
Essential Involvement of Neutrophil Elastase in Acute Acetaminophen Hepatotoxicity Using BALB/c Mice

Abstract
:1. Introduction
2. Results
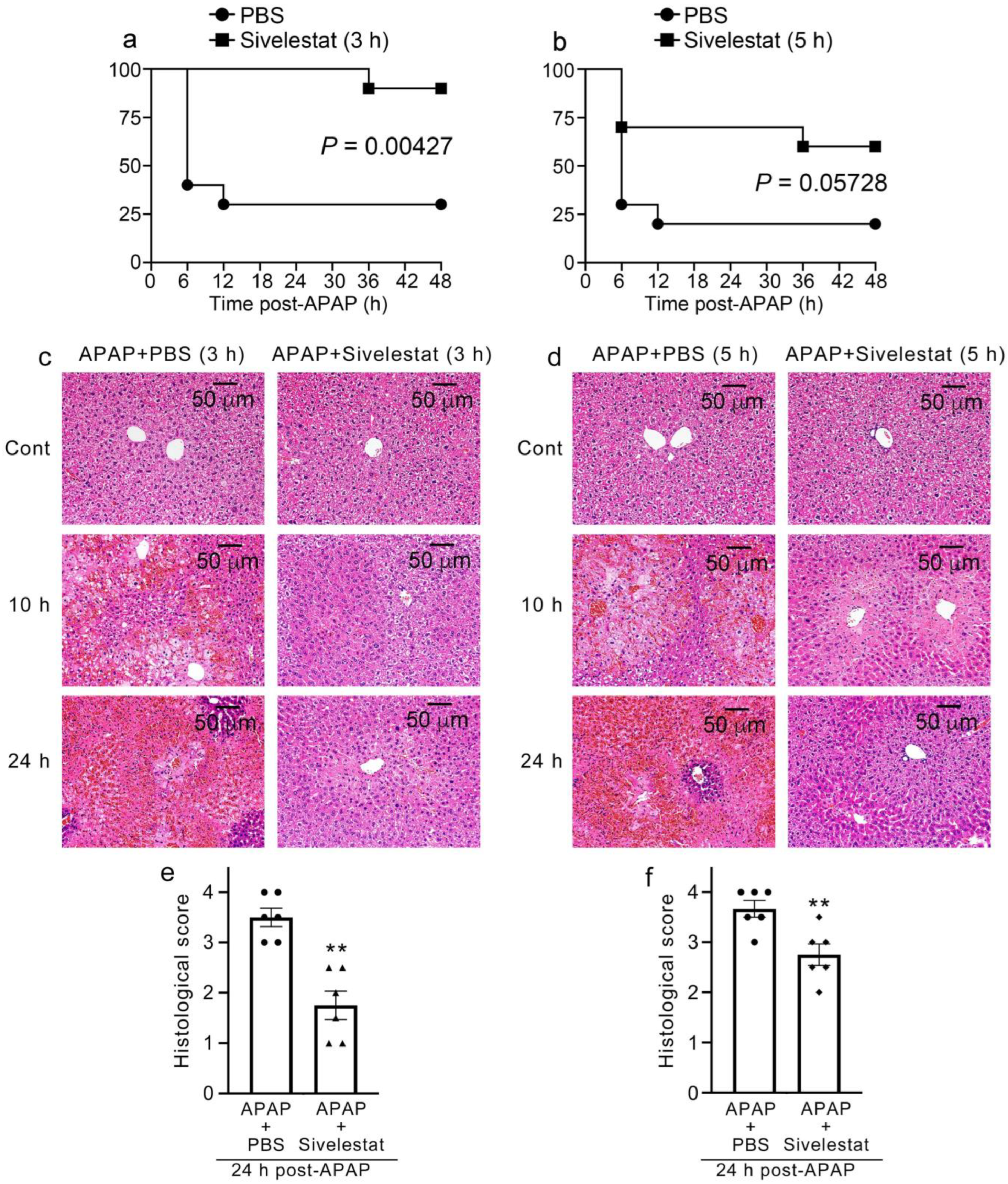
2.1. Improvement of Mortality in Sivelestat-Treated Mice after APAP Challenge
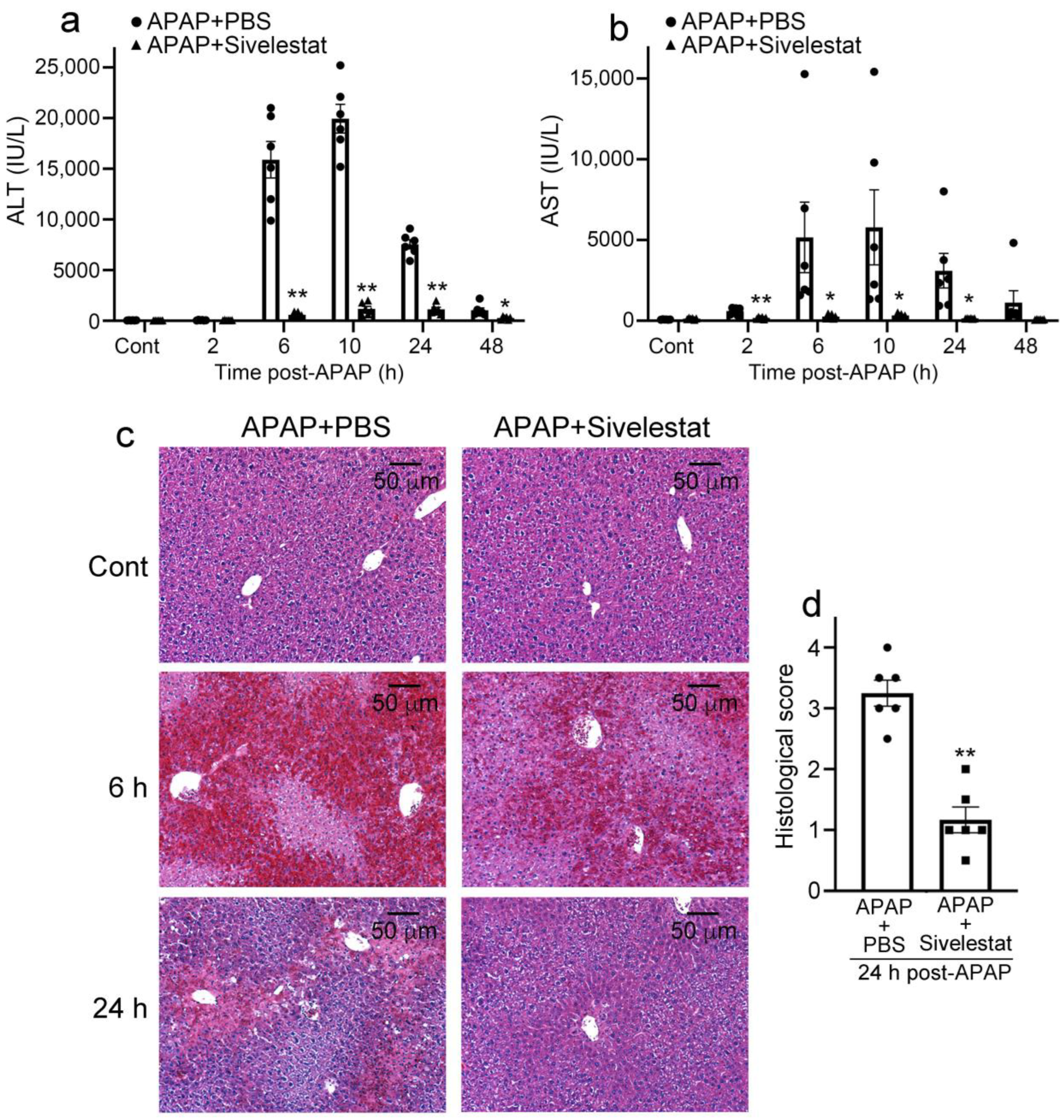
2.2. Sivelestat Treatment Attenuates Liver Damage Due to APAP Overdose
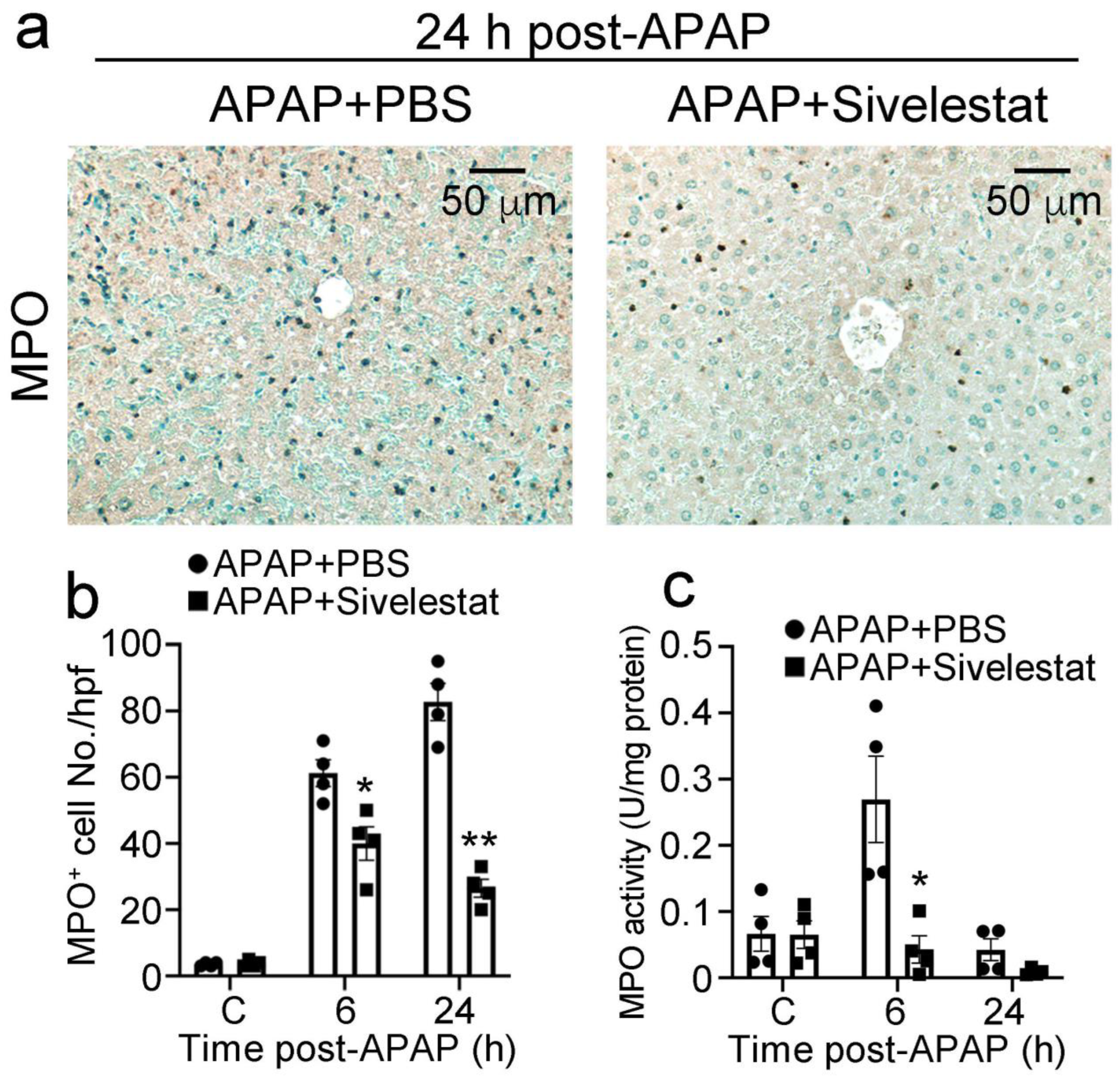
2.3. Treatment with Sivelestat Reduces Hepatic Neutrophil Infiltration after APAP Challenge
2.4. Attenuated Intrahepatic Gene Expression of Inflammatory Cytokines and Chemokines in Sivelestat-Treated Mice after APAP Challenge
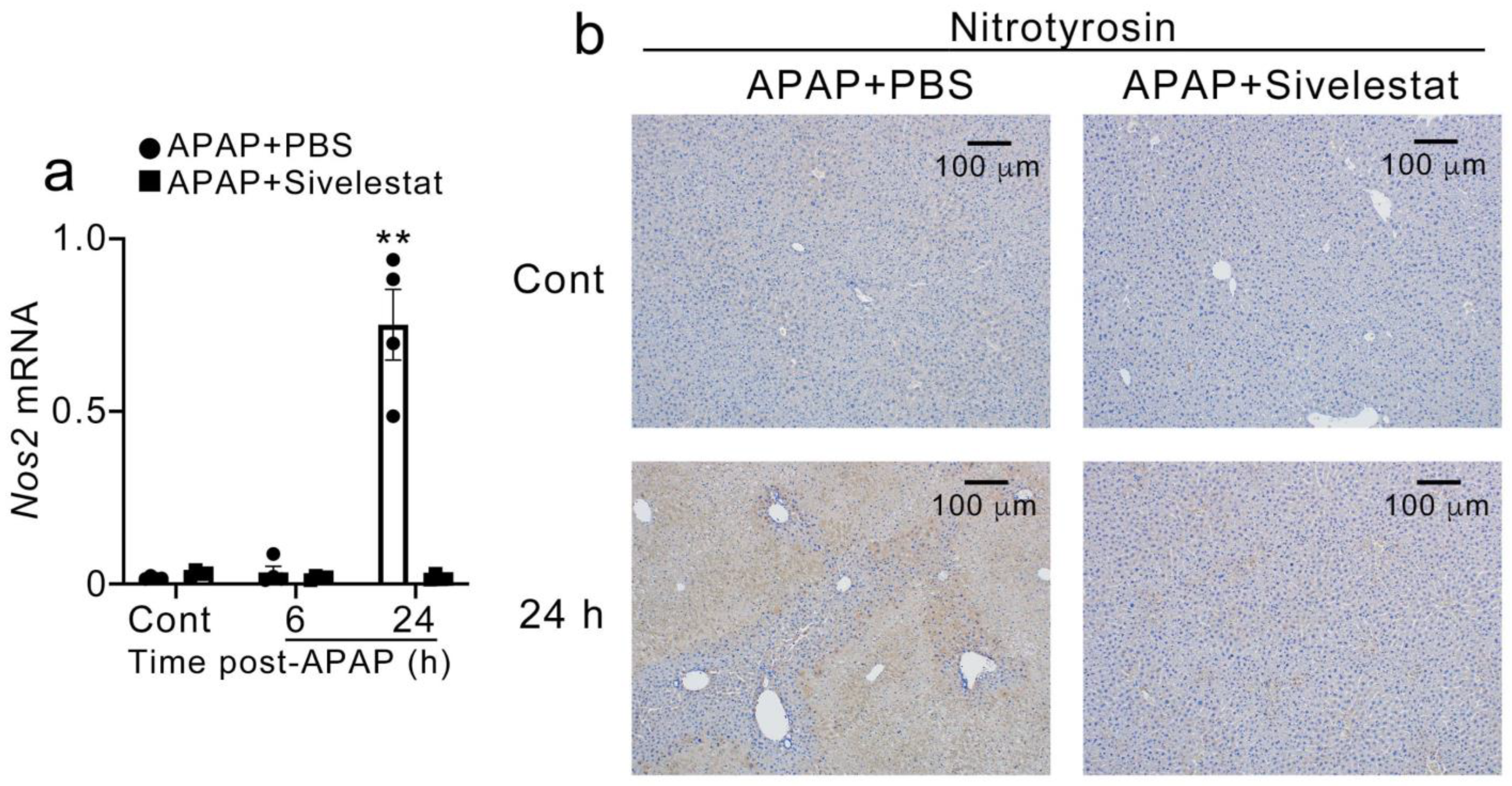
2.5. Sivelestat Treatment Reduced Hepatic Nos2 Expression and NO Production after APAP Challenge
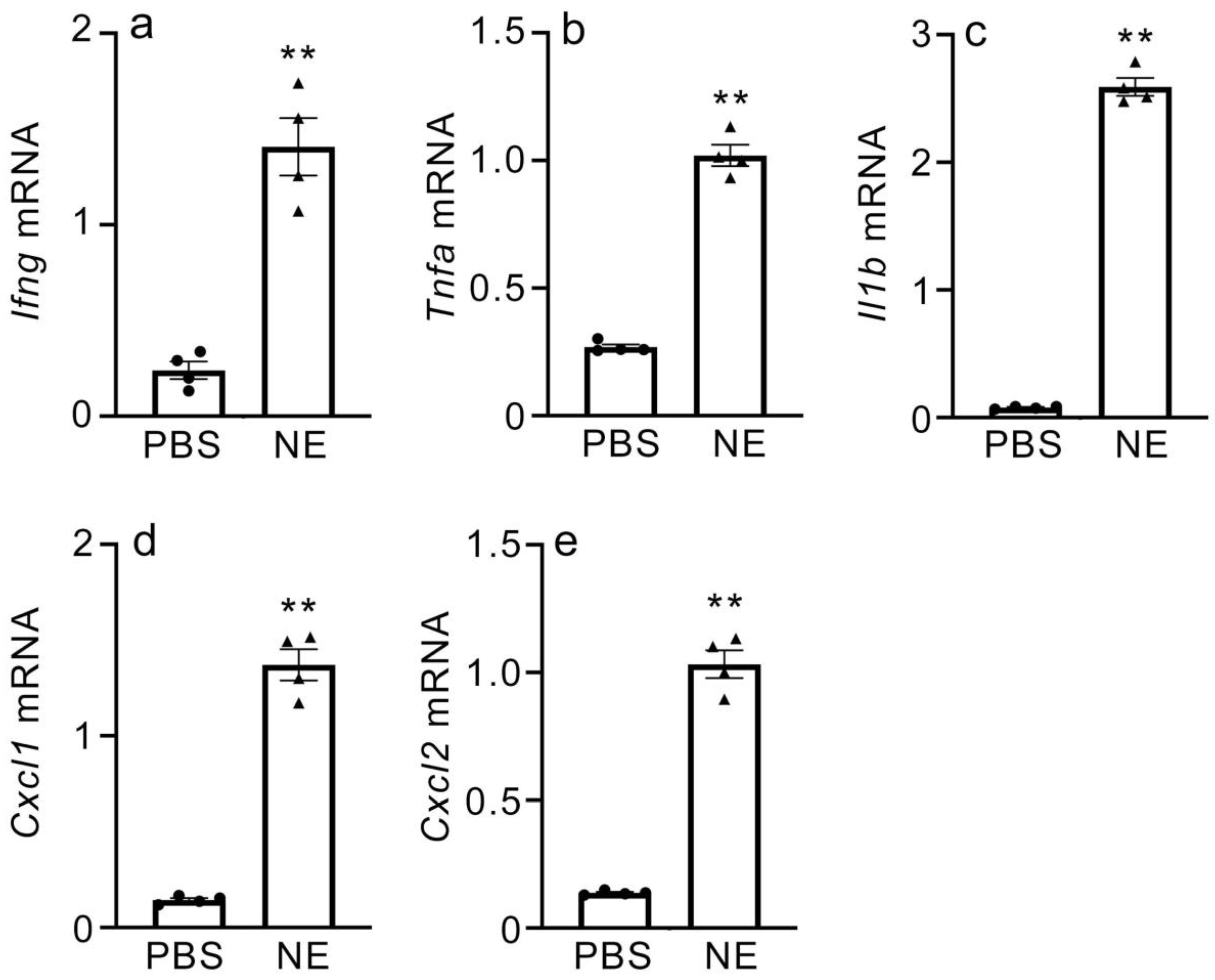
2.6. Gene Expression of Inflammatory Cytokines and CXC Chemokines in NE-Treated Macrophages
2.7. Sivelestat Therapy Is Effective in Improving Mortality Due to APAP Overdose in Mice
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies (Abs)
4.2. Mice
4.3. APAP-Induced Liver Injury
4.4. Determination of Serum Alanine Aminotransferase (ALT) and Aspartate Aminotransferase (AST) Levels
4.5. Histopathological Analysis
4.6. Immunohistochemical Analysis
4.7. ELISA
4.8. Myeloperoxidase Activity Assay
4.9. Quantitative RT-PCR Analysis
4.10. In Vitro Culture and Stimulation of Macrophages
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Larsen, F.S.; Wendon, J. Understanding paracetamol-induced liver failure. Intensiv. Care Med. 2014, 40, 888–890. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; Clinical Practice Guidelines Panel; Wendon, J.; Cordoba, J.; Dhawan, A.; Larsen, F.S.; Manns, M.; Nevens, F.; Samuel, D.; Simpson, K.J.; et al. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J. Hepatol. 2017, 66, 1047–1081. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune mechanisms in acetaminophen-induced acute liver failure. HepatoBiliary Surg. Nutr. 2014, 3, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 2011, 32, 8–20. [Google Scholar] [CrossRef]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiødt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- Chun, L.J.; Tong, M.J.; Busuttil, R.W.; Hiatt, J.R. Acetaminophen Hepatotoxicity and Acute Liver Failure. J. Clin. Gastroenterol. 