
Polydimethylsiloxane Organic–Inorganic Composite Drug Reservoir with Gliclazide




Abstract
:1. Introduction
2. Results
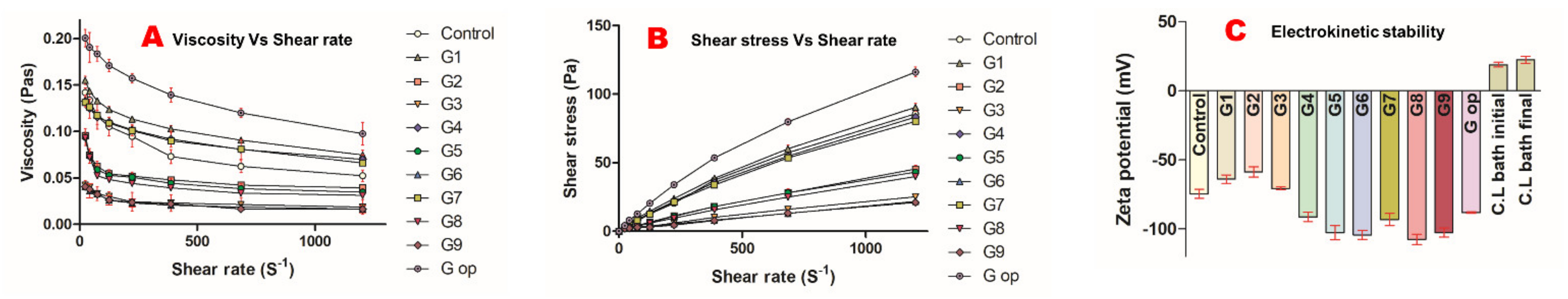
2.1. Rheological Studies
2.2. Electrochemical Stability
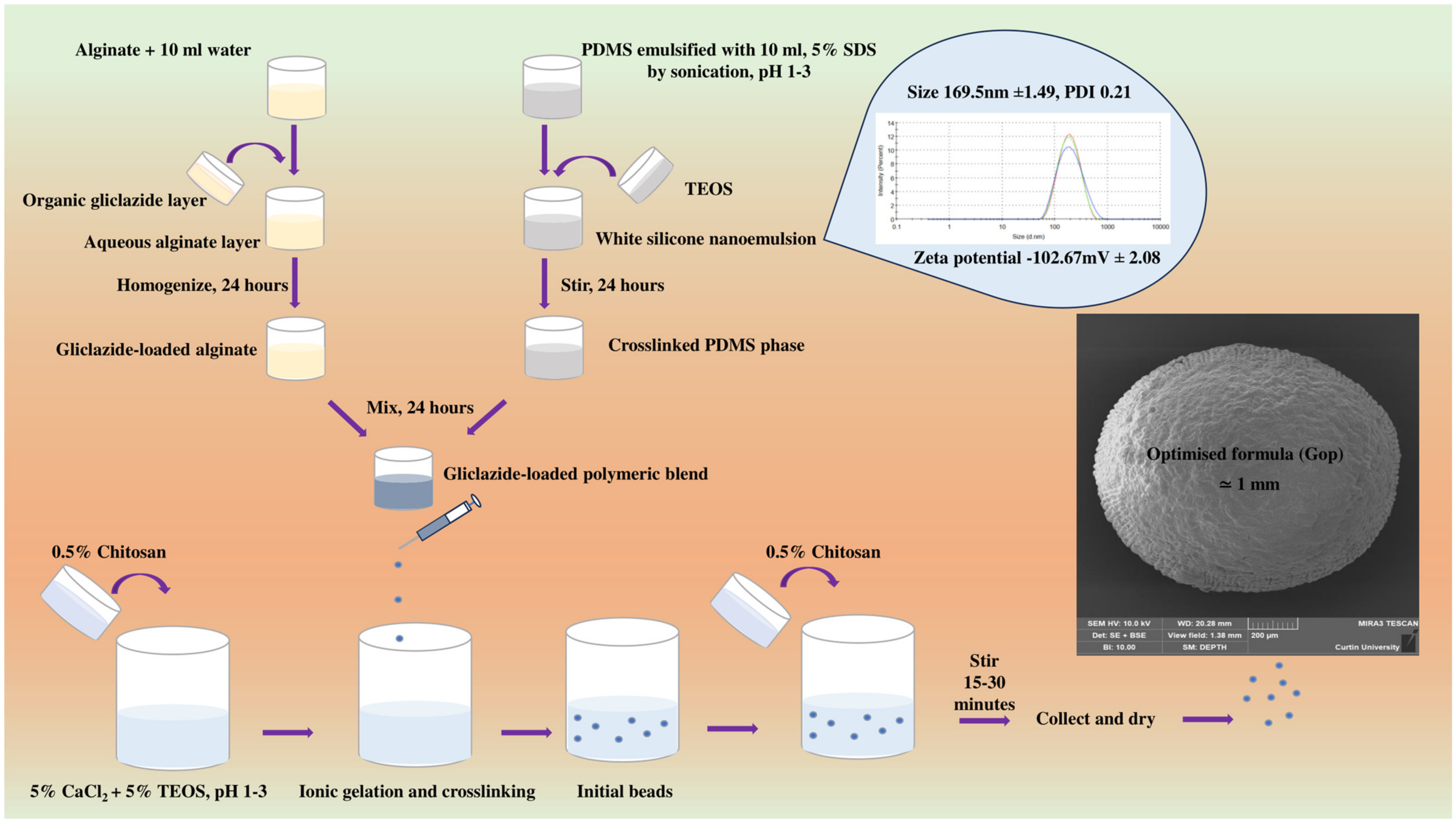
2.3. Characteristics of the Silicone Latex Employed in the Formulation of the Optimised Formula
2.4. Compressibility and Flow Properties of Formulated Beads
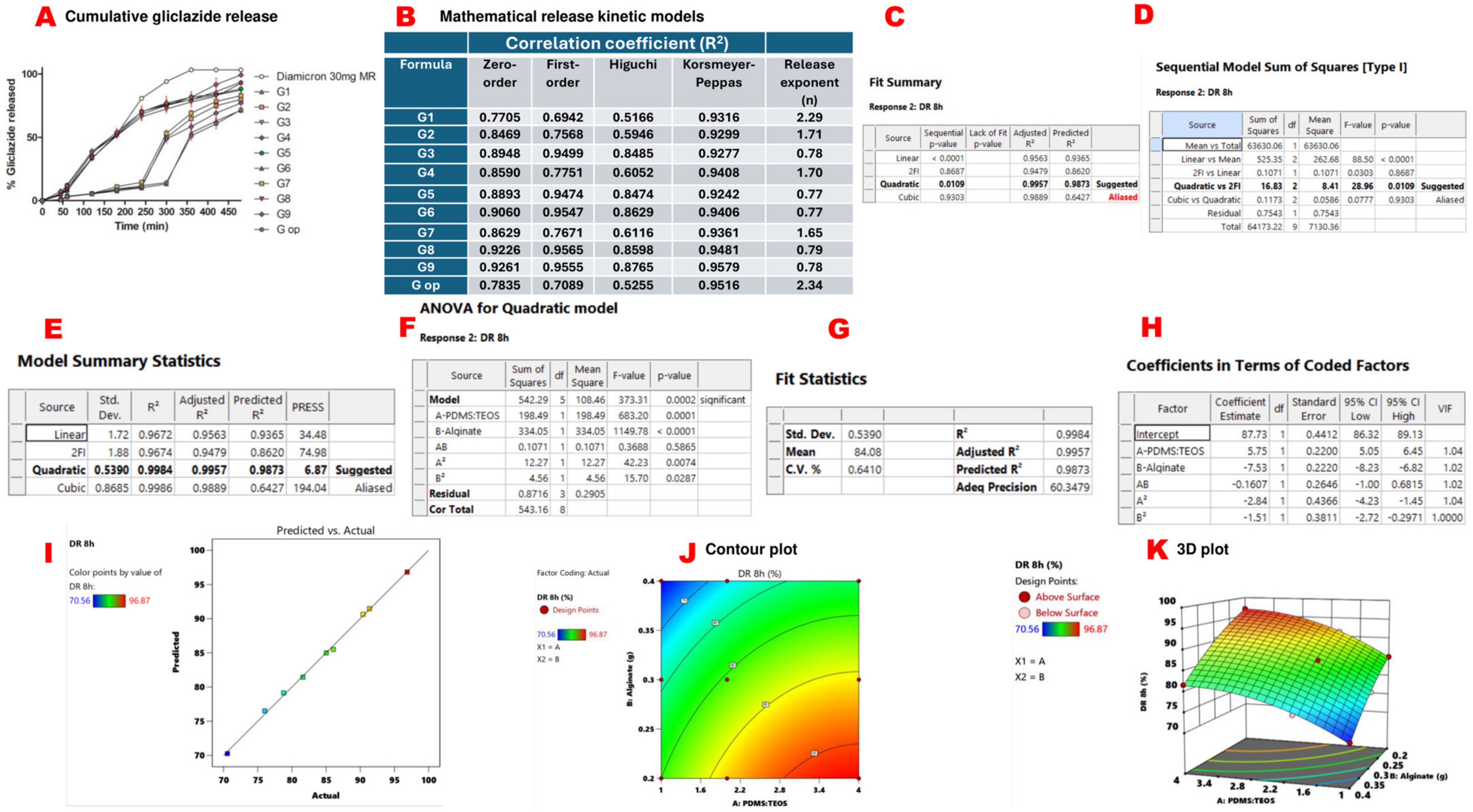
2.5. Bead Content of Gliclazide and Entrapment Efficiency (EE%) and Response Surface Methodology Results for Gliclazide’s EE%
2.6. Microcapsules’ Mechanical Integrity
2.7. Swelling Behaviour
2.8. In Vitro Gliclazide Release, Mathematical Models for Gliclazide Release and Response Surface Methodology Results of Cumulative Gliclazide Release over 8 h (DR 8 h)
2.9. Optical Microscopy and Size Determination of the Beads
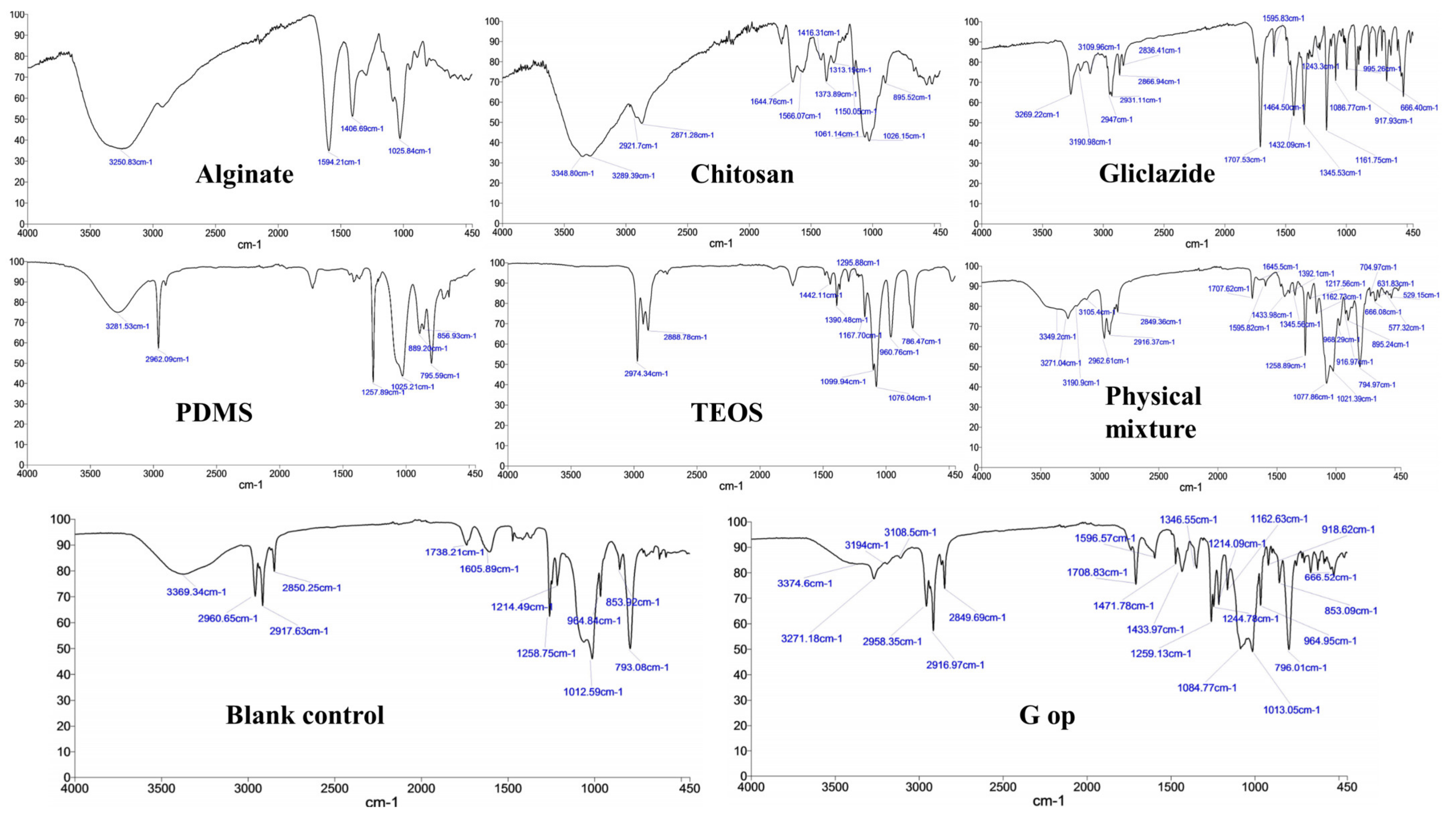
2.10. Bead FTIR Study
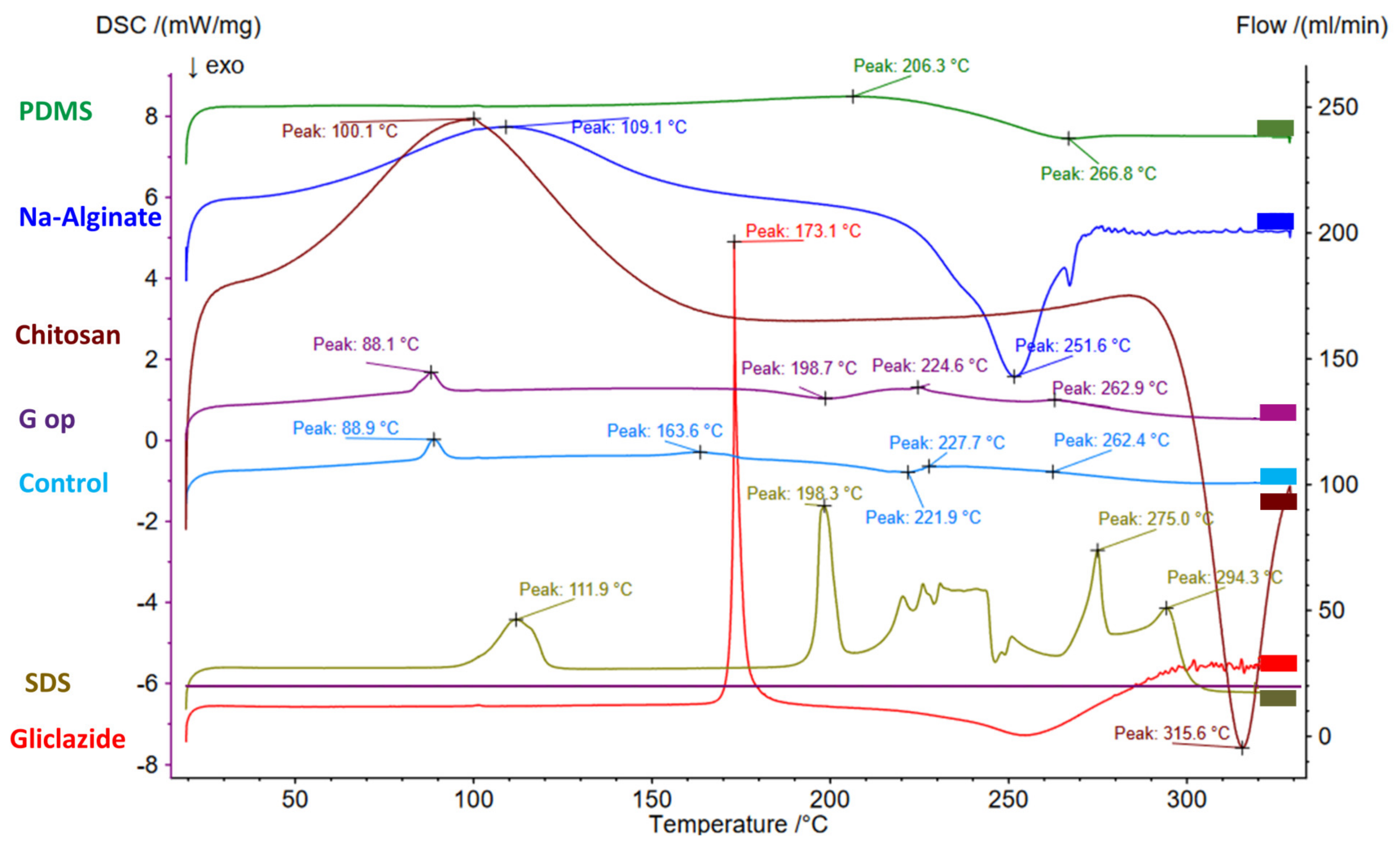
2.11. Bead Thermal Studies via DSC
2.12. SEM/EDXR
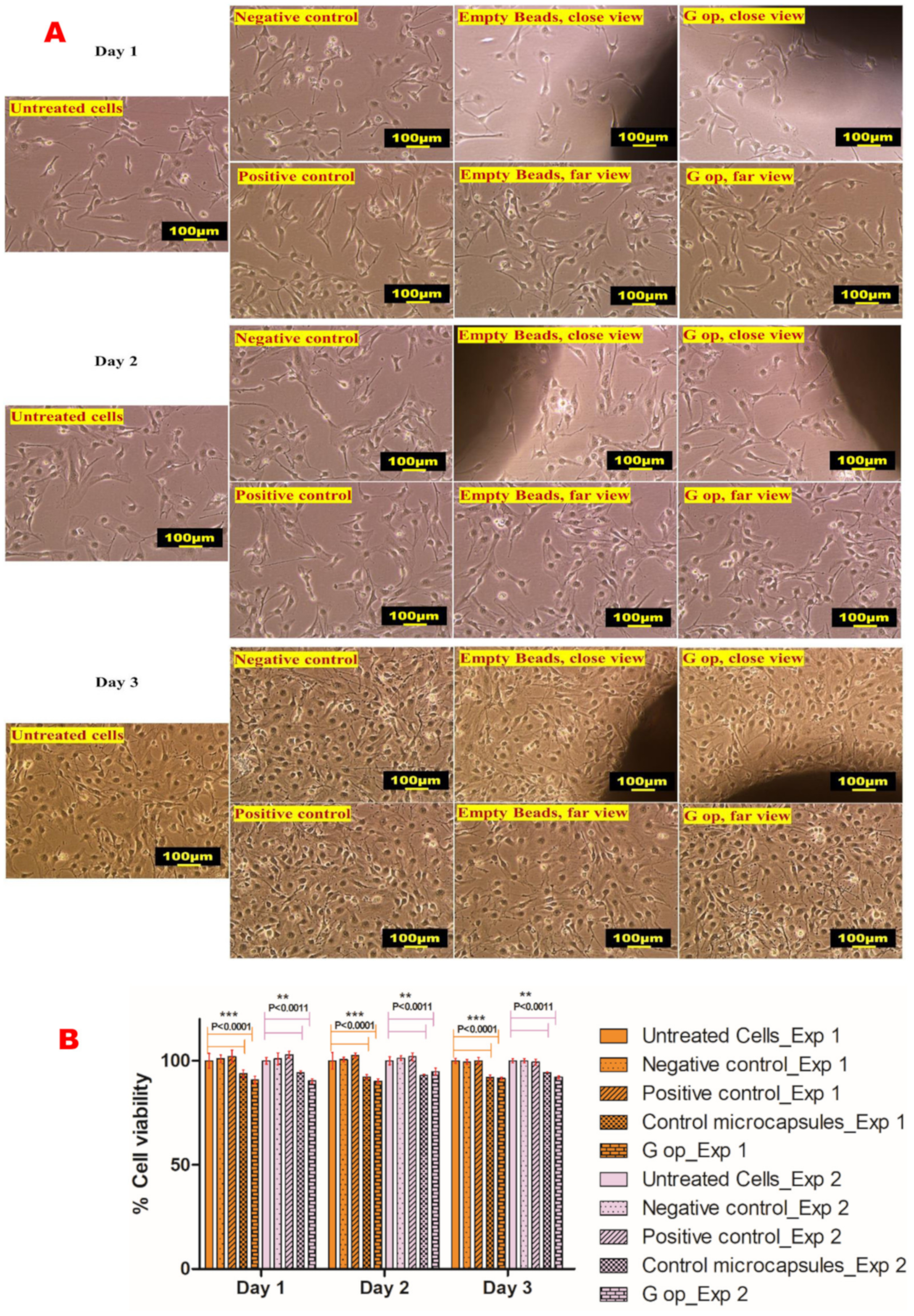
2.13. In Vitro Cell Viability Assay
3. Discussion
4. Materials and Methods
4.1. Microcapsule Preparation
4.2. Experimental Design
4.3. Pre-Encapsulation Assessment of Gliclazide-Loaded Silicone-Grafted Alginate
4.3.1. Rheological Properties
4.3.2. Electrokinetic Stability
4.3.3. Characterisation of the Silicone Latex (Nanoemulsion) Employed in the Optimised Formula (G op)
4.4. Post-Encapsulation Assessment of Gliclazide-Loaded Beads
4.4.1. Flow Properties and Compressibility of Microcapsules
4.4.2. Entrapment Efficiency and Gliclazide Loading/Content
4.4.3. Microcapsule Mechanical Integrity
4.4.4. Swelling Behaviour of Microcapsules
4.4.5. In Vitro Gliclazide Release and Release Kinetics
4.4.6. Optical Microscopy and Bead Size Determination
4.4.7. Fourier Transform Infrared Spectroscopy (FTIR)
4.4.8. Differential Scanning Calorimetry (DSC)
4.4.9. Electron Microscopy (SEM) and Energy Dispersive X-ray (EDXR)
4.4.10. Cell Culture and Viability Assay
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Owen, M. Why silicones behave funny. Chim. Nouv. 2004, 22, 27–33. [Google Scholar]
- Colas, A.; Rafidison, P. Silicones in new pharmaceutical developments, from formulations to manufacturing processes. Pharma. Chem. 2005, 4, 46–49. [Google Scholar]
- Mojsiewicz-Pieńkowska, K. Review of current pharmaceutical applications of polysiloxanes (Silicones). Handb. Polym. Pharm. Technol. Process. Appl. 2015, 2, 363–381. [Google Scholar]
- Pons-Faudoa, F.P.; Ballerini, A.; Sakamoto, J.; Grattoni, A. Advanced implantable drug delivery technologies: Transforming the clinical landscape of therapeutics for chronic diseases. Biomed. Microdevices 2019, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Mohtashami, Z.; Esmaili, Z.; Vakilinezhad, M.A.; Seyedjafari, E.; Akbari Javar, H. Pharmaceutical implants: Classification, limitations and therapeutic applications. Pharm. Dev. Technol. 2020, 25, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Magill, E.; Demartis, S.; Gavini, E.; Permana, A.D.; Thakur, R.R.S.; Adrianto, M.F.; Waite, D.; Glover, K.; Picco, C.J.; Korelidou, A. Solid implantable devices for sustained drug delivery. Adv. Drug Deliv. Rev. 2023, 199, 114950. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Doan, P.D.; Chitre, Y.; Helland, J.R. Method of Manufacturing a Drug-Eluting Endocardial Lead Utilizing Silicone Elastomer as a Drug Carrier. U.S. Patent 7,174,221, 6 February 2007. [Google Scholar]
