
Synthesis and Characterization of Amine and Aldehyde-Containing Copolymers for Enzymatic Crosslinking of Gelatine


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
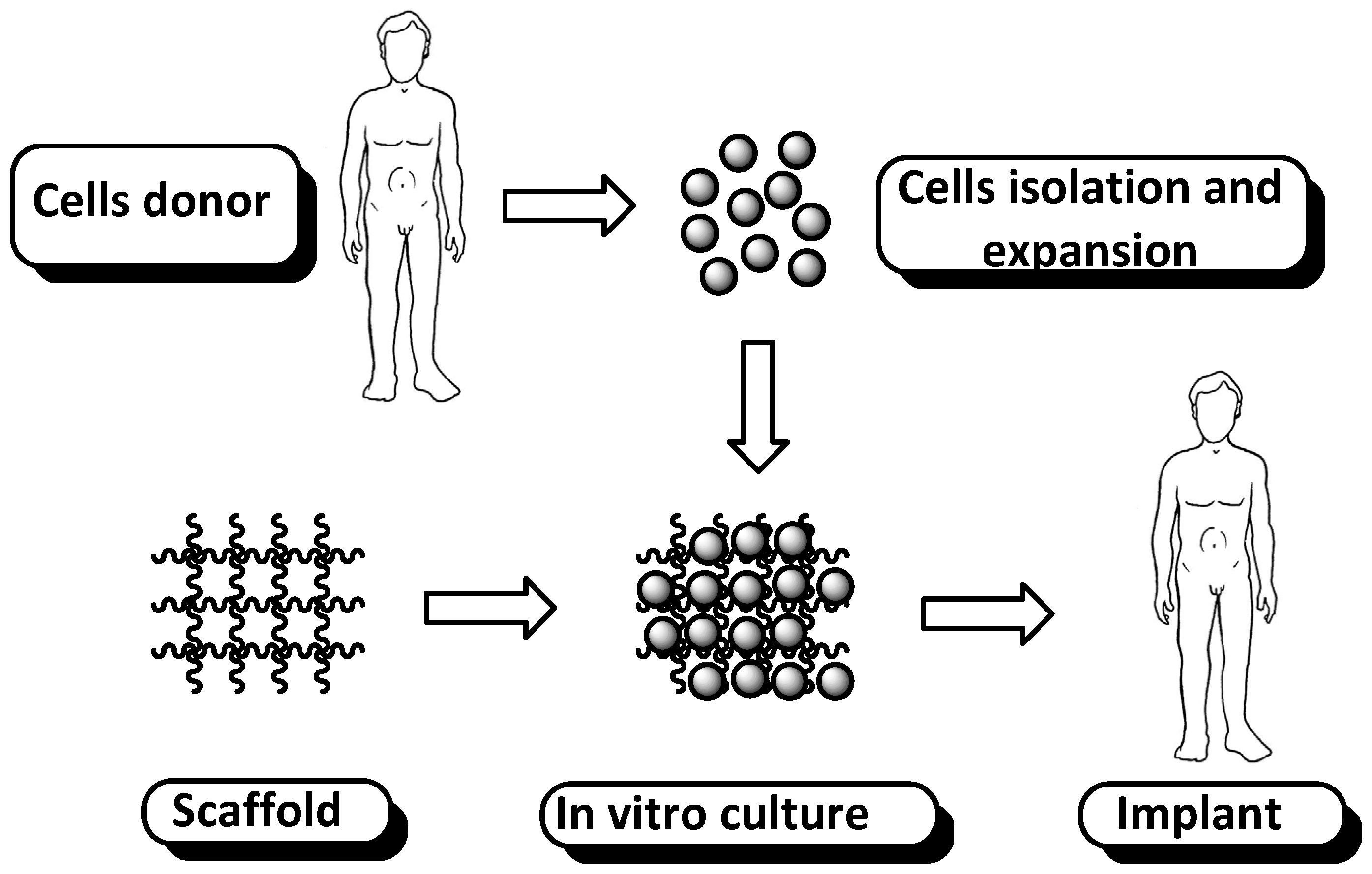
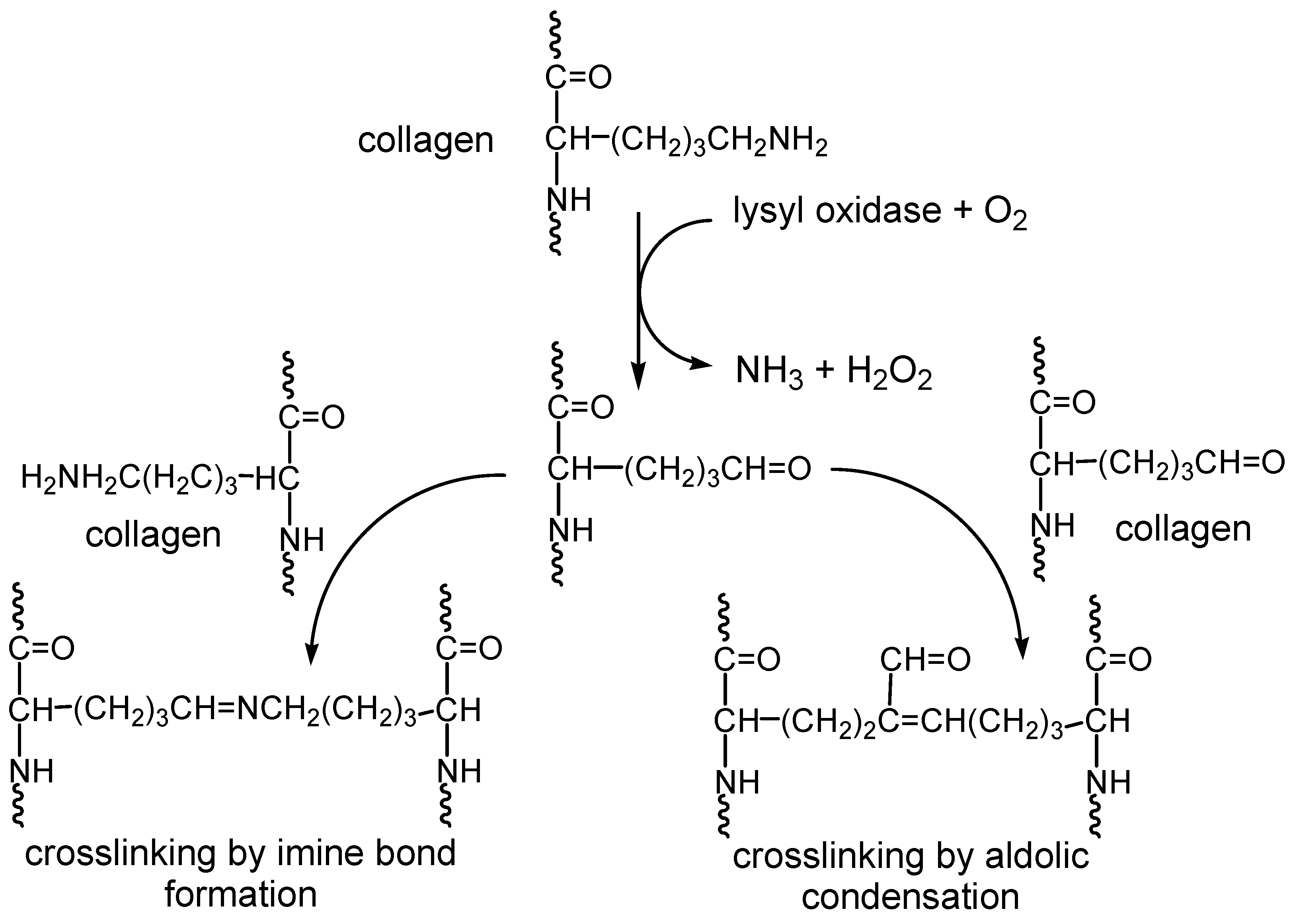
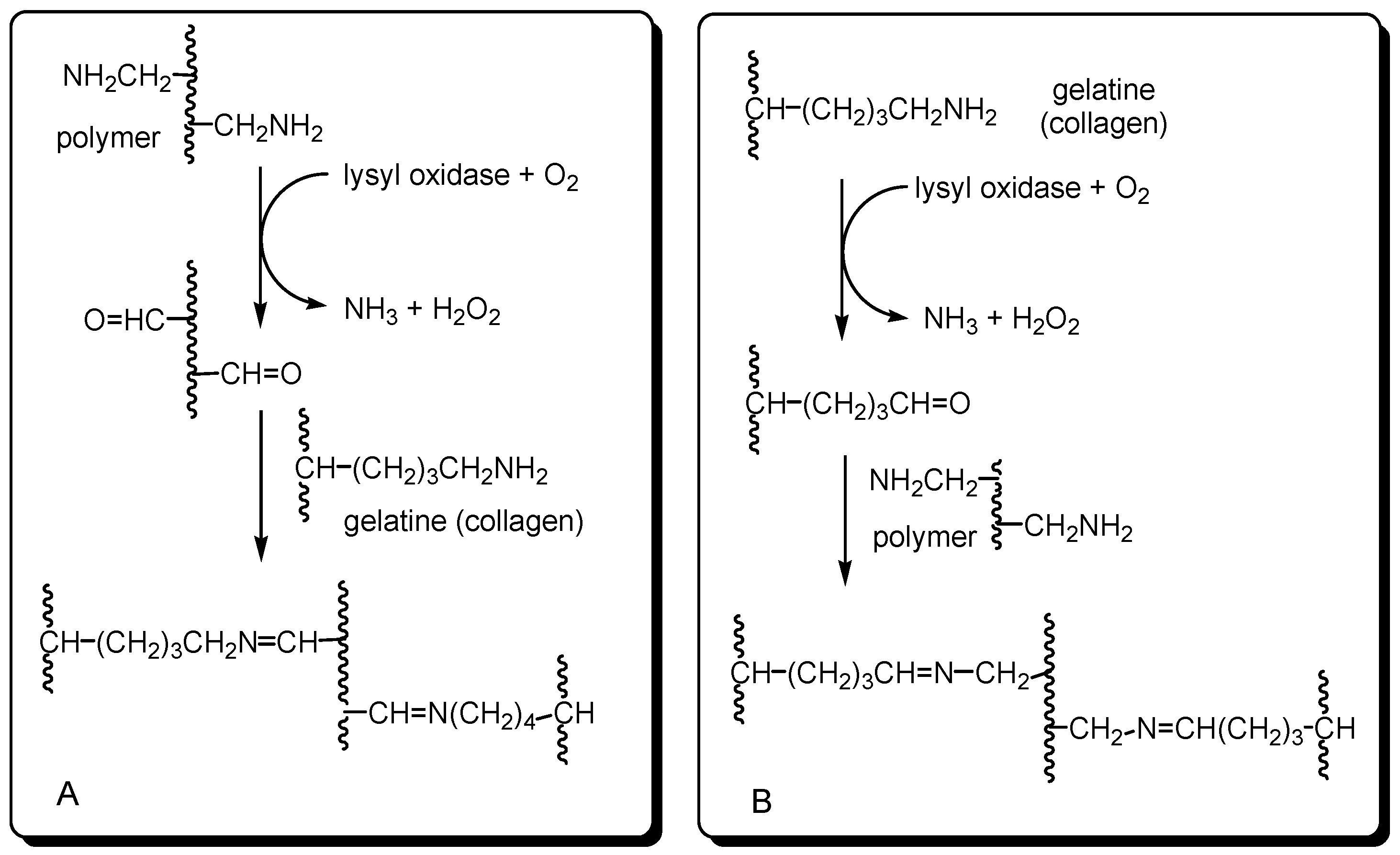