2009, 43, 342–349. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr. Pharmacol. Rep. 2018, 4, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Ishibe, T.; Kimura, A.; Ishida, Y.; Takayasu, T.; Hayashi, T.; Tsuneyama, K.; Matsushima, K.; Sakata, I.; Mukaida, N.; Kondo, T. Reduced acetaminophen-induced liver injury in mice by genetic disruption of IL-1 receptor antagonist. Lab. Investig. 2009, 89, 68–79. [Google Scholar] [CrossRef]
- Ishida, Y.; Kondo, T.; Kimura, A.; Tsuneyama, K.; Takayasu, T.; Mukaida, N. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur. J. Immunol. 2006, 36, 1028–1038. [Google Scholar] [CrossRef]
- Ishida, Y.; Kondo, T.; Tsuneyama, K.; Lu, P.; Takayasu, T.; Mukaida, N. The pathogenic roles of tumor necrosis factor receptor p55 in acetaminophen-induced liver injury in mice. J. Leukoc. Biol. 2004, 75, 59–67. [Google Scholar] [CrossRef]
- Ishida, Y.; Kondo, T.; Ohshima, T.; Fujiwara, H.; Iwakura, Y.; Mukaida, N. A pivotal involvement of IFN-’ in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J. 2002, 16, 1227–1236. [Google Scholar] [CrossRef]
- Liu, Z.-X.; Govindarajan, S.; Kaplowitz, N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 2004, 127, 1760–1774. [Google Scholar] [CrossRef]
- Ramaiah, S.K.; Jaeschke, H. Role of Neutrophils in the Pathogenesis of Acute Inflammatory Liver Injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Ávila, T.V.; Melgaço, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lan, T.; Mo, F.; Yang, J.; Wei, Y.; Wei, X. Antitumor and Radiosensitization Effects of a CXCR2 Inhibitor in Nasopharyngeal Carcinoma. Front. Cell Dev. Biol. 2021, 9, 689613. [Google Scholar] [CrossRef]
- Lacey, C.A.; Keleher, L.L.; Mitchell, W.J.; Brown, C.R.; Skyberg, J.A. CXCR2 Mediates Brucella-Induced Arthritis in Interferon γ–Deficient Mice. J. Infect. Dis. 2016, 214, 151–160. [Google Scholar] [CrossRef]
- Peres, R.S.; Menezes, G.B.; Teixeira, M.M.; Cunha, F.Q. Pharmacological opportunities to control inflammatory diseases through inhibition of the leukocyte recruitment. Pharmacol. Res. 2016, 112, 37–48. [Google Scholar] [CrossRef]
- Liu, Z.-X.; Han, D.; Gunawan, B.; Kaplowitz, N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006, 43, 1220–1230. [Google Scholar] [CrossRef]
