
Feline Calicivirus P39 Inhibits Innate Immune Responses by Autophagic Degradation of Retinoic Acid Inducible Gene I
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
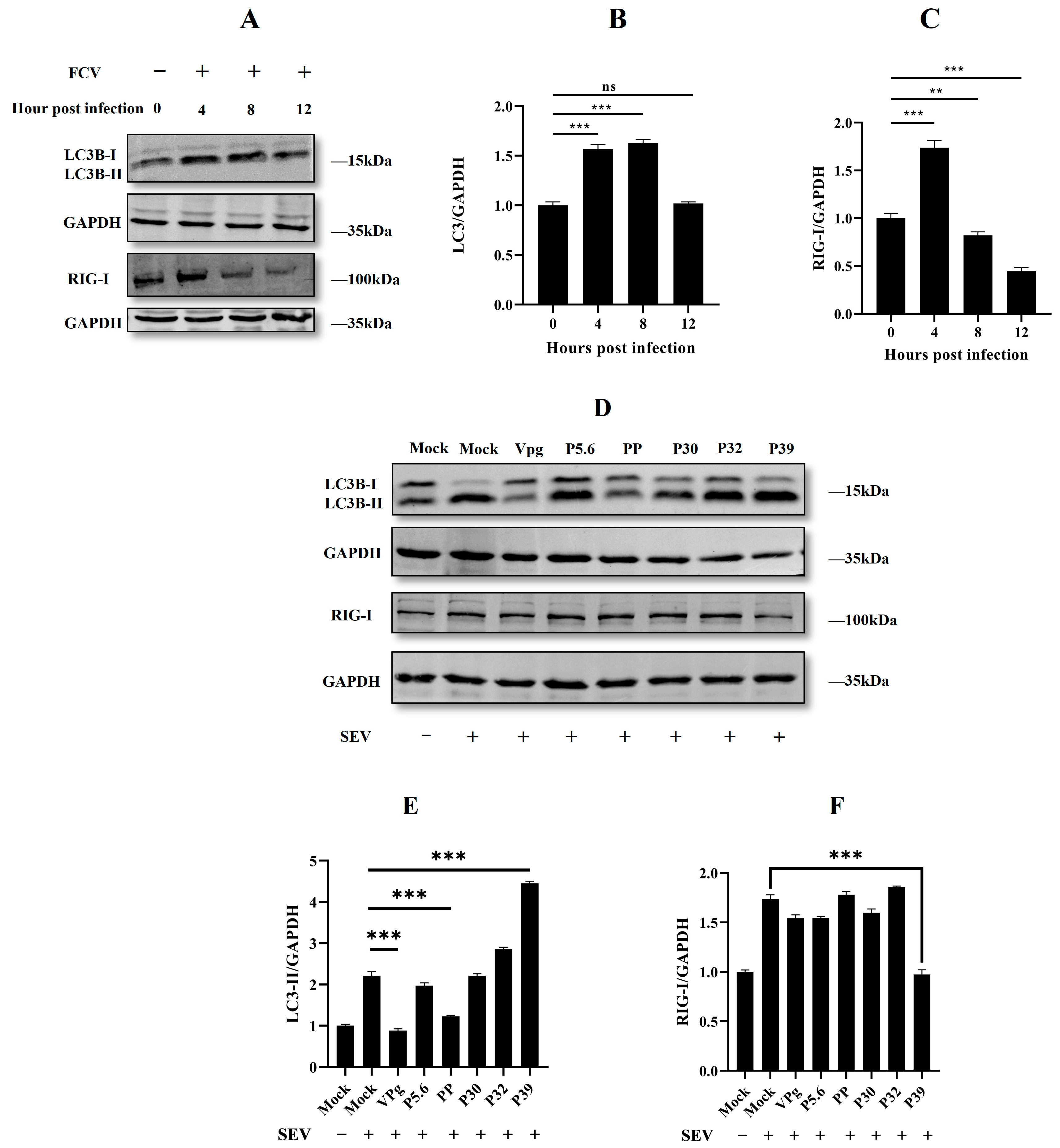
2.1. Feline Calicivirus Infection-Induced Autophagy