1. Introduction
2. Results and Discussion
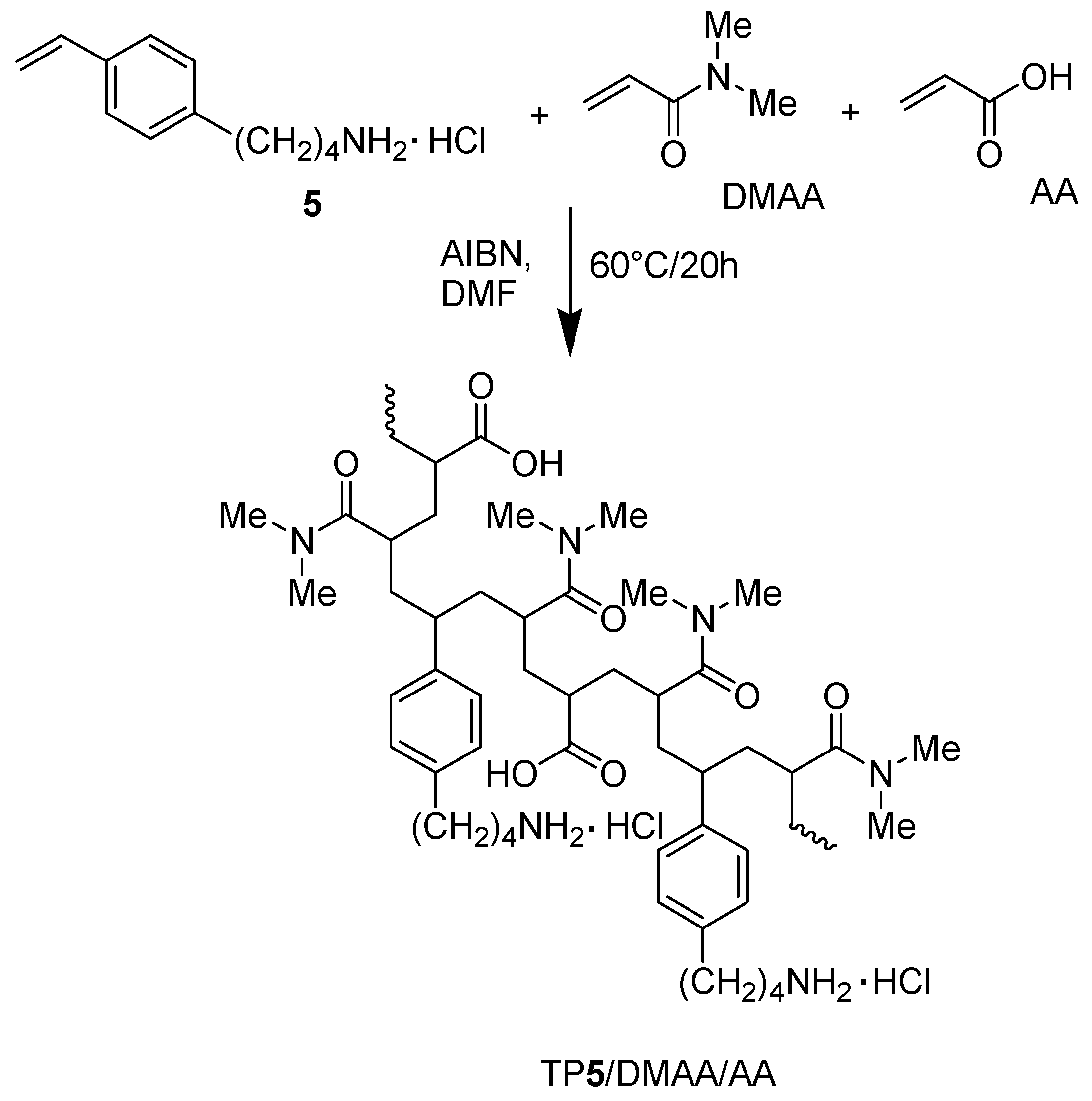
2.1. Synthesis and Characterization of Styrene-Based Monomer 5 and Its Copolymers
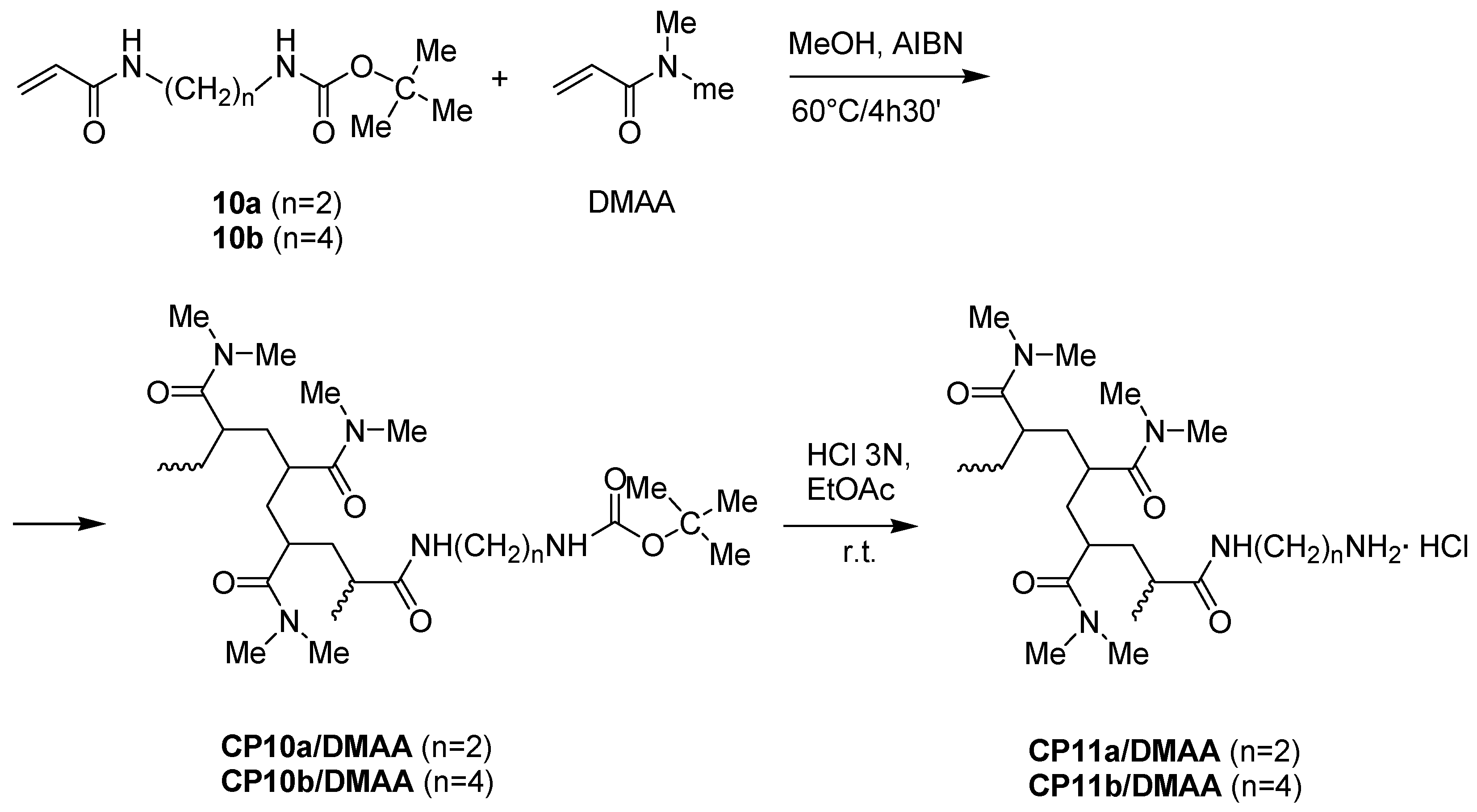
2.2. Synthesis and Characterization of Amidoamine Monomers 11a–c and Their Copolymers with DMAA