- Yao, W.; Han, X.; Guan, Y.; Guan, J.; Wu, S.; Chen, C.; Li, H.; Hei, Z. Neutrophil Elastase Inhibitors Suppress Oxidative Stress in Lung during Liver Transplantation. Oxidative Med. Cell. Longev. 2019, 2019, 7323986. [Google Scholar] [CrossRef]
- Chen, J.; Liang, B.; Bian, D.; Luo, Y.; Yang, J.; Li, Z.; Zhuang, Z.; Zang, S.; Shi, J. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem. Biophys. Res. Commun. 2019, 518, 691–697. [Google Scholar] [CrossRef]
- Lee, W.L.; Downey, G.P. Leukocyte elastase: Physiological functions and role in acute lung injury. Am. J. Respir. Crit. Care Med. 2001, 164, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Groutas, W.C.; Dou, D.; Alliston, K.R. Neutrophil elastase inhibitors. Expert Opin. Ther. Patents 2011, 21, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Mikumo, H.; Yanagihara, T.; Hamada, N.; Harada, E.; Ogata-Suetsugu, S.; Ikeda-Harada, C.; Arimura-Omori, M.; Suzuki, K.; Yokoyama, T.; Nakanishi, Y. Neutrophil elastase inhibitor sivelestat ameliorates gefitinib-naphthalene-induced acute pneumonitis in mice. Biochem. Biophys. Res. Commun. 2017, 486, 205–209. [Google Scholar] [CrossRef]
- Fujino, N.; Kubo, H.; Suzuki, T.; He, M.; Suzuki, T.; Yamada, M.; Takahashi, T.; Ota, C.; Yamaya, M. Administration of a specific inhibitor of neutrophil elastase attenuates pulmonary fibrosis after acute lung injury in mice. Exp. Lung Res. 2011, 38, 28–36. [Google Scholar] [CrossRef]
- Takemasa, A.; Ishii, Y.; Fukuda, T. A neutrophil elastase inhibitor prevents bleomycin-induced pulmonary fibrosis in mice. Eur. Respir. J. 2012, 40, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Inomata, T.; Okada, Y.; Shimbo, T.; Takahashi, M.; Akita, K.; Uesugi, Y.; Narumi, Y. Sivelestat sodium hydrate reduces radiation-induced lung injury in mice by inhibiting neutrophil elastase. Mol. Med. Rep. 2013, 7, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ma, L.; Zhang, F.; Sun, R.; Li, T. Neutrophil elastase ameliorates matrix metalloproteinase-9 to promote lipopolysaccharide-induced acute lung injury in mice 1. Acta Cir. Bras. 2016, 31, 382–388. [Google Scholar] [CrossRef]