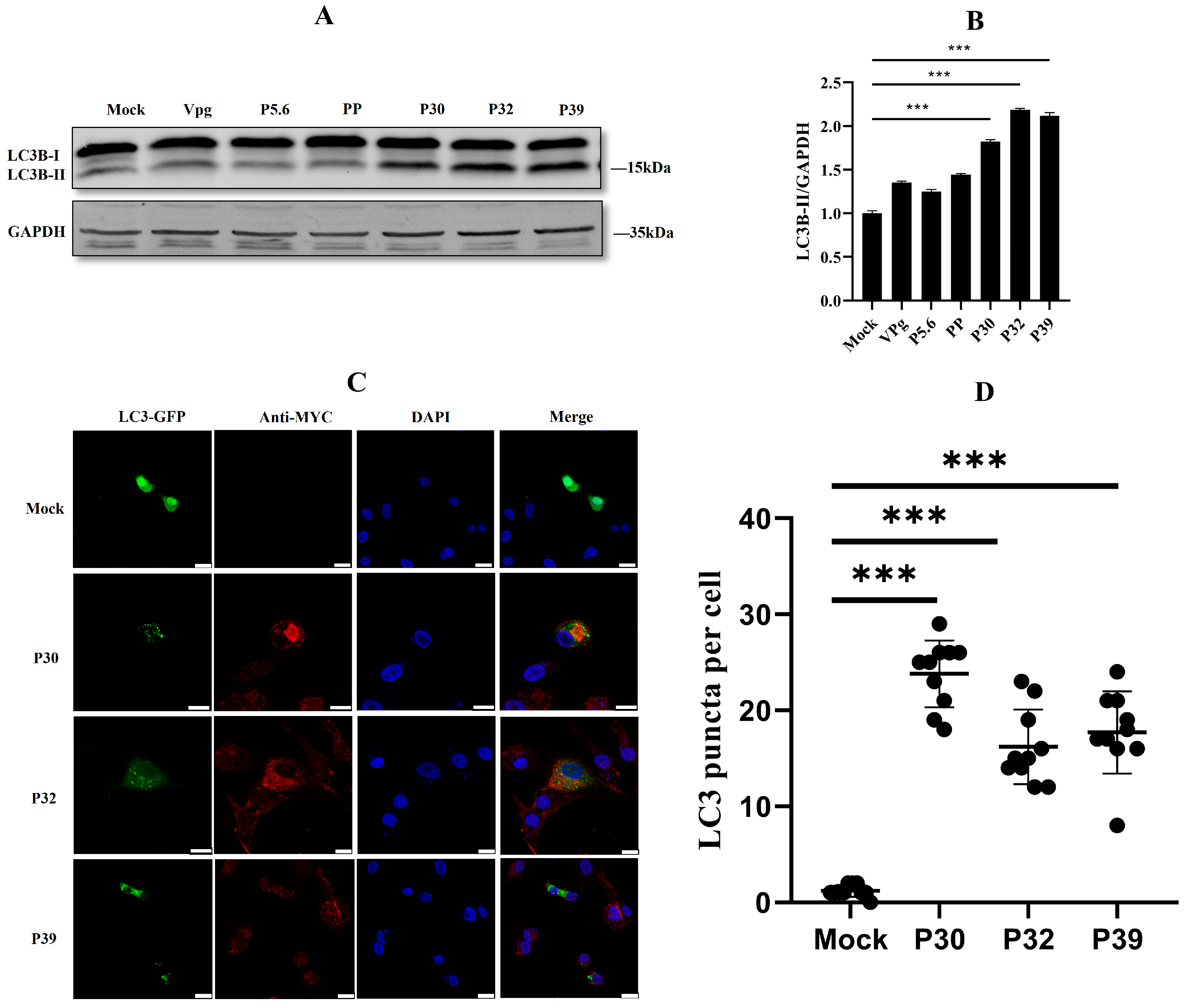
2.2. Non-Structural Protein P30, P32, and P39 Were Responsible for Autophagy Initiation