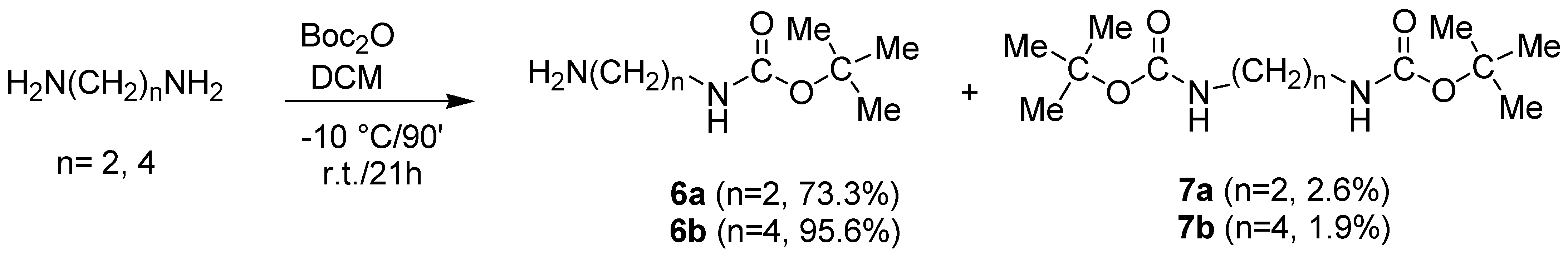
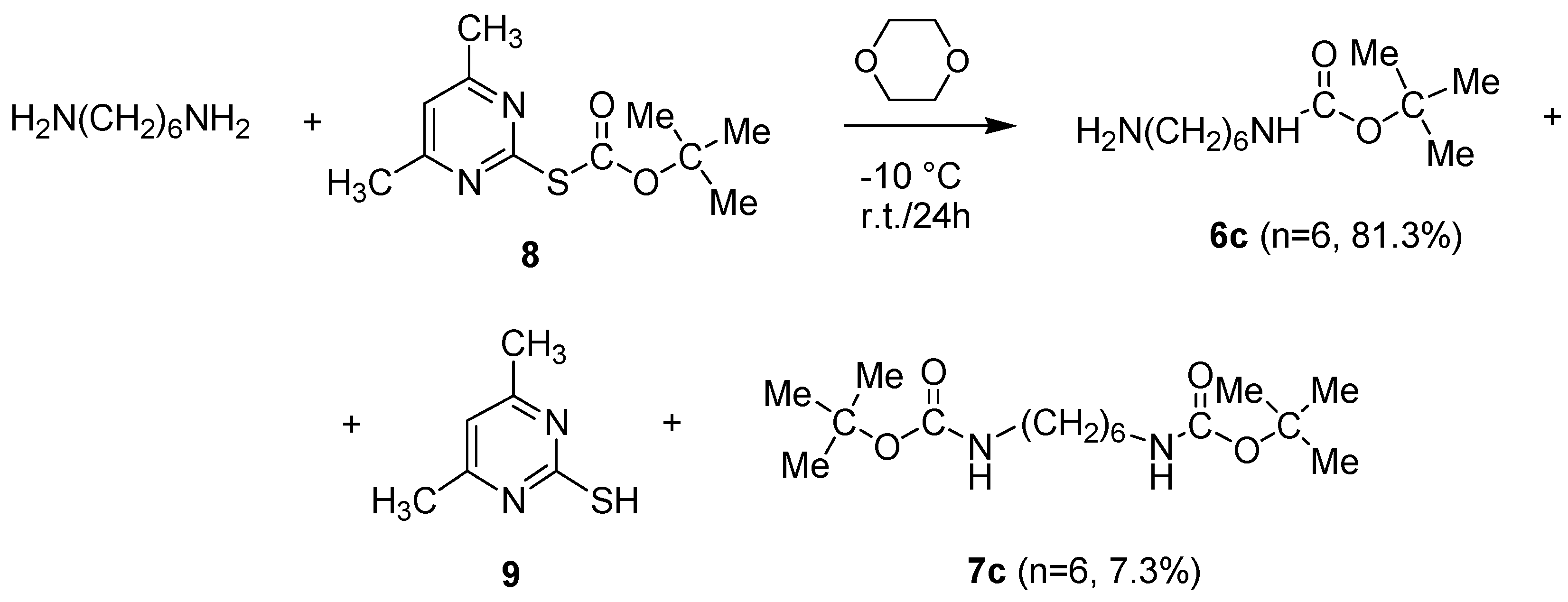
2.2.1. Synthesis of N-Boc-Protected α-ω-di-Aminoalkanes 6a–c
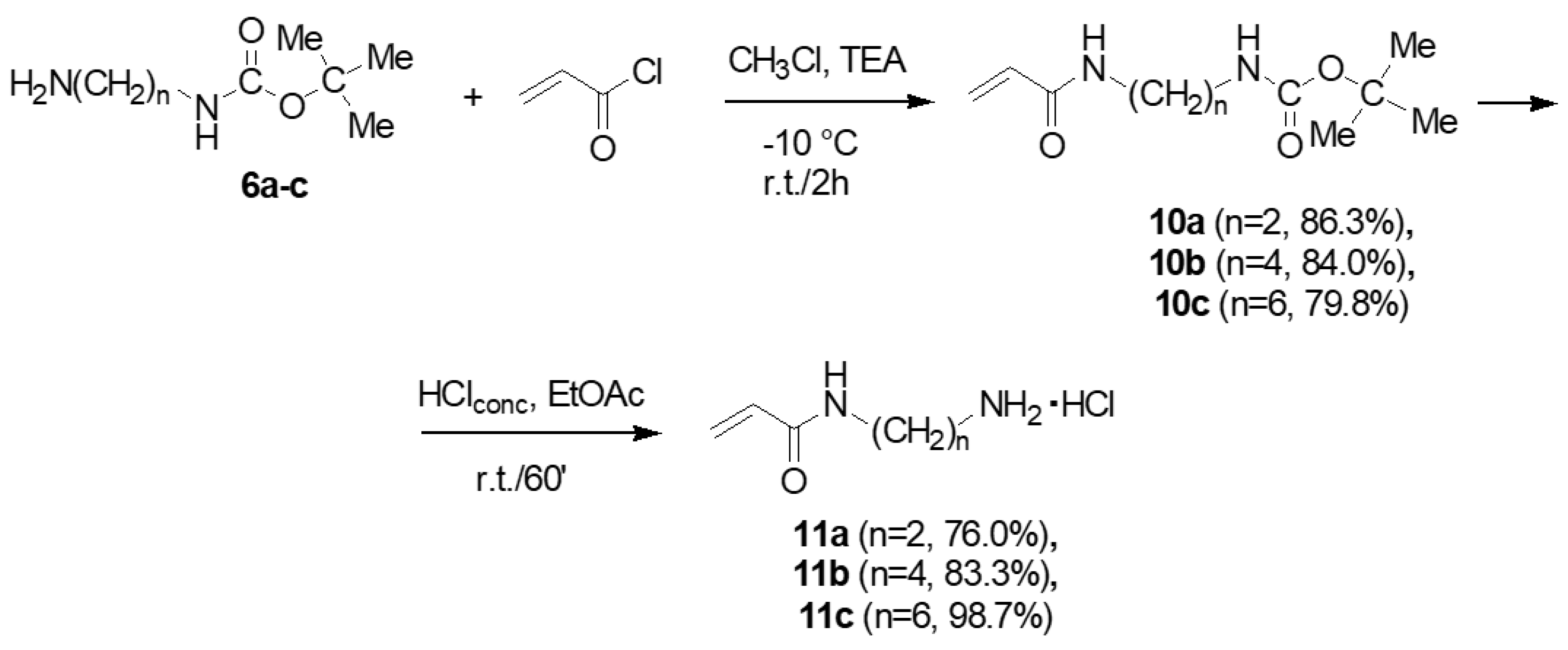
2.2.2. Synthesis of Acryloyl Monomers 11a–c
- Characterization of Intermediates 10a–c.