- Lee, J.; Yeo, C.D.; Lee, H.Y.; Rhee, C.K.; Kim, I.K.; Lee, D.G.; Lee, S.H.; Kim, J.W. Inhibition of neutrophil elastase contributes to attenuation of lipopolysaccharide-induced acute lung injury during neutropenia recovery in mice. J. Anesth. 2017, 31, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Takayama, T.; Moriguchi, M.; Hayashi, Y.; Mitsuka, Y.; Yoshida, N.; Higaki, T. Neutrophil Elastase Inhibitor Following Liver Resection: A Matched Cohort Study. Hepat. Mon. 2015, 15, e31235. [Google Scholar] [CrossRef]
- Raevens, S.; Van Campenhout, S.; Debacker, P.-J.; Lefere, S.; Verhelst, X.; Geerts, A.; Van Vlierberghe, H.; Colle, I.; Devisscher, L. Combination of sivelestat and N-acetylcysteine alleviates the inflammatory response and exceeds standard treatment for acetaminophen-induced liver injury. J. Leukoc. Biol. 2020, 107, 341–355. [Google Scholar] [CrossRef]
- Yang, T.; Wang, H.; Wang, X.; Li, J.; Jiang, L. The Dual Role of Innate Immune Response in Acetaminophen-Induced Liver Injury. Biology 2022, 11, 1057. [Google Scholar] [CrossRef]
- Guo, H.; Chen, S.; Xie, M.; Zhou, C.; Zheng, M. The complex roles of neutrophils in APAP-induced liver injury. Cell Prolif. 2021, 54, e13040. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Trautwein, C.; Tacke, F. Functional Role of Monocytes and Macrophages for the Inflammatory Response in Acute Liver Injury. Front. Physiol. 2012, 3, 56. [Google Scholar] [CrossRef]
- Tujios, S.; Fontana, R.J. Mechanisms of drug-induced liver injury: From bedside to bench. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Bajt, M.L.; Sharpe, M.R.; McGill, M.; Farhood, A.; Jaeschke, H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol. Appl. Pharmacol. 2014, 275, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Schulze-Osthoff, K. Mechanisms of Cell Death in Acute Liver Failure. Front. Physiol. 2012, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.E.; Oliveira, A.G.; Pereira, R.V.; David, B.A.; Gomides, L.F.; Saraiva, A.M.; Pires, D.A.; Novaes, J.T.; Patricio, D.O.; Cisalpino, D.; et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2014, 61, 348–360. [Google Scholar] [CrossRef]
- Hu, B.; Colletti, L.M. CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. Hepatology 2010, 52, 691–702. [Google Scholar] [CrossRef]