- Sano, A.; Maeda, H.; Kajihara, M.; Tani, S.; Sugie, T. Long Time Drug-Sustained Release Preparation. U.S. Patent No. 6,756,048, 29 June 2004. [Google Scholar]
- Mashak, A.; Rahimi, A. Silicone polymers in controlled drug delivery systems: A review. Iran. Polym. J. 2009, 18, 279–295. [Google Scholar]
- Watkins, E.S. From breakthrough to bust: The brief life of Norplant, the contraceptive implant. J. Women’s Hist. 2010, 22, 88–111. [Google Scholar] [CrossRef]
- Meirik, O. Implantable contraceptives for women. Contraception 2002, 65, 1–2. [Google Scholar] [CrossRef]
- Ferguson, T.; Needham, G.; Wagner, J. Compudose®: An implant system for growth promotion and feed efficiency in cattle. J. Control. Release 1988, 8, 45–54. [Google Scholar] [CrossRef]
- Hsieh, D.S.; Smith, N.; Chien, Y.W. Subcutaneous controlled delivery of estradiol by Compudose implants: In vitro and in vivo evaluations. Drug Dev. Ind. Pharm. 1987, 13, 2651–2666. [Google Scholar] [CrossRef]
- Cavalieri, J.; Kinder, J.; Fitzpatrick, L. Duration of ovulation suppression with subcutaneous silicone implants containing norgestomet in Bos indicus heifers and cows. Anim. Reprod. Sci. 1998, 51, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Keshvari, H.; Ourang, F.; Mirzadeh, H.; Khorasani, M.; Mansouri, P. Collagen immobilization onto acrylic acid laser-grafted silicone for using as artificial skin: In vitro. Iran. Polym. J. 2008, 17, 171–182. [Google Scholar]
- Okada, T.; Ikada, Y. Modification of silicone surface by graft polymerization of acrylamide with corona discharge. Die Makromol. Chem. Macromol. Chem. Phys. 1991, 192, 1705–1713. [Google Scholar] [CrossRef]
- Lopour, P.; Vondráček, P.; Janatova, V.; Šulc, J.; Vacik, J. Silicone rubber-hydrogel composites as polymeric biomaterials: II. Hydrophilicity and permeability to water-soluble low-molecular-weight compounds. Biomaterials 1990, 11, 397–402. [Google Scholar] [CrossRef]
- Mashak, A. In vitro drug release from silicone rubber–polyacrylamide composite. Silicon Chem. 2008, 3, 295–301. [Google Scholar] [CrossRef]
- Raus, R.A.; Nawawi, W.M.F.W.; Nasaruddin, R.R. Alginate and alginate composites for biomedical applications. Asian J. Pharm. Sci. 2021, 16, 280–306. [Google Scholar] [CrossRef]
- Landa, N.; Miller, L.; Feinberg, M.S.; Holbova, R.; Shachar, M.; Freeman, I.; Cohen, S.; Leor, J. Effect of injectable alginate implant on cardiac remodeling and function after recent and old infarcts in rat. Circulation 2008, 117, 1388–1396. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef]
- Rusdiputra, S.; Wibowo, A.; Asri, L.A.T.W.; Purwasasmita, B.S. Fabrication of Chitosan based-Scaffold as Potential Cornea Implant. IOP Conf. Ser. Mater. Sci. Eng. 2019, 547, 012062. [Google Scholar] [CrossRef]