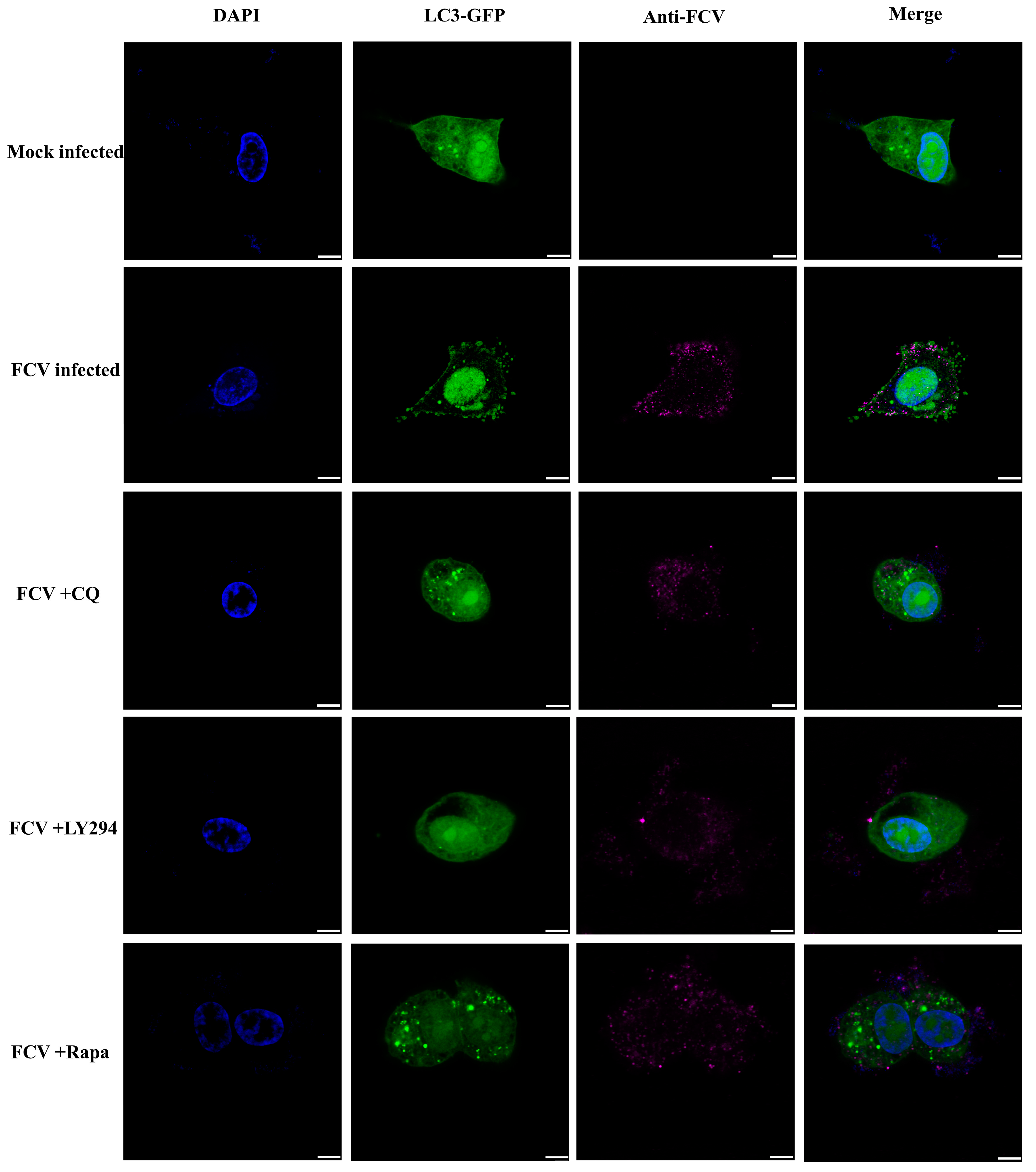
2.3. Autophagy Promoted Feline Calicivirus Replication