- Hogaboam, C.M.; Bone-Larson, C.L.; Steinhauser, M.L.; Lukacs, N.W.; Colletti, L.M.; Simpson, K.J.; Strieter, R.M.; Kunkel, S.L. Novel CXCR2-dependent liver regenerative qualities of ELR-containing CXC chemokines. FASEB J. 1999, 13, 1565–1574. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating Monocyte-Derived Macrophages and Resident Kupffer Cells Display Different Ontogeny and Functions in Acute Liver Injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.S.; Nadig, D.E.; Kokoska, E.R.; Solomon, H.; Tiniakos, D.G.; Miller, T.A. Role of Neutrophils in Hepatotoxicity Induced by Oral Acetaminophen Administration in Rats. J. Surg. Res. 1998, 80, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; Ofrecio, J.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Mansuy-Aubert, V.; Zhou, Q.L.; Xie, X.; Gong, Z.; Huang, J.-Y.; Khan, A.R.; Aubert, G.; Candelaria, K.; Thomas, S.; Shin, D.-J.; et al. Imbalance between Neutrophil Elastase and its Inhibitor α1-Antitrypsin in Obesity Alters Insulin Sensitivity, Inflammation, and Energy Expenditure. Cell Metab. 2013, 17, 534–548. [Google Scholar] [CrossRef]
- Uchida, Y.; Freitas, M.C.S.; Zhao, D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. The Protective Function of Neutrophil Elastase Inhibitor in Liver Ischemia/Reperfusion Injury. Transplantation 2010, 89, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, N.; Obara, H.; Suda, K.; Takeuchi, H.; Miyasho, T.; Kawasako, K.; Du, W.; Yamada, S.; Ono, S.; Matsumoto, K.; et al. Neutrophil elastase inhibitor improves survival rate after ischemia reperfusion injury caused by supravisceral aortic clamping in rats. J. Surg. Res. 2012, 180, e31–e36. [Google Scholar] [CrossRef]
- Tsujii, S.; Okabayashi, T.; Shiga, M.; Takezaki, Y.; Sugimoto, T.; Kobayashi, M.; Hanazaki, K. The Effect of the Neutrophil Elastase Inhibitor Sivelestat on Early Injury after Liver Resection. World J. Surg. 2012, 36, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Kambe, M.; Bessho, R.; Fujii, M.; Ochi, M.; Shimizu, K. Sivelestat reduces myocardial ischemia and reperfusion injury in rat hearts even when administered after onset of myocardial ischemia. Interact. Cardiovasc. Thorac. Surg. 2009, 8, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Matayoshi, H.; Hirata, T.; Yamashita, S.; Ishida, K.; Mizukami, Y.; Gondo, T.; Matsumoto, M.; Sakabe, T. Neutrophil elastase inhibitor attenuates hippocampal neuronal damage after transient forebrain ischemia in rats. Brain Res. 2009, 1259, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A.-H.; Qiu, Z. Neutrophil elastase inhibitor prevents endotoxin-induced liver injury following experimental partial hepatectomy. Br. J. Surg. 2007, 94, 609–619. [Google Scholar] [CrossRef]