- Leedy, M.R.; Martin, H.J.; Norowski, P.A.; Jennings, J.A.; Haggard, W.O.; Bumgardner, J.D. Use of chitosan as a bioactive implant coating for bone-implant applications. Chitosan Biomater. II 2011, 244, 129–165. [Google Scholar] [CrossRef]
- Gedawy, A.; Dass, C.R.; Al-Salami, H. Polydimethylsiloxane-customized nanoplatform for delivery of antidiabetic drugs. Ther. Deliv. 2020, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Gedawy, A.; Luna, G.; Martinez, J.; Brown, D.; Al-Salami, H.; Dass, C.R. Novel Silicone-Grafted Alginate as a Drug Delivery Scaffold: Pharmaceutical Characterization of Gliclazide-Loaded Silicone-Based Composite Microcapsules. Pharmaceutics 2023, 15, 530. [Google Scholar] [CrossRef] [PubMed]
- Gedawy, A.; Al-Salami, H.; Dass, C.R. Role of metformin in various pathologies: State-of-the-art microcapsules for improving its pharmacokinetics. Ther. Deliv. 2020, 11, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Abdelbary, A.; El-Gendy, N.; Hosny, A. Microencapsulation approach for orally extended delivery of glipizide: In vitro and in vivo evaluation. Indian J. Pharm. Sci. 2012, 74, 319–330. [Google Scholar] [PubMed]
- Wittaya-areekul, S.; Kruenate, J.; Prahsarn, C. Preparation and in vitro evaluation of mucoadhesive properties of alginate/chitosan microparticles containing prednisolone. Int. J. Pharm. 2006, 312, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gåserød, O.; Smidsrød, O.; Skjåk-Bræk, G. Microcapsules of alginate-chitosan–I: A quantitative study of the interaction between alginate and chitosan. Biomaterials 1998, 19, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Gåserød, O.; Sannes, A.; Skjåk-Bræk, G. Microcapsules of alginate–chitosan. II. A study of capsule stability and permeability. Biomaterials 1999, 20, 773–783. [Google Scholar] [CrossRef]
- Mackenzie, J.D.; Chung, Y.; Hu, Y. Rubbery ormosils and their applications. J. Non Cryst. Solids 1992, 147, 271–279. [Google Scholar] [CrossRef]
- Mackenzie, J.D.; Bescher, E.P. Mechanical Properties of Organic–Inorganic Hybrids. Handb. Sol-Gel Sci. Technol. 2004, 2, 1547–1563. [Google Scholar] [CrossRef]
- Tellez, L.; Rubio, J.; Rubio, F.; Morales, E.; Oteo, J. Synthesis of inorganic-organic hybrid materials from TEOS, TBT and PDMS. J. Mater. Sci. 2003, 38, 1773–1780. [Google Scholar] [CrossRef]
- Hinzmann, A.; Stricker, M.; Gröger, H. Immobilization of aldoxime dehydratases and their use as biocatalysts in aqueous reaction media. Catalysts 2020, 10, 1073. [Google Scholar] [CrossRef]
- Hernández-González, A.C.; Téllez-Jurado, L.; Rodríguez-Lorenzob, L.M. Synthesis of in-situ silica-alginate hybrid hydrogels by a sol-gel route. Carbohydr. Polym. 2020, 250, 116877. [Google Scholar] [CrossRef]
- Pemble, O.J.; Bardosova, M.; Povey, I.M.; Pemble, M.E. A Slot-Die Technique for the Preparation of Continuous, High-Area, Chitosan-Based Thin Films. Polymers 2021, 13, 1566. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-B.; You, J.-O.; Park, H.-Y.; Haam, S.J.; Kim, W.-S. A novel pH-sensitive membrane from chitosan—TEOS IPN; preparation and its drug permeation characteristics. Biomaterials 2001, 22, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Budnyak, T.M.; Pylypchuk, I.V.; Tertykh, V.A.; Yanovska, E.S.; Kolodynska, D. Synthesis and adsorption properties of chitosan-silica nanocomposite prepared by sol-gel method. Nanoscale Res. Lett. 2015, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Essel, T.Y.; Koomson, A.; Seniagya, M.