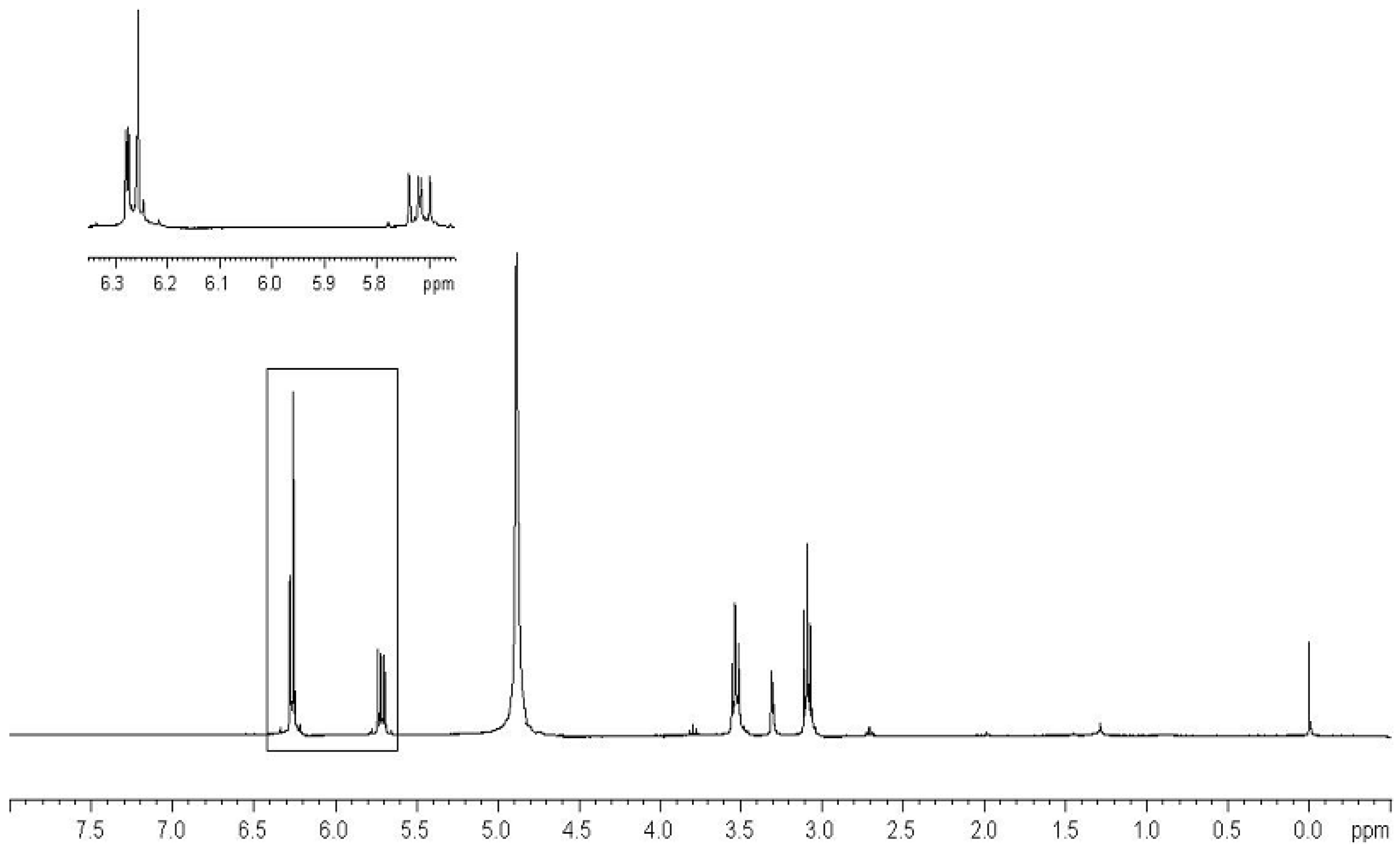
- Characterization of Monomers 11a–c.
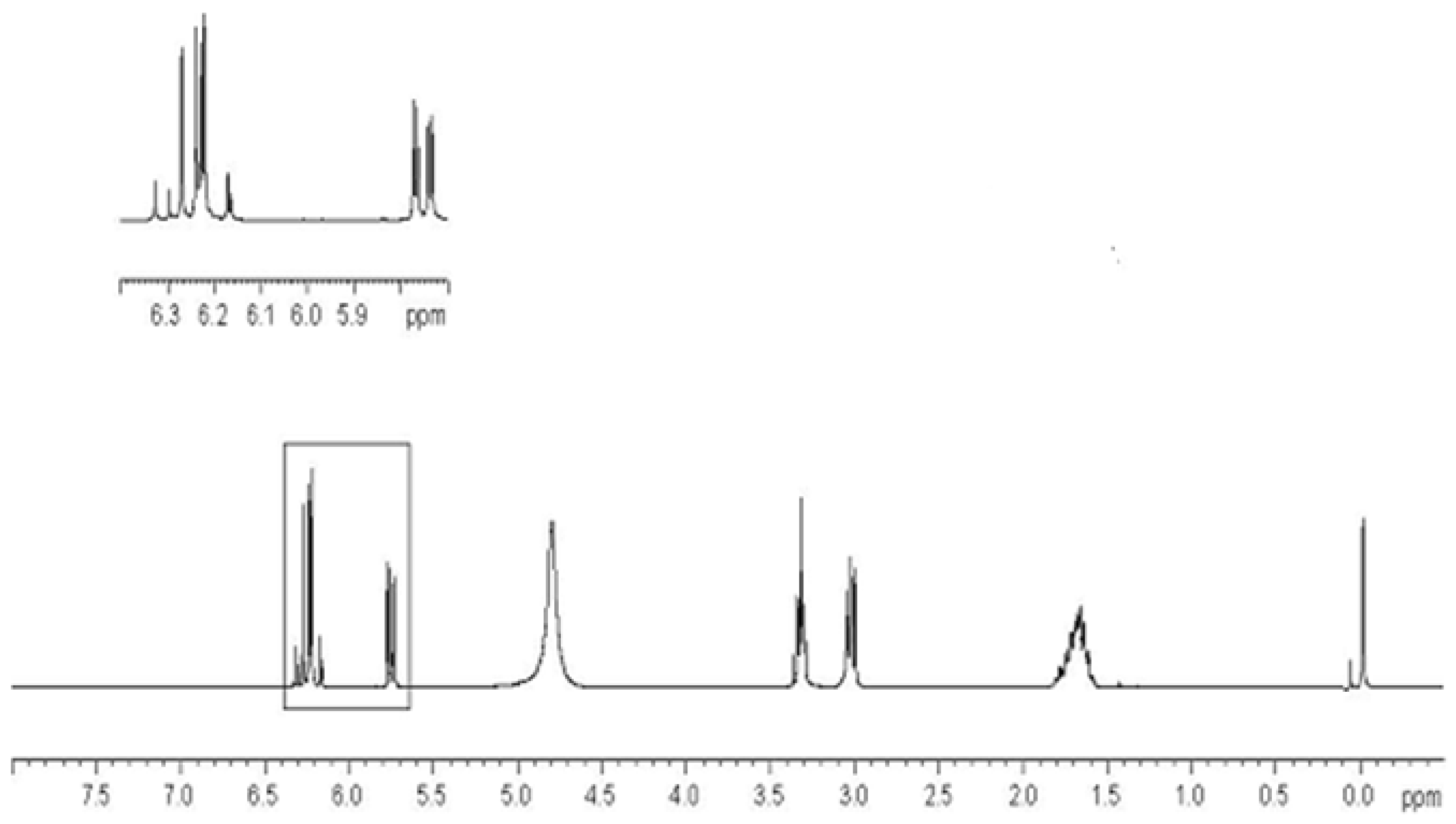
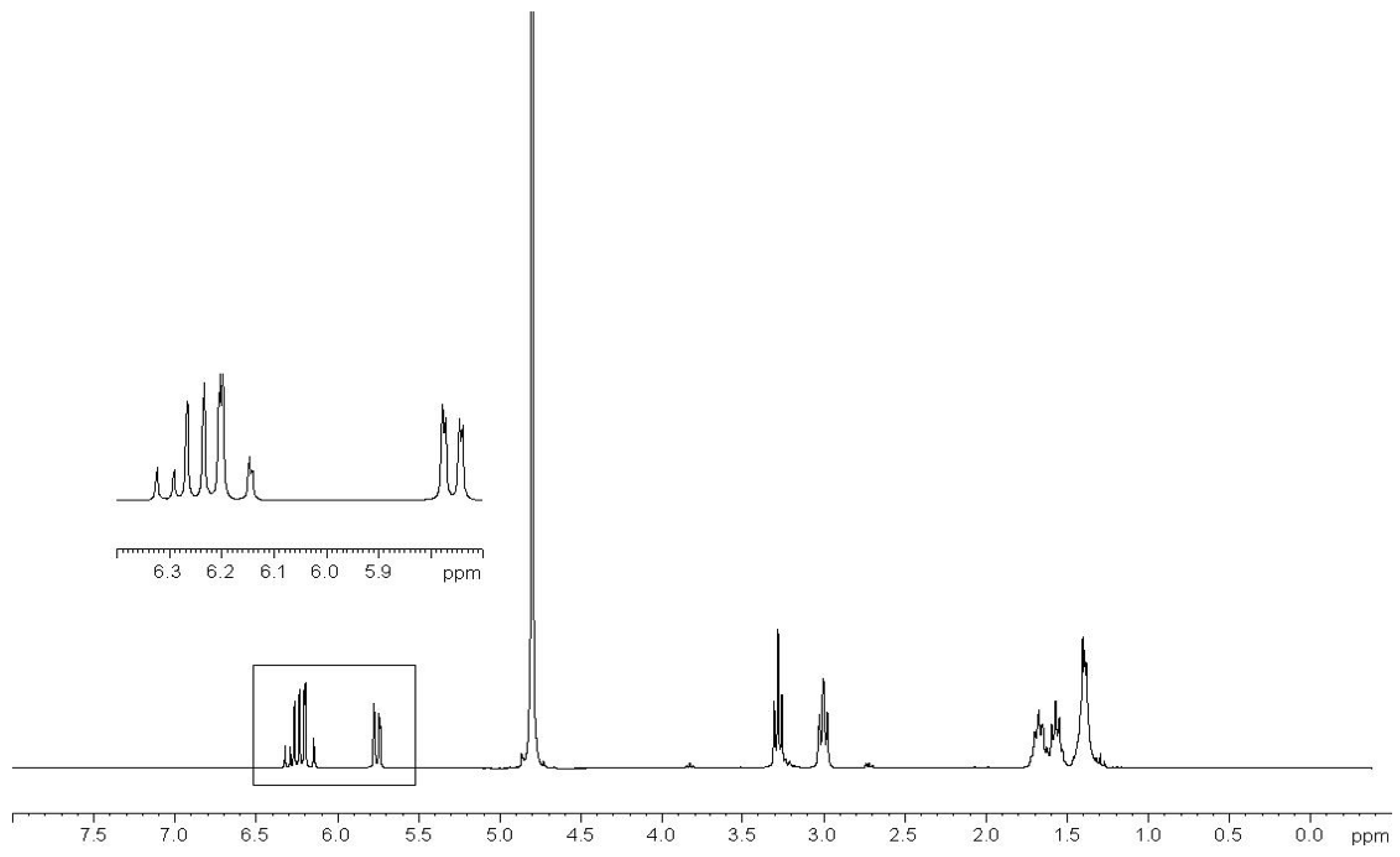
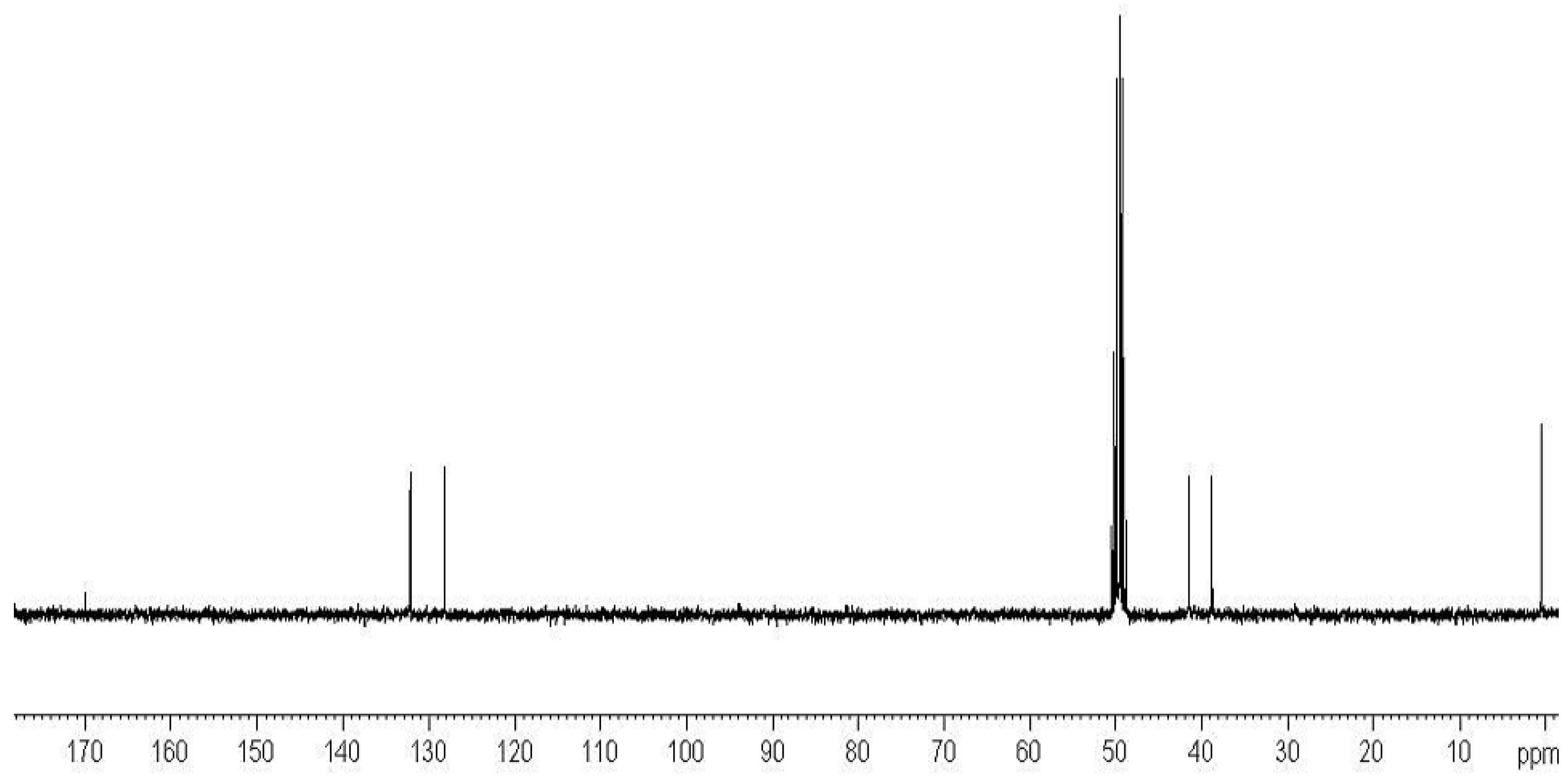
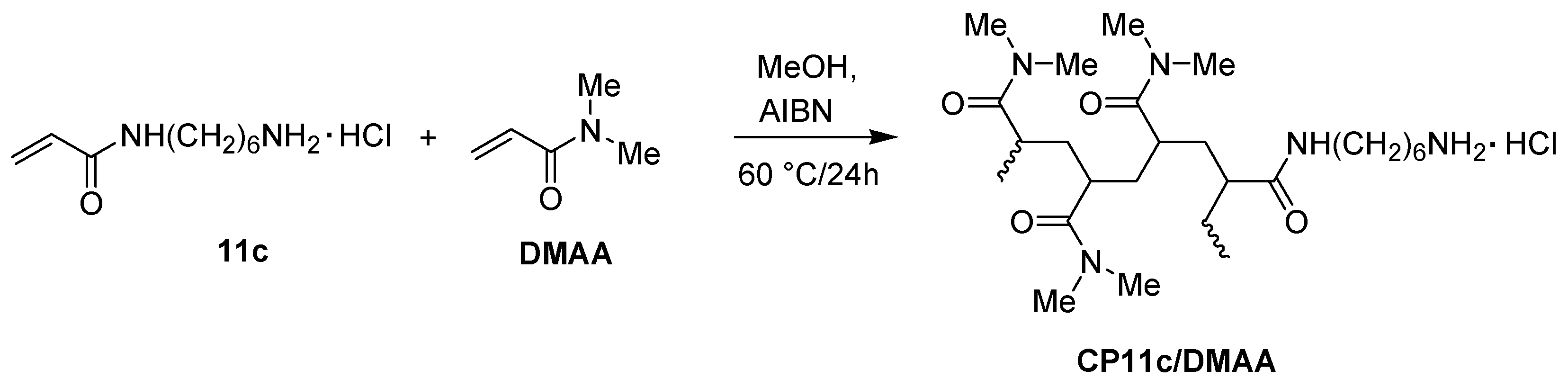
2.3. Preparation of Copolymers of 11a–c with DMAA (CP11a–c/DMAA)
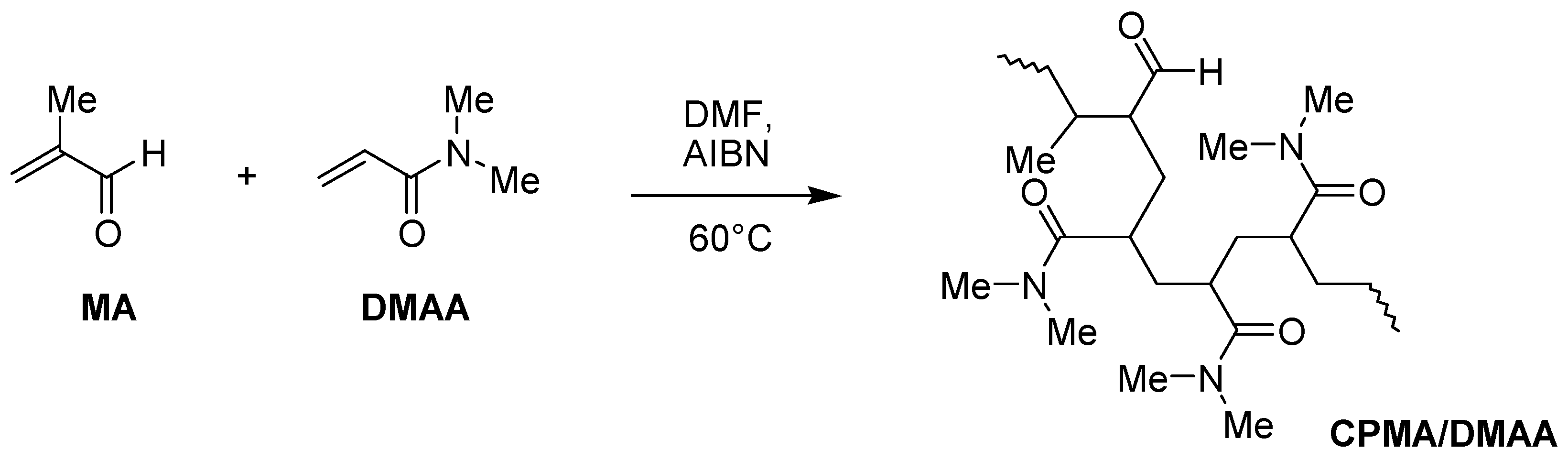
2.4. Preparation of Methacrolein (MA)/DMAA Copolymer (CPMA/DMAA)
2.5. Experiments of Gelatine Crosslinking
2.5.1. Gelatine
2.5.2. Enzymes
2.5.3. Enzymatic Oxidation Assays
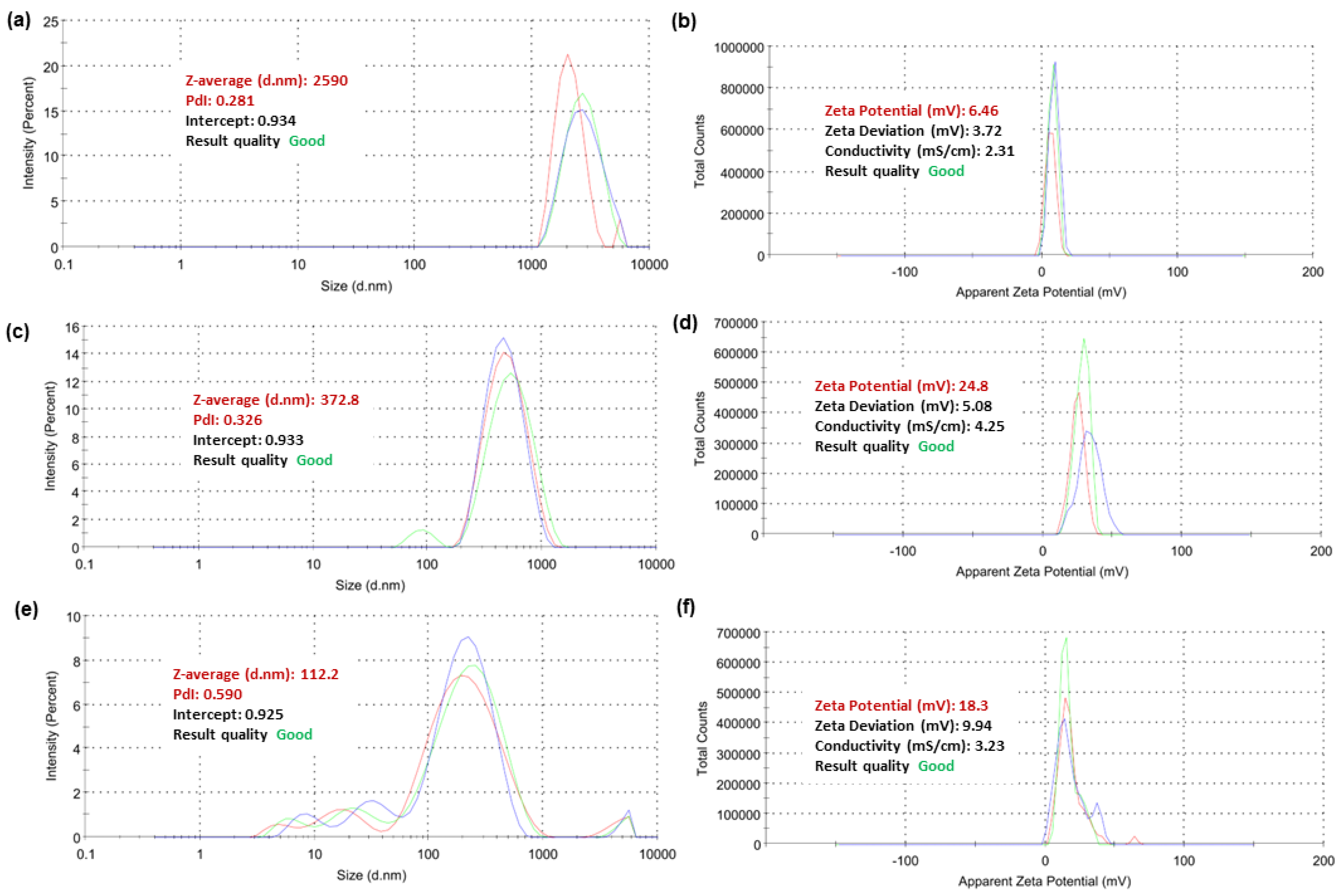
2.5.4. Dynamic Light Scattering (DLS) Analyses of Selected Copolymers
2.5.5. Gelatine Crosslinking Experiments
2.5.6. Titrations of Crosslinked Gelatines
2.5.7. Cytotoxicity Experiment on P5 Structural Analogous of CP5/DMAA
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. Synthesis and Characterization of Styrene-Based Monomer 5 and Its Copolymers
Preparation of 5/DMAA/Acrylic Acid (AA) Terpolymer (TP5/DMAA/AA)
3.3. Synthesis and Characterization of Acryloyl Monomers (11a–c) and Their Copolymers (CP11a–c/DMAA)
3.3.1. Synthesis and Characterization of N-t-Butoxy Carbonyl-Diaminoalkanes (6a–c)
