathione transferases (Figure 7).
Figure 6.
A proposed general mechanism leading to an identical set of DHP-derived DNA adduct formation from metabolism of the three major classes of carcinogenic pyrrolizidine alkaloids.
Figure 6.
A proposed general mechanism leading to an identical set of DHP-derived DNA adduct formation from metabolism of the three major classes of carcinogenic pyrrolizidine alkaloids.
Nevertheless, the best strategy of reduction of risk posed by pyrrolizidine alkaloids should be reduction of exposure. Thus, it is important to determine the sources of human exposure and to assess human health risk posed by these compounds. It is known that herbal medicines and dietary supplements, such as comfrey and coltsfoot may contain toxic pyrrolizidine alkaloids. Consequently, since the use of dietary supplements has increased tremendously in the United States and many other countries, they may pose human health risk.
Figure 7.
Invention of pyrrolizidine alkaloid induced carcinogenesis by enzyme modulation.
References
- International Agency for Research in Cancer (IARC). IARC Monograph on the evaluation of carcinogenic risk of chemicals to man - Some naturally occurring substance; International Agency for Research in Cancer: Lyon, France, 1976. [Google Scholar]
- Mattocks, A. R. Nature 1968, 217, 723–728.
- Mattocks, A. R. Academic Press: London, NY, 1986.
- Roeder, E. Pharmazie 1995, 50, 83–98.
- Stegelmeier, B. L.; Edgar, J. A.; Colegate, S. M.; Gardner, D. R.; Schoch, T. K. J. Nat. Toxins 1999, 8, 95–116.
- Woo, Y.-T.; Lai, D. Y.; Arcos, J. C.; Argus, M. F. Academic Press Inc.: San Diego, 1988.
- Roeder, E. Pharmazie 2000, 55, 711–726.
- Smith, L. W.; Culvenor, C. C. J. Nat. Prod. 1981, 44, 129–152. [CrossRef]
- Betz, J. M.; Eppley, R. M.; Taylor, W. C.; Andrzejewski, D. J. Pharm. Sci. 1994, 83, 649–653. [CrossRef]
- Huxtable, R. J. Biol. Med. 1980, 24, 1–14.
- Hirono, I.; Mori, H.; Culvenor, C. C. Gann 1976, 67, 125–129.
- Winship, K. A. Adverse Drug React. Toxicol. Rev. 1991, 10, 47–59.
- Byron, J. Food Chem. News 1998, 14, 6–7.
- Prakash, A. S.; Pereira, T. N.; Reilly, P. E.; Seawright, A. A. Mutat. Res. 1999, 443, 53–67.
- Yang, Y. C.; Yan, J.; Doerge, D. R.; Chan, P. C.; Fu, P. P. Chem. Res. Toxicol. 2001, 14, 101–109. [CrossRef]
- White, I.N.H.; Mattocks, A.R. Biochem. J. 1972, 128, 291–297.
- Eastman, D.F.; Dimenna, G.P.; Segall, H.J. Drug Metab. Disp. 1982, 10, 236–240.
- Petry, T. W.; Bowden, G. T.; Huxtable, R. J.; Sipes, I. G. Cancer Res. 1984, 44, 1505–1509.
- Fu, P.P.; Chou, M.W.; Xia, Q.; Yang, Y.-C.; Yan, J.; Dorege, D.R.; Chan, P.C. Environ. Carcinogen Ecotoxicol. Rev. 2001, C19 (2), 353.
- Coulombe, R. A., Jr.; Drew, G. L.; Stermitz, F. R. Toxicol. Appl. Pharmacol. 1999, 154, 198–202. [CrossRef]
- Hincks, J. R.; Kim, H. Y.; Segall, H. J.; Molyneux, R. J.; Stermitz, F. R. Toxicol. Appl. Pharmacol. 1991, 111, 90–98. [CrossRef]
- Kim, H. Y.; Stermitz, F. R.; Li, J. K.; Coulombe, R. A. Food Chem. Toxicol. 1999, 37, 619–625.
- Pereira, T. N.; Webb, R. I.; Reilly, P. E.; Seawright, A. A.; Prakash, A. S. Nucleic Acids Res. 1998, 26, 5441–5447.
- Reed, R. L.; Ahern, K. G.; Pearson, G. D.; Buhler, D. R. Carcinogenesis 1988, 9, 1355–1361.
- Hincks, J. R.; Coulombe, R. A. Environ. Mol. Mutag. 1989, 13, 211–217. [CrossRef]
- Kim, H. Y.; Stermitz, F. R.; Coulombe, R. A. Carcinogenesis 1995, 16, 2691–2697.