-P.O.; Cobbold, G.P.; Kwofie, S.K.; Asimeng, B.O.; Arthur, P.K.; Awandare, G.; Tiburu, E.K. Chitosan composites synthesized using acetic acid and tetraethylorthosilicate respond differently to methylene blue adsorption. Polymers 2018, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.; Alcock, E.; Buttimer, F.; Schmidt, M.; Clarke, D.; Pemble, M.; Bardosova, M. Synthesis and characterisation of cross-linked chitosan composites functionalised with silver and gold nanoparticles for antimicrobial applications. Sci Technol. Adv. Mater. 2017, 18, 528–540. [Google Scholar] [CrossRef]
- Ryan, C.; Delezuk, J.; Pavinatto, A.; Oliveira, O.; Fudouzi, H.; Pemble, M.; Bardosova, M. Silica-based photonic crystals embedded in a chitosan-TEOS matrix: Preparation, properties and proposed applications. J. Mater. Sci. 2016, 51, 5388–5396. [Google Scholar] [CrossRef]
- Ryan, C.; Bardosova, M.; Pemble, M. Structural and mechanical properties of a range of chitosan-based hybrid networks loaded with colloidal silica and polystyrene particles. J. Mater. Sci. 2017, 52, 8338–8347. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of skeletal muscle in insulin resistance and glucose uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Goodyear, L.J. Exercise and type 2 diabetes: Molecular mechanisms regulating glucose uptake in skeletal muscle. Adv. Physiol. Educ. 2014, 38, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Koistinen, H.A.; Zierath, J. Regulation of glucose transport in human skeletal muscle. Ann. Med. 2002, 34, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Singh, J.; Dey, C.S. PPAR-γ expression modulates insulin sensitivity in C2C12 skeletal muscle cells. Br. J. Pharmacol. 2004, 143, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Dey, C.S. Gliclazide increases insulin receptor tyrosine phosphorylation but not p38 phosphorylation in insulin-resistant skeletal muscle cells. J. Exp. Biol. 2002, 205, 3739–3746. [Google Scholar] [CrossRef] [PubMed]
- Pulido, N.; Suarez, A.; Casanova, B.; Romero, R.; Rodriguez, E.; Rovira, A. Gliclazide treatment of streptozotocin diabetic rats restores GLUT4 protein content and basal glucose uptake in skeletal muscle. Metabolism 1997, 46, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Pulido, N.; Casla, A.; Suarez, A.; Casanova, B.; Arrieta, F.; Rovira, A. Sulphonylurea stimulates glucose uptake in rats through an ATP-sensitive K+ channel dependent mechanism. Diabetologia 1996, 39, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Pulido, N.; Romero, R.; Suárez, A.I.; Rodríguez, E.; Casanova, B.; Rovira, A. Sulfonylureas stimulate glucose uptake through GLUT4 transporter translocation in rat skeletal muscle. Biochem. Biophys. Res Commun. 1996, 228, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.J.; Brogden, R.N. Gliclazide: An update of its pharmacological properties and therapeutic efficacy in non-insulin-dependent diabetes mellitus. Drugs 1993, 46, 92–125. [Google Scholar] [CrossRef]