- General Procedure to Synthesize N-t-Butoxy Carbonyl-Diaminoalkanes (6a,b)
- Procedure to Synthesize N-t-Butoxycarbonyl-1,6-diaminohexane (6c)
- Characterization of Compounds 6a–c and 7a–c
3.3.2. Synthesis and Characterization of N-t-Butoxy Carbonyl-N’-Acryloyl-Diaminoalkanes (10a–c)
- General Procedure to Synthesize N-t-Butoxy Carbonyl-N’-Acryloyl-Diaminoalkanes (10a–c)
- Characterization of Compounds 10a–c
3.3.3. Synthesis and Characterization of Acryloyl Monomers (N-Acryloyl-1,6-diaminoalkane Hydrochlorides) (11a–c)
- General Procedure to Synthesize N-Acryloyl-1,6-diaminoalkane hydrochlorides) (11a–c)
- Characterization of Compounds 11a–c
3.3.4. Preparation of DMAA Copolymers of Compounds 11a–c (CP11a–c/DMAA)
- General Procedure to Polymerize 10a and 10b as well as 11c with DMAA
- Characterization of Copolymers
- Fractioning of Copolymers CP10a, b/DMAA
- General Procedure to Deprotect N-Boc protected Copolymers CP10a, b/DMAA
- Characterization of Copolymers
3.4. Synthesis and Characterization of Methacrolein (MA) Copolymer with DMAA (CPMA/DMAA)
3.5. Enzymatic Oxidation Assays of the Prepared Copolymers
3.6. Particle Size, Zeta Potential (ζ-p), and Polydispersity Index (PDI) of Selected Copolymers
3.7. Gelatine Crosslinking Tests
3.8. Titration of Gelatine and Crosslinked Gelatine
3.8.1. Acid/Base Titrations of Gelatine
3.8.2. UV Titrations of Gelatine
3.9. Cytotoxicity Experiments
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marler, J.J.; Upton, J.; Langer, R.; Vacanti, J.P. Transplantation of Cells in Matrices for Tissue Regeneration. Adv. Drug Deliv. Rev. 1998, 33, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Langer, R. Biomaterials in Drug Delivery and Tissue Engineering: One Laboratory’s Experience. Acc. Chem. Res. 2000, 33, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Jo, S.; Mikos, A.G. Biomimetic Materials for Tissue Engineering. Biomaterials 2003, 24, 4353–4364. [Google Scholar] [CrossRef]
- Cancedda, R.; Dozin, B.; Giannoni, P.; Quarto, R. Tissue Engineering and Cell Therapy of Cartilage and Bone. Matrix Biol. 2003, 22, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Viola, J.; Lal, B.; Grad, O. The Emergence of Tissue Engineering as a Research Field; National Science Foundation: Alexandria, VA, USA, 2003.