- Rubiolo, P.; Pieters, L.; Calomme, M.; Bicchi, C.; Vlietinck, A.; et al. Mutat. Res. 1992, 281, 143–147. [CrossRef] [PubMed]
- Kuhara, K.; Takanashi, H.; Hirono, I.; Furuya, T.; Asada, Y. Cancer Lett. 1980, 10, 117–122.
- Schoental, R. Cancer Res. 1975, 35, 2020–2024.
- Schoental, R.; Cavanagh, J.B. J. Natl. Cancer Inst. 1972, 49, 665.
- Crout, D. H. J. Chem. Soc. [Perkin 1] 1972, 13, 1602–1607. [CrossRef]
- Schoental, R.; Fowler, M.E.; Coady, A. Cancer res. 1970, 30, 2127.
- Schoental, R.; Hard, G. C.; Gibbard, S. J. Natl. Cancer Inst. 1971, 47, 1037–1044.
- Schoental, R.; Head, M.A.; Peacock, P.R. Br. J. Cancer 1954, 8, 458. [CrossRef]
- Cook, J. W.; Duffy, E.; Schoental, R. Br. J. Cancer 1950, 4, 405–410. [CrossRef] [Green Version]
- Svoboda, D. J.; Reddy, J. K. Cancer Res. 1972, 32, 908–913.
- Rao, M. S.; Reddy, J. K. Br. J. Cancer 1978, 37, 289–293. [CrossRef]
- Hirono, I.; Ueno, I.; Aiso, S.; Yamaji, T.; Haga, M. Cancer Lett. 1983, 20, 191–198. [CrossRef] [PubMed]
- Harris, P. N.; Chen, K. K. Cancer Res. 1970, 30, 2881–2886.
- Chan, P. C. NIH Publication No. 94-3350. 1993.
- Hirono, I.; Mori, H.; Haga, M. J. Natl. Cancer Inst. 1978, 61, 865–868.
- Hirono, I.; Haga, M.; Fujii, M.; Matsuura, S.; Matsubara, N.; et al. J. Natl. Cancer Inst. 1979, 63, 469–472. [PubMed]
- Brink, N.G. Mutat. Res. 1982, 104, 105.
- Jago, M. V.; Edgar, J. A.; Smith, L. W.; Culvenor, C. C. Mol. Pharmacol. 1970, 6, 402–406.
- Mattocks, A. R.; White, I. N. Chem. Biol. Interact. 1971, 3, 383–396. [CrossRef]
- Eastman, D. F.; Segall, H. J. Toxicol Lett 1981, 8, 217–222.
- Williams, D. E.; Reed, R. L.; Kedzierski, B.; Dannan, G. A.; Guengerich, F. P. Drug Metab. Dispos. 1989, 17, 387–392.
- Miranda, C. L.; Reed, R. L.; Guengerich, F. P.; Buhler, D. R. Carcinogenesis 1991, 12, 515–519.
- Chung, W. G.; Buhler, D. R. Toxicol. Appl. Pharmacol. 1994, 127, 314–319. [CrossRef]
- Reid, M. J.; Lame, M. W.; Morin, D.; Wilson, D. W.; Segall, H. J. J. Biochem. Mol. Toxicol. 1998, 12, 157–166.
- Kasahara, Y.; Kiyatake, K.; Tatsumi, K.; Sugito, K.; Kakusaka, I.; et al. J. Cardiovasc. Pharmacol. 1997, 30, 124–129. [CrossRef]
- Chung, W. G.; Miranda, C. L.; Buhler, D. R. Xenobiotica 1995, 25, 929–939.
- Buhler, D. R.; Kedzierski, B. Adv. Exp. Med. Biol. 1986, 197, 611–620.
- Dueker, S. R.; Lame, M. W.; Morin, D.; Wilson, D. W.; Segall, H. J. Drug Metab. Dispos. 1992, 20, 275–280.
- Lame, M. W.; Jones, A. D.; Morin, D.; Segall, H. J. Drug Metab. Dispos. 1991, 19, 516–524.
- Kedzierski, B.; Buhler, D. R. Toxicol. Lett. 1985, 25, 115–119.
- Kedzierski, B.; Buhler, D. R. Chem. Biol. Interact. 1986, 57, 217–222. [CrossRef]
- Segall, H. J.; Wilson, D. W.; Dallas, J. L.; Haddon, W. F. Science 1985, 229, 472–475.