- Sarkar, A.; Tiwari, A.; Bhasin, P.S.; Mitra, M. Pharmacological and pharmaceutical profile of gliclazide: A review. J. Appl. Pharm. Sci. 2011, 1, 11–19. [Google Scholar]
- Al-Omary, F.A. Gliclazide. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 42, pp. 125–192. [Google Scholar]
- McGavin, J.K.; Perry, C.M.; Goa, K.L. Gliclazide modified release. Drugs 2002, 62, 1357–1364. [Google Scholar] [CrossRef]
- Remedi, M.S.; Nichols, C.G. Chronic antidiabetic sulfonylureas in vivo: Reversible effects on mouse pancreatic β-cells. PLoS Med. 2008, 5, 1473–1485. [Google Scholar] [CrossRef]
- Bordoloi, R.; Ahmed, A.B.; Bhattacharya, K. Pharmacoscintigraphic evaluation and antidiabetic efficacy of gliclazide-loaded 99mTc-labelled mucoadhesive microspheres. Future J. Pharm. Sci. 2021, 7, 229. [Google Scholar] [CrossRef]
- Elsayed, E.W.; El-Ashmawy, A.A.; Mahmoud, K.M.; Mursi, N.M.; Emara, L.H. Modulating gliclazide release and bioavailability utilizing multiparticulate drug delivery systems. J. Pharm. Innov. 2022, 17, 674–689. [Google Scholar] [CrossRef]
- Kuo, A.C. Poly (dimethylsiloxane). Polym. Data Handb. 1999, 2, 411–435. [Google Scholar]
- Sundararajan, S.; Samui, A.B.; Kulkarni, P.S. Crosslinked polymer networks of poly (ethylene glycol)(PEG) and hydroxyl terminated poly (dimethyl siloxane)(HTPDMS) as polymeric phase change material for thermal energy storage. Sol. Energy 2019, 181, 187–194. [Google Scholar] [CrossRef]
- Hadela, A.; Lakić, M.; Potočnik, M.; Košak, A.; Gutmaher, A.; Lobnik, A. Novel reusable functionalized magnetic cobalt ferrite nanoparticles as oil adsorbents. Adsorp. Sci. Technol. 2020, 38, 168–190. [Google Scholar] [CrossRef]
- De, G.; Karmakar, B.; Ganguli, D. Hydrolysis–condensation reactions of TEOS in the presence of acetic acid leading to the generation of glass-like silica microspheres in solution at room temperature. J. Mater. Chem. 2000, 10, 2289–2293. [Google Scholar] [CrossRef]
- Yasmeen, S.; Kabiraz, M.; Saha, B.; Qadir, M.; Gafur, M.; Masum, S. Chromium (VI) ions removal from tannery effluent using chitosan-microcrystalline cellulose composite as adsorbent. Int. Res. J. Pure Appl. Chem. 2016, 10, 1–14. [Google Scholar] [CrossRef]
- Azhary, S.; Purnama, D.; Florena, F.; Vanitha, M.; Panatarani, C.; Joni, I. Synthesis and characterization of chitosan: SiO2 nanocomposite by ultrasonic spray drying. IOP Conf. Ser. Mater. Sci. Eng. 2019, 550, 012037. [Google Scholar] [CrossRef]
- Suratman, A.; Oktaviani, A.; Aprilita, N.; Wibowo, A. Alginate-chitosan hydrogel as controlled release of NPK macronutrient. IOP Conf. Ser. Mater. Sci. Eng. 2019, 578, 012074. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, M.; Lu, L.; Yin, X.; Chen, J.; Jiang, Z. Intensifying esterification reaction between lactic acid and ethanol by pervaporation dehydration using chitosan–TEOS hybrid membranes. Chem. Eng. J. 2009, 155, 800–809. [Google Scholar] [CrossRef]