- Lavik, E.; Langer, R. Tissue Engineering: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2004, 65, 1–8. [Google Scholar] [CrossRef]
- Chan, G.; Mooney, D.J. New Materials for Tissue Engineering: Towards Greater Control over the Biological Response. Trends Biotechnol. 2008, 26, 382–392. [Google Scholar] [CrossRef]
- Skalak, R.; Fox, C.F. Tissue Engineering. In Proceedings of the Tissue Engineering, Granlibakken, Lake Tahoe, CA, USA, 26 February 1988; Alan R. Liss, Inc.: New York, NY, USA, 1988. [Google Scholar]
- Langer, R.; Vacanti, J.P. Tissue Engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef]
- Jones, I.; Currie, L.; Martin, R. A Guide to Biological Skin Substitutes. Br. J. Plast. Surg. 2002, 55, 185–193. [Google Scholar] [CrossRef]
- Atala, A.; Bauer, S.B.; Soker, S.; Yoo, J.J.; Retik, A.B. Tissue-Engineered Autologous Bladders for Patients Needing Cystoplasty. Lancet 2006, 367, 1241–1246. [Google Scholar] [CrossRef]
- Tenreiro, M.F.; Louro, A.F.; Alves, P.M.; Serra, M. Next Generation of Heart Regenerative Therapies: Progress and Promise of Cardiac Tissue Engineering. NPJ Regen. Med. 2021, 6, 30. [Google Scholar] [CrossRef]
- Mayo Clinic. Neuroregenerative Medicine. Available online: https://www.Mayo.Edu/Research/Documents/Neuroregenerative-Medicine-Booklet/Doc-20092381 (accessed on 26 February 2024).
- Khan, M.U.A.; Aslam, M.A.; Bin Abdullah, M.F.; Hasan, A.; Shah, S.A.; Stojanović, G.M. Recent Perspective of Polymeric Biomaterial in Tissue Engineering—A Review. Mater. Today Chem. 2023, 34, 101818. [Google Scholar] [CrossRef]
- Dimitriou, R.; Jones, E.; McGonagle, D.; Giannoudis, P.V. Bone Regeneration: Current Concepts and Future Directions. BMC Med. 2011, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Chen, J.; Liu, S.; Jin, Y. Stem Cell-Based Bone and Dental Regeneration: A View of Microenvironmental Modulation. Int. J. Oral Sci. 2019, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Puleo, D. Understanding and Controlling the Bone–Implant Interface. Biomaterials 1999, 20, 2311–2321. [Google Scholar] [CrossRef]
- Ducheyne, P.; Qiu, Q. Bioactive Ceramics: The Effect of Surface Reactivity on Bone Formation and Bone Cell Function. Biomaterials 1999, 20, 2287–2303. [Google Scholar] [CrossRef]
- Benezra Rosen, V.; Hobbs, L.W.; Spector, M. The Ultrastructure of Anorganic Bovine Bone and Selected Synthetic Hyroxyapatites Used as Bone Graft Substitute Materials. Biomaterials 2002, 23, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Singla, A.; Lee, Y. Biomedical Applications of Collagen. Int. J. Pharm. 2001, 221, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Place, E.S.; George, J.H.; Williams, C.K.; Stevens, M.M. Synthetic Polymer Scaffolds for Tissue Engineering. Chem. Soc. Rev. 2009, 38, 1139. [Google Scholar] [CrossRef]
- Seal, B. Polymeric Biomaterials for Tissue and Organ Regeneration. Mater. Sci. Eng. R Rep. 2001, 34, 147–230. [Google Scholar] [CrossRef]
- Kyriacou, H.; Kamaraj, A.; Khan, W.S. Developments in Antibiotic-Eluting Scaffolds for the Treatment of Osteomyelitis. Appl. Sci. 2020, 10, 2244. [Google Scholar] [CrossRef]
- Janoušková, O. Synthetic Polymer Scaffolds for Soft Tissue Engineering. Physiol. Res. 2018, 67, S335–S348. [Google Scholar] [CrossRef]
- Hajebi, S.; Mohammadi Nasr, S.; Rabiee, N.; Bagherzadeh, M.; Ahmadi, S.; Rabiee, M.; Tahriri, M.; Tayebi, L.; Hamblin, M.R. Bioresorbable Composite Polymeric Materials for Tissue Engineering Applications. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 926–940. [Google Scholar] [CrossRef]
- Ilia, G. Phosphorus Containing Hydrogels. Polym. Adv. Technol. 2009, 20, 707–722. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Pomahač, B.; Svensjö, T.; Yao, F.; Brown, H.; Eriksson, E. Tissue Engineering of Skin. Crit. Rev. Oral Biol. Med. 1998, 9, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Said, G.; Tadie, M. Regrowth of the Rostral Spinal Axons into the Caudal Ventral Roots through a Collagen Tube Implanted into Hemisected Adult Rat Spinal Cord. Neurosurgery 2001, 49, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Voytik-Harbin, S.L.; Brightman, A.O.; Waisner, B.Z.; Robinson, J.P.; Lamar, C.H. Small Intestinal Submucosa: A Tissue-Derived Extracellular Matrix That Promotes Tissue-Specific Growth and Differentiation of Cells In Vitro. Tissue Eng. 1998, 4, 157–174. [Google Scholar] [CrossRef]
- Othmer, K. Encyclopedia of Chemical Technology; Wiley: Hoboken, NJ, USA, 2001; ISBN 9780471484943. [Google Scholar]
- Young, S.; Wong, M.; Tabata, Y.; Mikos, A.G. Gelatin as a Delivery Vehicle for the Controlled Release of Bioactive Molecules. J. Control. Release 2005, 109, 256–274. [Google Scholar] [CrossRef]
- Gurumurthy, B.; Janorkar, A.V. Improvements in Mechanical Properties of Collagen-Based Scaffolds for Tissue Engineering. Curr. Opin. Biomed. Eng. 2021, 17, 100253. [Google Scholar] [CrossRef]
- Madaghiele, M.; Piccinno, A.; Saponaro, M.; Maffezzoli, A.; Sannino, A. Collagen- and Gelatine-Based Films Sealing Vascular Prostheses: Evaluation of the Degree of Crosslinking for Optimal Blood Impermeability. J. Mater. Sci. Mater. Med. 2009, 20, 1979–1989. [Google Scholar] [CrossRef]
- Barbani, N.; Giusti, P.; Lazzeri, L.; Polacco, G.; Pizzirani, G. Bioartificial Materials Based on Collagen: 1. Collagen Cross-Linking with Gaseous Glutaraldehyde. J. Biomater. Sci. Polym. Ed. 1996, 7, 461–469. [Google Scholar] [CrossRef]
- Olde Damink, L.H.H.; Dijkstra, P.J.; van Luyn, M.J.A.; van Wachem, P.B.; Nieuwenhuis, P.; Feijen, J. Cross-Linking of Dermal Sheep Collagen Using a Water-Soluble Carbodiimide. Biomaterials 1996, 17, 765–773. [Google Scholar] [CrossRef]
- Dalev, P.; Vassileva, E.; Mark, J.E.; Fakirov, S. Enzymatic Degradation of Formaldehyde-Crosslinked Gelatin. Biotechnol. Tech. 1998, 12, 889–892. [Google Scholar] [CrossRef]
- Lien, S.-M.; Li, W.-T.; Huang, T.-J. Genipin-Crosslinked Gelatin Scaffolds for Articular Cartilage Tissue Engineering with a Novel Crosslinking Method. Mater. Sci. Eng. C 2008, 28, 36–43. [Google Scholar] [CrossRef]
- Sung, H.W.; Huang, D.M.; Chang, W.H.; Huang, R.N.; Hsu, J.C. Evaluation of Gelatin Hydrogel Crosslinked with Various Crosslinking Agents as Bioadhesives: In Vitro Study. J. Biomed. Mater. Res. 1999, 46, 520–530. [Google Scholar] [CrossRef]
- Bigi, A.; Cojazzi, G.; Panzavolta, S.; Rubini, K.; Roveri, N. Mechanical and Thermal Properties of Gelatin Films at Different Degrees of Glutaraldehyde Crosslinking. Biomaterials 2001, 22, 763–768. [Google Scholar] [CrossRef]
- Liang, H.; Chang, W.; Liang, H.; Lee, M.; Sung, H. Crosslinking Structures of Gelatin Hydrogels Crosslinked with Genipin or a Water-soluble Carbodiimide. J. Appl. Polym. Sci. 2004, 91, 4017–4026. [Google Scholar] [CrossRef]