- Nakano, Y.; Kondo, T.; Matsuo, R.; Murata, S.; Fukunaga, K.; Ohkohchi, N. Prevention of Leukocyte Activation by the Neutrophil Elastase Inhibitor, Sivelestat, in the Hepatic Microcirculation After Ischemia-Reperfusion. J. Surg. Res. 2009, 155, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Van Thuy, T.T.; Thuy, L.T.T.; Yoshizato, K.; Kawada, N. Possible Involvement of Nitric Oxide in Enhanced Liver Injury and Fibrogenesis during Cholestasis in Cytoglobin-deficient Mice. Sci. Rep. 2017, 7, srep41888. [Google Scholar] [CrossRef]
- McKim, S.E.; Gäbele, E.; Isayama, F.; Lambert, J.C.; Tucker, L.M.; Wheeler, M.D.; Connor, H.D.; Mason, R.P.; Doll, M.A.; Hein, D.W.; et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: Studies with knockout mice. Gastroenterology 2003, 125, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Ohmori, M.; Takimoto, M.; Ota, H.; Yoshida, T. Cocaine-induced liver injury in mice is mediated by nitric oxide and reactive oxygen species. Eur. J. Pharmacol. 1997, 336, 43–49. [Google Scholar] [CrossRef]
- Morio, L.A.; Chiu, H.; Sprowles, K.A.; Zhou, P.; Heck, D.E.; Gordon, M.K.; Laskin, D.L. Distinct Roles of Tumor Necrosis Factor-α and Nitric Oxide in Acute Liver Injury Induced by Carbon Tetrachloride in Mice. Toxicol. Appl. Pharmacol. 2001, 172, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Razavi, H.M.; Wang, L.F.; Weicker, S.; Rohan, M.; Law, C.; McCormack, D.G.; Mehta, S. Pulmonary neutrophil infiltration in murine sepsis: Role of inducible nitric oxide synthase. Am. J. Respir. Crit. Care Med. 2004, 170, 227–233. [Google Scholar] [CrossRef]
- Ajuebor, M.N.; Virág, L.; Flower, R.J.; Perretti, M.; Szabó, C. Role of inducible nitric oxide synthase in the regulation of neutrophil migration in zymosan-induced inflammation. Immunology 1998, 95, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Taneja, R.; Razavi, H.M.; Law, C.; Gillis, C.; Mehta, S. Specific Role of Neutrophil Inducible Nitric Oxide Synthase in Murine Sepsis-Induced Lung Injury In Vivo. Shock 2012, 37, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-W.; Hsu, C.