- Lawrie, G.; Keen, I.; Drew, B.; Chandler-Temple, A.; Rintoul, L.; Fredericks, P.; Grøndahl, L. Interactions between alginate and chitosan biopolymers characterized using FTIR and XPS. Biomacromolecules 2007, 8, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, T.I.; Montaser, A.; Li, S. Effect of cellulose nanocrystals on scaffolds comprising chitosan, alginate and hydroxyapatite for bone tissue engineering. Int. J. Biol. Macromol. 2019, 121, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ramay, H.R.; Hauch, K.D.; Xiao, D.; Zhang, M. Chitosan–alginate hybrid scaffolds for bone tissue engineering. Biomaterials 2005, 26, 3919–3928. [Google Scholar] [CrossRef] [PubMed]
- Katuwavila, N.P.; Perera, A.; Samarakoon, S.R.; Soysa, P.; Karunaratne, V.; Amaratunga, G.A.; Karunaratne, D. Chitosan-alginate nanoparticle system efficiently delivers doxorubicin to MCF-7 cells. J. Nanomater. 2016, 2016, 3178904. [Google Scholar] [CrossRef]
- Hanrahan, G.; Lu, K. Application of factorial and response surface methodology in modern experimental design and optimization. Crit. Rev. Anal. Chem. 2006, 36, 141–151. [Google Scholar] [CrossRef]
- Khuri, A.I.; Mukhopadhyay, S. Response surface methodology. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 128–149. [Google Scholar] [CrossRef]
- Grbic, S.; Parojcic, J.; Ibric, S.; Djuric, Z. In vitro–in vivo correlation for gliclazide immediate-release tablets based on mechanistic absorption simulation. AAPS PharmSciTech 2011, 12, 165–171. [Google Scholar] [CrossRef]
- Zhou, B.; Teng, D.; Li, J.; Zhang, Y.; Qi, M.; Hong, M.; Ren, G.-B. Development of a gliclazide ionic liquid and its mesoporous silica particles: An effective formulation strategy to improve oral absorption properties. RSC Adv. 2022, 12, 1062–1076. [Google Scholar] [CrossRef]
- Manca, M.L.; Loy, G.; Zaru, M.; Fadda, A.M.; Antimisiaris, S.G. Release of rifampicin from chitosan, PLGA and chitosan-coated PLGA microparticles. Colloids Surf. B Biointerfaces 2008, 67, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Heichal-Segal, O.; Rappoport, S.; Braun, S. Immobilization in alginate-silicate sol-gel matrix protects β-glucosidase against thermal and chemical denaturation. Bio/Technology 1995, 13, 798–800. [Google Scholar] [CrossRef]
- Ge, M.; Li, X.; Li, Y.; Jahangir Alam, S.; Gui, Y.; Huang, Y.; Cao, L.; Liang, G.; Hu, G. Preparation of Magadiite-Sodium Alginate Drug Carrier Composite by Pickering-Emulsion-Templated-Encapsulation Method and Its Properties of Sustained Release Mechanism by Baker–Lonsdale and Korsmeyer–Peppas Model. J. Polym. Environ. 2022, 30, 3890–3900. [Google Scholar] [CrossRef]
- Gooneh-Farahani, S.; Naghib, S.M.; Naimi-Jamal, M.R. A novel and inexpensive method based on modified ionic gelation for pH-responsive controlled drug release of homogeneously distributed chitosan nanoparticles with a high encapsulation efficiency. Fibers Polym. 2020, 21, 1917–1926. [Google Scholar] [CrossRef]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Singhvi, G.; Singh, M. In-vitro drug release characterization models. Int. J. Pharm. Stud. Res. 2011, 2, 77–84. [Google Scholar]
- Nasr, M.; Almawash, S.; Al Saqr, A.; Bazeed, A.Y.; Saber, S.; Elagamy, H.I. Bioavailability and antidiabetic activity of gliclazide-loaded cubosomal nanoparticles. Pharmaceuticals 2021, 14, 786. [Google Scholar] [CrossRef] [PubMed]