- Zeugolis, D.I.; Paul, G.R.; Attenburrow, G. Cross-linking of Extruded Collagen Fibers—A Biomimetic Three-dimensional Scaffold for Tissue Engineering Applications. J. Biomed. Mater. Res. A 2009, 89, 895–908. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Beckman, E.J.; Piesco, N.P.; Agarwal, S. A New Peptide-Based Urethane Polymer: Synthesis, Biodegradation, and Potential to Support Cell Growth in Vitro. Biomaterials 2000, 21, 1247–1258. [Google Scholar] [CrossRef]
- Bonzani, I.C.; Adhikari, R.; Houshyar, S.; Mayadunne, R.; Gunatillake, P.; Stevens, M.M. Synthesis of Two-Component Injectable Polyurethanes for Bone Tissue Engineering. Biomaterials 2007, 28, 423–433. [Google Scholar] [CrossRef]
- Schacht, E.; Bogdanov, B.; Van Den Bulcke, A.; De Rooze, N. Hydrogels Prepared by Crosslinking of Gelatin with Dextran Dialdehyde. React. Funct. Polym. 1997, 33, 109–116. [Google Scholar] [CrossRef]
- Draye, J.-P.; Delaey, B.; Van de Voorde, A.; Van Den Bulcke, A.; Bogdanov, B.; Schacht, E. In Vitro Release Characteristics of Bioactive Molecules from Dextran Dialdehyde Cross-Linked Gelatin Hydrogel Films. Biomaterials 1998, 19, 99–107. [Google Scholar] [CrossRef]
- Yu, H.; Xiao, C. Synthesis and Properties of Novel Hydrogels from Oxidized Konjac Glucomannan Crosslinked Gelatin for in Vitro Drug Delivery. Carbohydr. Polym. 2008, 72, 479–489. [Google Scholar] [CrossRef]
- Chen, T.; Embree, H.D.; Brown, E.M.; Taylor, M.M.; Payne, G.F. Enzyme-Catalyzed Gel Formation of Gelatin and Chitosan: Potential for in Situ Applications. Biomaterials 2003, 24, 2831–2841. [Google Scholar] [CrossRef]
- Ahmady, A.; Abu Samah, N.H. A Review: Gelatine as a Bioadhesive Material for Medical and Pharmaceutical Applications. Int. J. Pharm. 2021, 608, 121037. [Google Scholar] [CrossRef]
- Zuccari, G.; Russo, E.; Villa, C.; Zorzoli, A.; Marimpietri, D.; Marchitto, L.; Alfei, S. Preparation and Characterization of Amorphous Solid Dispersions for the Solubilization of Fenretinide. Pharmaceuticals 2023, 16, 388. [Google Scholar] [CrossRef]
- Bertini, V.; Alfei, S.; Pocci, M.; Lucchesini, F.; Picci, N.; Iemma, F. Monomers Containing Substrate or Inhibitor Residues for Copper Amine Oxidases and Their Hydrophilic Beaded Resins Designed for Enzyme Interaction Studies. Tetrahedron 2004, 60, 11407–11414. [Google Scholar] [CrossRef]
- Alfei, S.; Piatti, G.; Caviglia, D.; Schito, A. Synthesis, Characterization, and Bactericidal Activity of a 4-Ammoniumbuthylstyrene-Based Random Copolymer. Polymers 2021, 13, 1140. [Google Scholar] [CrossRef]
- Mushtaq, F.; Raza, Z.A.; Batool, S.R.; Zahid, M.; Onder, O.C.; Rafique, A.; Nazeer, M.A. Preparation, Properties, and Applications of Gelatin-Based Hydrogels (GHs) in the Environmental, Technological, and Biomedical Sectors. Int. J. Biol. Macromol. 2022, 218, 601–633. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, M.; Zhou, D.; Chen, F.; Cai, Q.; Yan, X.; Li, J. Gelatine Methacrylamide-Based Multifunctional Bilayer Hydrogels for Accelerating Diabetic Wound Repair. Mater. Des. 2022, 218, 110687. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Valenti, G.; Domenicotti, C. Synthesis of Polystyrene-Based Cationic Nanomaterials with Pro-Oxidant Cytotoxic Activity on Etoposide-Resistant Neuroblastoma Cells. Nanomaterials 2021, 11, 977. [Google Scholar] [CrossRef]
- Valenti, G.E.; Marengo, B.; Milanese, M.; Zuccari, G.; Brullo, C.; Domenicotti, C.; Alfei, S. Imidazo-Pyrazole-Loaded Palmitic Acid and Polystyrene-Based Nanoparticles: Synthesis, Characterization and Antiproliferative Activity on Chemo-Resistant Human Neuroblastoma Cells. Int. J. Mol. Sci. 2023, 24, 15027. [Google Scholar] [CrossRef]
- Jacobson, A.R.; Makris, A.N.; Sayre, L.M. Monoacylation of Symmetrical Diamines. J. Org. Chem. 1987, 52, 2592–2594. [Google Scholar] [CrossRef]
- Stahl, G.L.; Walter, R.; Smith, C.W. General Procedure for the Synthesis of Mono-N-Acylated 1,6-Diaminohexanes. J. Org. Chem. 1978, 43, 2285–2286. [Google Scholar] [CrossRef]
- Nagasawa, T.; Kuroiwa, K.; Narita, K.; Isowa, Y. New Agents for t-Butyloxycarbonylation and p-Methoxybenzyloxycarbonylation of Amino Acids. Bull. Chem. Soc. Jpn. 1973, 46, 1269–1272. [Google Scholar] [CrossRef]
- Kenchington, A.W.; Ward, A.G. The Titration Curve of Gelatin. Biochem. J. 1954, 58, 202–207. [Google Scholar] [CrossRef]
- Cannan, R.K. The Acid-Base Titration of Proteins. Chem. Rev. 1942, 30, 395–412. [Google Scholar] [CrossRef]
- Bertoldo, M.; Bronco, S.; Gragnoli, T.; Ciardelli, F. Modification of Gelatin by Reaction with 1,6-Diisocyanatohexane. Macromol. Biosci. 2007, 7, 328–338. [Google Scholar] [CrossRef]
- Ofner, I.C.M.; Bubnis, W.A. Chemical and Swelling Evaluations of Amino Group Crosslinking in Gelatin and Modified Gelatin Matrices. Pharm. Res. 1996, 13, 1821–1827. [Google Scholar] [CrossRef]
- Idini, B. Sintesi Di Glicopolimeri Nanostrutturati e Loro Funzionalizzazione in Studi Di Interazione Con Le Amminossidasi Contenenti Ram; Università di Genova: Genova, Italy; Available online: https://opac.bncf.firenze.sbn.it/Record/TD17067534 (accessed on 10 February 2024).
- Kagan, H.M.; Cai, P. Oxidase. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1995; pp. 122–132. [Google Scholar]
- Trackman, P.C.; Zoski, C.G.; Kagan, H.M. Development of a Peroxidase-Coupled Fluorometric Assay for Lysyl Oxidase. Anal. Biochem. 1981, 113, 336–342. [Google Scholar] [CrossRef]
- Guilbault, G.G.; Kramer, D.N.; Hackley, E.B. New Substrate for Fluorometric Determination of Oxidative Enzymes. Anal. Chem. 1967, 39, 271. [Google Scholar] [CrossRef]
- Courtenay, J.C.; Johns, M.A.; Galembeck, F.; Deneke, C.; Lanzoni, E.M.; Costa, C.A.; Scott, J.L.; Sharma, R.I. Surface Modified Cellulose Scaffolds for Tissue Engineering. Cellulose 2017, 24, 253–267. [Google Scholar] [CrossRef]
- Shankar, K.G.; Gostynska, N.; Montesi, M.; Panseri, S.; Sprio, S.; Kon, E.; Marcacci, M.; Tampieri, A.; Sandri, M. Investigation of Different Cross-Linking Approaches on 3D Gelatin Scaffolds for Tissue Engineering Application: A Comparative Analysis. Int. J. Biol. Macromol. 2017, 95, 1199–1209. [Google Scholar] [CrossRef]
- Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles. Nanomaterials 2022, 12, 2743. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfei, S.; Pintaudi, F.; Zuccari, G. Synthesis and Characterization of Amine and Aldehyde-Containing Copolymers for Enzymatic Crosslinking of Gelatine. Int. J. Mol. Sci. 2024, 25, 2897. https://doi.org/10.3390/ijms25052897
Alfei S, Pintaudi F, Zuccari G. Synthesis and Characterization of Amine and Aldehyde-Containing Copolymers for Enzymatic Crosslinking of Gelatine. International Journal of Molecular Sciences. 2024; 25(5):2897. https://doi.org/10.3390/ijms25052897
Chicago/Turabian StyleAlfei, Silvana, Federica Pintaudi, and Guendalina Zuccari. 2024. "Synthesis and Characterization of Amine and Aldehyde-Containing Copolymers for Enzymatic Crosslinking of Gelatine" International Journal of Molecular Sciences 25, no. 5: 2897. https://doi.org/10.3390/ijms25052897