- Miranda, C. L.; Reed, R. L.; Cheeke, P. R.; Buhler, D. R. Toxicol. Appl. Pharmacol. 1981, 59, 424–430. [CrossRef]
- Lin, G.; Cui, Y. Y.; Hawes, E. M. Drug Metab. Disps. 1998, 26, 181–184.
- Yan, C. C.; Huxtable, R. J. Toxicol. Appl. Pharmacol. 1995, 130, 132–139. [CrossRef]
- Yan, C. C.; Huxtable, R. J. Toxicon 1995, 33, 627–634. [CrossRef] [PubMed]
- White, I. N. H. Chem.-Bio. Interact. 1976, 13, 333–342. [CrossRef]
- Lin, G.; Cui, Y. Y.; Hawes, E. M. Drug Metab. Dispos. 2000, 28, 1475–1483.
- Williams, D. E.; Reed, R. L.; Kedzierski, B.; Ziegler, D. M.; Buhler, D. R. Drug Metab. Dispos. 1989, 17, 380–386.
- Miranda, C. L.; Chung, W.; Reed, R. E.; Zhao, X.; Henderson, M. C. Biochem. Biophys. Res. Commun. 1991, 178, 546–552.
- McLean, E.K. Pharmacol. Rev. 1970, 22, 429.
- Huan, J.-Y.; Miranda, C.L.; Buhler, D.R.; Cheeke, P.R. Toxical. Appl. Pharmacol. 1998, 151, 229. [CrossRef]
- White, I. N. H.; Mattocks, A. R.; Butler, W. H. Chem.-Biol. Interact. 1973, 6, 207–218. [CrossRef] [PubMed]
- Robertson, K. A. Cancer. Res. 1982, 42, 8–14.
- Hincks, J.R.; Kim, H.-Y.; Segall, H.J.; Molyneux, R.J.; Stermitz, F.R.; Coulombe, R.A., jr. Toxicol. Appl. Pharmacol. 1991, 111, 90–98. [CrossRef] [PubMed]
- Kim, H.-Y.; Stermitz, F.R.; Molyneux, R.J.; wilson, D.W.; Taylor, D.; Coulombe, R.A., Jr. Toxicol. Appl. Pharmacol. 1993, 122, 61–69. [CrossRef] [PubMed]
- Yang, Y.; Yan, J.; Churchwell, M.; Beger, R.; Chan, P. Chem. Res. Toxicol. 2001, 14, 91–100. [CrossRef]
- Yan, J.; Yang, Y-C.; Williams, L.; Xia, Q.; Doerge, D. R.; Chou, M. W.; Fu, Peter P. 222nd National Meeting of the American Chemical Society, Chicago, USA, August 26-30, 2001.
- Chou, M. W.; Yan, J.; Nichols, J.; Xia, Q.; Beland, F. A.; Fu, P.P.; Chan, P.C. Proc. Am. Assoc. Cancer Res. 2002, 43, 1718.
- Chou, M. W.; Yan, J.; Nichols, J.; Xia, Q.; Beland, F. A.; Fu, P.P.; Chan, P.C. Cancer Lett. (Submitted).
- Xia, Q.; Chou, M. W.; Kadlubar, F. F.; Chan, Po-Cheun; Fu, P. P. Proc. Am. Assoc. Cancer Res. 2002, 43, 1722.
- Yan, J.; Jasyl Nichols, J.; Yang, Y.-C.; Fu, P.P.; Chou, M.W. Int. J. Mol. Sci. submitted for publication.
- Segall, H.J. Science 1985, 229, 472–475.
© 2002 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Fu, P.P.; Xia, Q.; Lin, G.; Chou, M.W. Genotoxic Pyrrolizidine Alkaloids — Mechanisms Leading to DNA Adduct Formation and Tumorigenicity. Int. J. Mol. Sci. 2002, 3, 948-964. https://doi.org/10.3390/i3090948
AMA Style
Fu PP, Xia Q, Lin G, Chou MW. Genotoxic Pyrrolizidine Alkaloids — Mechanisms Leading to DNA Adduct Formation and Tumorigenicity. International Journal of Molecular Sciences. 2002; 3(9):948-964. https://doi.org/10.3390/i3090948
Chicago/Turabian StyleFu, Peter P., Qingsu Xia, Ge Lin, and Ming W. Chou. 2002. "Genotoxic Pyrrolizidine Alkaloids — Mechanisms Leading to DNA Adduct Formation and Tumorigenicity" International Journal of Molecular Sciences 3, no. 9: 948-964. https://doi.org/10.3390/i3090948