- Dudek, G.; Turczyn, R. New type of alginate/chitosan microparticle membranes for highly efficient pervaporative dehydration of ethanol. RSC Adv. 2018, 8, 39567–39578. [Google Scholar] [CrossRef] [PubMed]
- Brawer, A. Silicon and matrix macromolecules: New research opportunities for old diseases from analysis of potential mechanisms of breast implant toxicity. Med. Hypotheses 1998, 51, 27–35. [Google Scholar] [CrossRef]
- Brawer, A. Is silicone breast implant toxicity an extreme form of a more generalized toxicity adversely affecting the population as a whole. Int. Ann. Med. 2017, 1, 1–7. Available online: https://www.explantcostarica.com/wp-content/uploads/2017/11/Vague-Syndromes.pdf (accessed on 30 December 2023). [CrossRef]
- Herdman, R.; Ernster, V.; Bondurant, S. Safety of Silicone Breast Implants; National Academy Press: Washington, DC, USA, 1999. [Google Scholar]
- Sreekantan, S.; Hassan, M.; Sundera Murthe, S.; Seeni, A. Biocompatibility and Cytotoxicity Study of Polydimethylsiloxane (PDMS) and Palm Oil Fuel Ash (POFA) sustainable super-hydrophobic coating for biomedical applications. Polymers 2020, 12, 3034. [Google Scholar] [CrossRef] [PubMed]
- Miranda, I.; Souza, A.; Sousa, P.; Ribeiro, J.; Castanheira, E.M.; Lima, R.; Minas, G. Properties and applications of PDMS for biomedical engineering: A review. J. Funct. Biomater. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Khadivi, P.; Salami-Kalajahi, M.; Roghani-Mamaqani, H. Evaluation of in vitro cytotoxicity and properties of polydimethylsiloxane-based polyurethane/crystalline nanocellulose bionanocomposites. J. Biomed. Mater. Res. A 2019, 107, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Baipaywad, P.; Kim, Y.; Wi, J.-S.; Paik, T.; Park, H. Size-controlled synthesis, characterization, and cytotoxicity study of monodisperse poly (dimethylsiloxane) nanoparticles. J. Ind. Eng. Chem. 2017, 53, 177–182. [Google Scholar] [CrossRef]
- Victor, A.; Ribeiro, J.; Araújo, F.F. Study of PDMS characterization and its applications in biomedicine: A review. J. Mechan. Eng. Biomechan. 2019, 4, 1–9. [Google Scholar] [CrossRef]
- Onnekink, C.; Kappel, R.M.; Boelens, W.C.; Pruijn, G.J. Low molecular weight silicones induce cell death in cultured cells. Sci. Rep. 2020, 10, 9558. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Ghosh, I.; Mukherjee, A. Poly (dimethylsiloxane) Induces Cytotoxicity and Genotoxicity in Human Lymphocytes. Proc. Zool. Soc. 2020, 73, 82–85. [Google Scholar] [CrossRef]
- Gedawy, A.; Al-Salami, H.; Dass, C.R. Development and validation of a new analytical HPLC method for simultaneous determination of the antidiabetic drugs, metformin and gliclazide. J. Food Drug Anal. 2019, 27, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Gedawy, A.; Al-Salami, H.; Dass, C.R. Advanced and multifaceted stability profiling of the first-line antidiabetic drugs metformin, gliclazide and glipizide under various controlled stress conditions. Saudi Pharm. J. 2020, 28, 362–368. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- ISO10993-5:2009; Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. ISO: Geneva, Switzerland, 2009. Available online: https://www.iso.org/obp/ui/#iso:std:iso:10993:-5:ed-3:v1:en (accessed on 30 December 2023).