-M.; Wang, J.-S.; Chen, H.-L.; Chen, J.-S. Inhibition of inducible nitric oxide synthase (iNOS) prevents lung neutrophil deposition and damage in burned rats. Shock 2001, 15, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Hwang, S.-Y.; Hwang, J.-S.; Lee, J.-W.; Oh, E.-S.; Han, I.-O. C6 Glioma Cell Insoluble Matrix Components Enhance Interferon-γ-stimulated Inducible Nitric-oxide Synthase/Nitric Oxide Production in BV2 Microglial Cells. J. Biol. Chem. 2008, 283, 2526–2533. [Google Scholar] [CrossRef]
- Feng, C.-W.; Chen, N.-F.; Chan, T.-F.; Chen, W.-F. Therapeutic Role of Protein Tyrosine Phosphatase 1B in Parkinson’s Disease via Antineuroinflammation and Neuroprotection In Vitro and In Vivo. Park. Dis. 2020, 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Cingolani, F.; Malik, S.A.; Wen, J.; Liu, Y.; Czaja, M.J. Sex-Specific Regulation of Interferon-γ Cytotoxicity in Mouse Liver by Autophagy. Hepatology 2021, 74, 2745–2758. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Koerber, K.; Bang, R.; Guehring, H.; Tiegs, G. Inducible nitric oxide synthase is critical for immune-mediated liver injury in mice. J. Clin. Investig. 2001, 107, 439–447. [Google Scholar] [CrossRef]
- Dos Santos Tenório, M.C.; Graciliano, N.G.; Moura, F.; de Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Fisher, E.S.; Curry, S.C. Evaluation and treatment of acetaminophen toxicity. Adv. Pharmacol. 2019, 85, 263–272. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Sugiyama, S.; Horie, T. Th1/Th2 cytokine balance as a determinant of acetaminophen-induced liver injury. Chem. Interact. 2009, 179, 273–279. [Google Scholar] [CrossRef]
- Casley, W.L.; Menzies, A.; Mousseau, N.; Girard, M.; Moon, T.W.; Whitehouse, L.W. Increased basal expression of hepatic Cyp1a1 and Cyp 1a2 genes in inbred mice selected for susceptibility to acetaminophen-induced hepatotoxicity. Pharmacogenetics 1997, 7, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Welch, K.D.; Wen, B.; Goodlett, D.R.; Yi, E.C.; Lee, H.; Reilly, T.P.; Nelson, S.D.; Pohl, L.R. Proteomic Identification of Potential Susceptibility Factors in Drug-Induced Liver Disease. Chem. Res. Toxicol. 2005, 18, 924–933. [Google Scholar] [CrossRef]