- Gola, J. Quality control of biomaterials—Overview of the relevant technologies. In Stem Cells and Biomaterials for Regenerative Medicine; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Code | PDMS: TEOS (g: g) | Alginate (g) | EE% (Mean ± SD, n = 3) | %DR 8 h (Mean ± SD, n = 3) | Carr’s Index (Mean ± SD, n = 3) | Hausner Ratio (Mean ± SD, n = 3) |
|---|---|---|---|---|---|---|
| Control | 0.8: 0.8 | 0.4 | - | - | 11.07 ± 1.28 | 1.12 ± 0.016 |
| G1 | 0.8: 0.8 | 0.4 | 93.20 ± 0.24 | 70.56 ± 0.36 | 12.29 ± 1.21 | 1.14 ± 0.015 |
| G2 | 0.8: 0.8 | 0.3 | 86.29 ± 0.13 | 78.79 ± 0.82 | 11.16 ± 2.25 | 1.13 ± 0.028 |
| G3 | 0.8: 0.8 | 0.2 | 80.18 ± 0.19 | 85.04 ± 0.19 | 11.89 ± 0.49 | 1.14 ± 0.006 |
| G4 | 0.8: 0.4 | 0.4 | 88.03 ± 0.31 | 76.03 ± 0.60 | 12.18 ± 0.88 | 1.14 ± 0.011 |
| G5 | 0.8: 0.4 | 0.3 | 80.93 ± 0.60 | 86.07 ± 0.15 | 11.05 ± 1.59 | 1.12 ± 0.020 |
| G6 | 0.8: 0.4 | 0.2 | 75.15 ± 0.18 | 91.36 ± 0.11 | 11.66 ± 1.58 | 1.13 ± 0.021 |
| G7 | 0.8: 0.2 | 0.4 | 82.88 ± 0.33 | 81.62 ± 0.83 | 11.82 ± 1.13 | 1.13 ± 0.014 |
| G8 | 0.8: 0.2 | 0.3 | 76.01 ± 0.24 | 90.41 ± 0.59 | 11.75 ± 1.33 | 1.13 ± 0.017 |
| G9 | 0.8: 0.2 | 0.2 | 72.32 ± 0.47 | 96.87 ± 0.16 | 11.90 ± 0.57 | 1.14 ± 0.007 |
| G op | 0.8: 0.437 | 0.493 | 96.98 Predicted/estimated | 64.55 Predicted/estimated | - | - |
| G op (Practical) | 93.48 ± 0.19 Observed/practical | 70.29 ± 0.18 Observed/practical | 12.21 ± 0.31 | 1.14 ± 0.004 | ||
| −3.61% | 8.89% | - | - | |||
| Ingredient | Functional Group | Appeared as FTIR Peak/Band at | Reference |
|---|---|---|---|
| Na-alginate | -OH stretching vibration from carboxyl group | 3250.83 cm−1 (broad peak) | [25,56,57] |
| Carboxylate stretching vibration, asymmetric | 1594.21 cm−1 | ||
| Carboxylate stretching vibration, symmetric | 1406.69 cm−1 | ||
| stretching vibration (-C-O-C-) | 1025.84 cm−1 | ||
| Chitosan | -OH stretching | 3348.80 cm−1 | [38,62,63] |
| -NH stretching | 3289.39 cm−1 | ||
| Aliphatic -CH stretching | 2921.7 cm−1, 2871.28 cm−1 | ||
| Amide I band (-C=O stretching) | 1644.76 cm−1 | ||
| Amide II band (bending vibration/deformation of -NH2) | 1566.07 cm−1 | ||
| -CH bending/wagging | 1416.31 cm−1 | ||
| -OH bending | 1373.89 cm−1 | ||
| Amide III band (-CN stretching) | 1313.19 cm−1 | ||
| Asymmetric stretching vibration of glycosidic linkage (-C-O-C-) | 1150.05 cm−1 | ||
| Skeletal vibration involving (-CO stretching) | 1061.14 cm−1 and 1026.15 cm−1 | ||
| Related to saccharide structure | 895.52 cm−1 | ||
| PDMS elastomer | Silanol (-Si-OH) | 3281.53 cm−1, broad peak | [25,58,59] |
| (-Si-CH3) | 2962.09 cm−1, sharp peak | ||
| (-Si (CH3)2), symmetric stretching | 1257.89 cm−1, sharp peak | ||
| (-Si-O-Si-), asymmetric stretching | 1025.21 cm−1 | ||
| Silanol (-Si-OH) | 889.2 cm−1 | ||
| (-Si-CH3) | 856.93 cm−1 | ||
| Asymmetric bending (-Si-(CH3)2) | 795.59 cm−1, sharp | ||
| TEOS | Aliphatic (-CH stretching) in the ester group | 2974.34 cm−1 and 2888.78 cm−1 | [25,60,61] |
| Asymmetric wagging/bending (-CH) | Small peaks at 1442.11 cm−1, 1390.48 cm−1 and 1295.88 cm−1 | ||
| CH3 rocking | 1167.7 cm−1 | ||
| Asymmetric stretching of Si attached ethoxy group (Si-O-C-O-) | 1099.94 cm−1 | ||
| -CH rocking | 960.76 cm−1 | ||
| Gliclazide | -NH | 3269.22 cm−1 | [25,56,57] |
| -CH, aromatic | 3190.98 cm−1 and 3109.96 cm−1 | ||
| Aliphatic perhydro-cyclopenta pyrrole ring (-CH stretching) | Peaks in the range 2947 cm−1–2836.41 cm−1 | ||
| Carbonyl stretch -C=O | 1707.53 cm−1, sharp peak | ||
| -NH bending | 1595.83 cm−1 | ||
| C=C stretching, aromatic | 1432.09 cm−1 | ||
| Sulfonyl stretching (-S=O), asymmetric | 1345.53 cm−1 | ||
| Heterocyclic C-N ring stretch | 1243.3 cm−1 | ||
| Sulfonyl vibration(-S=O), symmetric | 1161.75 cm−1 | ||
| -C-O stretching | 1086.77 cm−1 | ||
| C=C bending | 995.26 cm−1 | ||
| Phenyl, aromatic P substitution | 917.93 cm−1 | ||
| Aromatic ring | 666.40 cm−1 |
| Ingredient | DSC Thermogram Peaks |
|---|---|
| PDMS | Endothermic peak at 206.3 °C Exothermic peak at 266.8 °C |
| SDS | Early endothermic peak at 111.9 °C (dehydration) Sharp endothermic melting peak at 198.3 °C Late peaks at 275 °C and 294.3 °C (decomposition) |
| Na-alginate | Broad endothermic peak at 109.1 °C (dehydration) Exothermic peak at 251.6 °C (polymer decomposition) |
| Chitosan | Broad endothermic peak at 100.1 °C (dehydration) Exothermic peak at 315.6 °C (polymer decomposition) |
| Gliclazide | Sharp endothermic melting peak at 173.1 °C |
| Control microcapsule | Endothermic peaks at 88.1 °C, 224.6 °C and 262.9 °C Exothermic peak at 198.7 °C |
| G op | Endothermic peaks at 88.9 °C, 163.6 °C, 227.7 °C and 262.4 °C Exothermic peak at 221.9 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gedawy, A.; Al-Salami, H.; Dass, C.R. Polydimethylsiloxane Organic–Inorganic Composite Drug Reservoir with Gliclazide. Int. J. Mol. Sci. 2024, 25, 3991. https://doi.org/10.3390/ijms25073991
Gedawy A, Al-Salami H, Dass CR. Polydimethylsiloxane Organic–Inorganic Composite Drug Reservoir with Gliclazide. International Journal of Molecular Sciences. 2024; 25(7):3991. https://doi.org/10.3390/ijms25073991
Chicago/Turabian StyleGedawy, Ahmed, Hani Al-Salami, and Crispin R. Dass. 2024. "Polydimethylsiloxane Organic–Inorganic Composite Drug Reservoir with Gliclazide" International Journal of Molecular Sciences 25, no. 7: 3991. https://doi.org/10.3390/ijms25073991
APA StyleGedawy, A., Al-Salami, H., & Dass, C. R. (2024). Polydimethylsiloxane Organic–Inorganic Composite Drug Reservoir with Gliclazide. International Journal of Molecular Sciences, 25(7), 3991. https://doi.org/10.3390/ijms25073991