- Cover, C.; Liu, J.; Farhood, A.; Malle, E.; Waalkes, M.P.; Bajt, M.L.; Jaeschke, H. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2006, 216, 98–107. [Google Scholar] [CrossRef]
- Morohoshi, Y.; Matsuoka, K.; Chinen, H.; Kamada, N.; Sato, T.; Hisamatsu, T.; Okamoto, S.; Inoue, N.; Takaishi, H.; Ogata, H.; et al. Inhibition of neutrophil elastase prevents the development of murine dextran sulfate sodium-induced colitis. J. Gastroenterol. 2006, 41, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Kuninaka, Y.; Nosaka, M.; Furuta, M.; Kimura, A.; Taruya, A.; Yamamoto, H.; Shimada, E.; Akiyama, M.; Mukaida, N.; et al. CCL2-Mediated Reversal of Impaired Skin Wound Healing in Diabetic Mice by Normalization of Neovascularization and Collagen Accumulation. J. Investig. Dermatol. 2019, 139, 2517–2527.e5. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Sequence |
|---|---|
| Ifng | (F) 5′-CGGCACAGTCATTGAAAGCCTA-3′ (R) 5′-GTTGCTGATGGCCTGATTGTC-3′ |
| Tnf | (F) 5′-AAGCCTGTAGCCCACGTCGTA-3′ (R) 5′-GGCACCACTAGTTGGTTGTCTTTG-3′ |
| Il1b | (F) 5′-TCCAGGATGAGGACATGAGCAC-3′ (R) 5′-GAACGTCACACACCAGCAGGTTA-3′ |
| Cxcl1 | (F) 5′-TGCACCCAAACCGAAGTC-3′ (R) 5′-GTCAGAAGCCAGCGTTCACC-3′ |
| Cxcl2 | (F) 5’-AAAGTTTGCCTTGACCCTGAA-3’ (R) 5’-CTCAGACAGCGAGGCACATC-3’ |
| Actb | (F) 5′-CATCCGTAAAGACCTCTATGCCAAC-3′ (R) 5′-ATGGAGCCACCGATCCACA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishida, Y.; Zhang, S.; Kuninaka, Y.; Ishigami, A.; Nosaka, M.; Harie, I.; Kimura, A.; Mukaida, N.; Kondo, T. Essential Involvement of Neutrophil Elastase in Acute Acetaminophen Hepatotoxicity Using BALB/c Mice. Int. J. Mol. Sci. 2023, 24, 7845. https://doi.org/10.3390/ijms24097845
Ishida Y, Zhang S, Kuninaka Y, Ishigami A, Nosaka M, Harie I, Kimura A, Mukaida N, Kondo T. Essential Involvement of Neutrophil Elastase in Acute Acetaminophen Hepatotoxicity Using BALB/c Mice. International Journal of Molecular Sciences. 2023; 24(9):7845. https://doi.org/10.3390/ijms24097845
Chicago/Turabian StyleIshida, Yuko, Siying Zhang, Yumi Kuninaka, Akiko Ishigami, Mizuho Nosaka, Isui Harie, Akihiko Kimura, Naofumi Mukaida, and Toshikazu Kondo. 2023. "Essential Involvement of Neutrophil Elastase in Acute Acetaminophen Hepatotoxicity Using BALB/c Mice" International Journal of Molecular Sciences 24, no. 9: 7845. https://doi.org/10.3390/ijms24097845