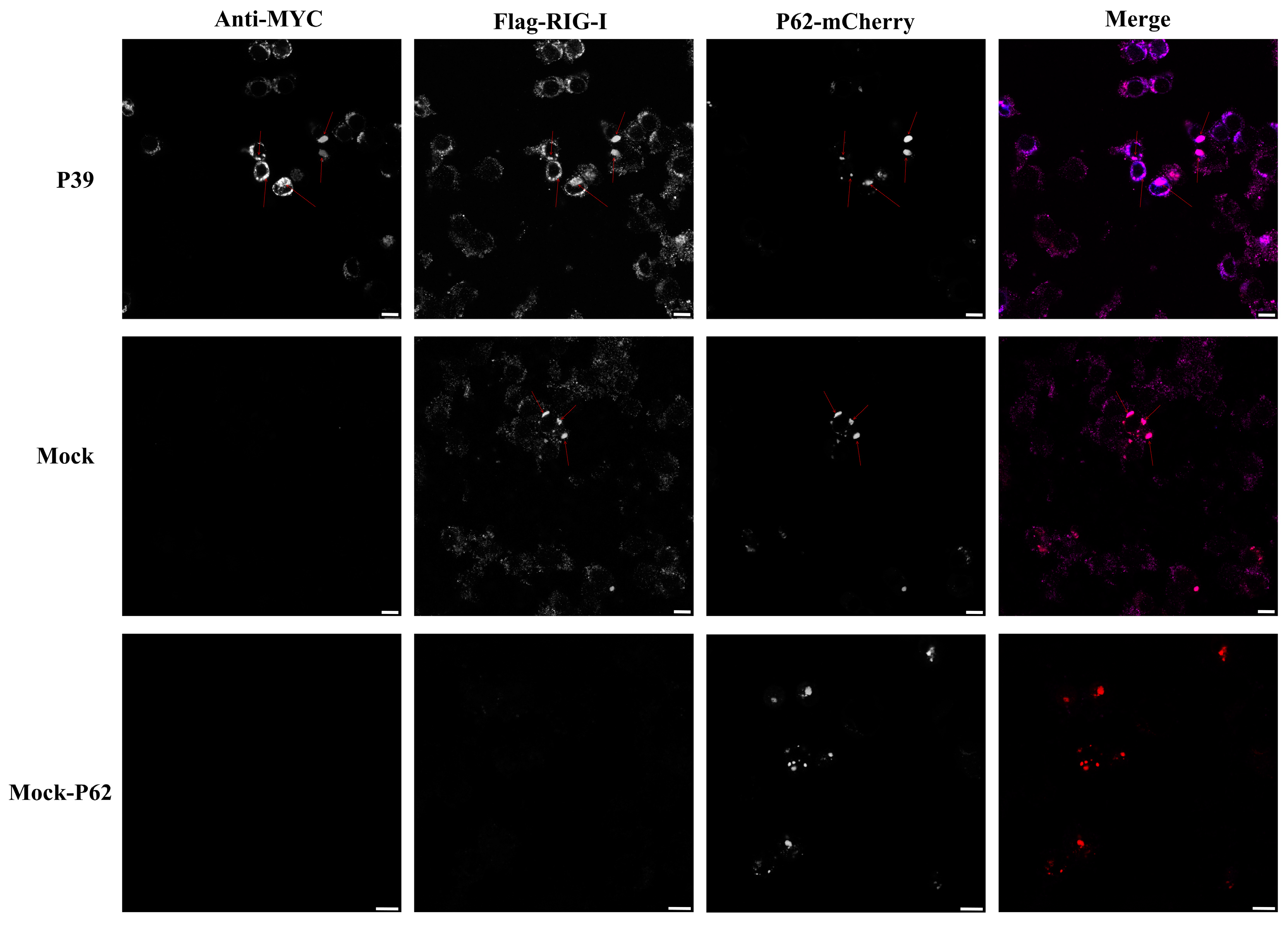
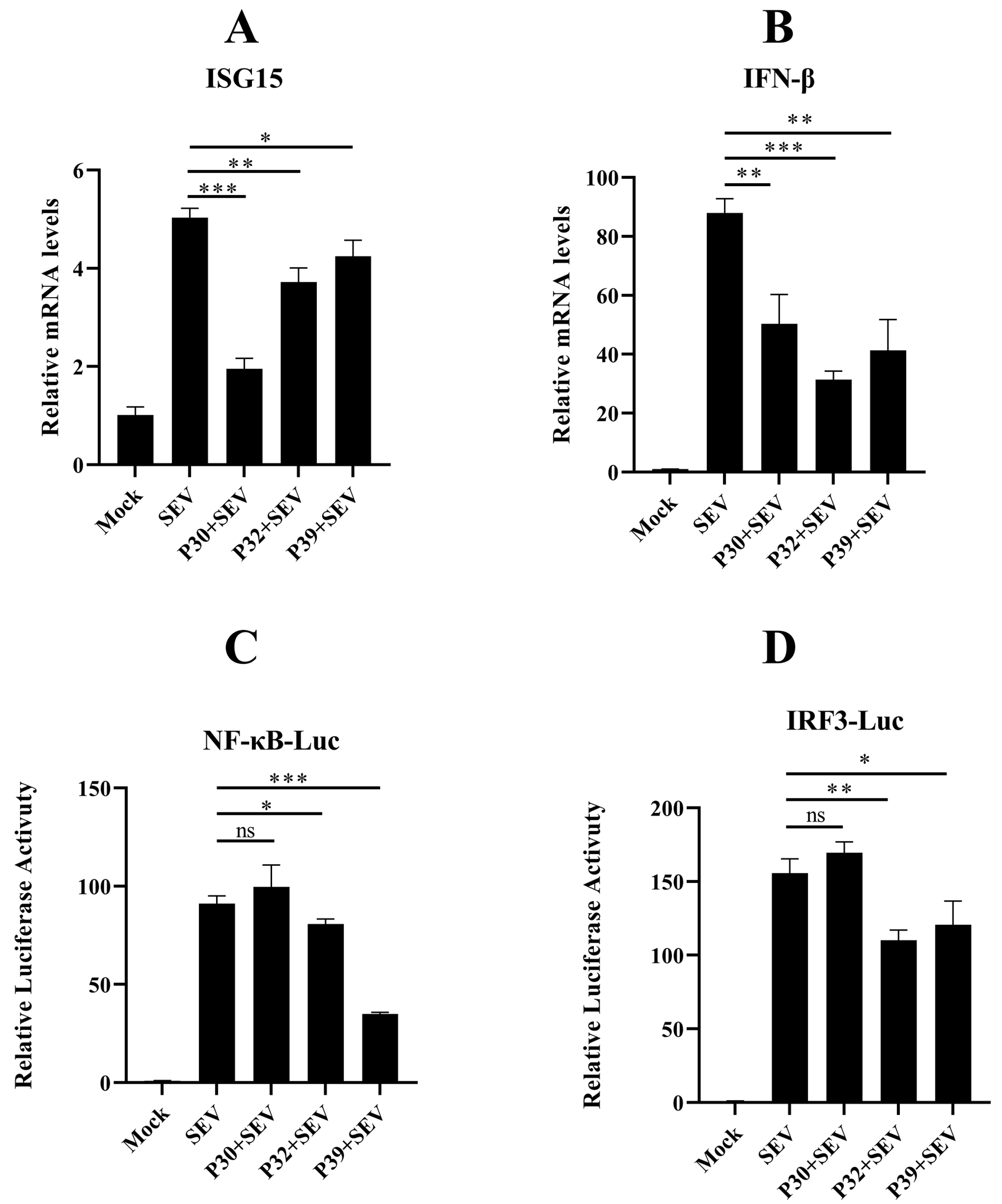
2.4. Feline Calicivirus P39 Promotes Autophagy and Degrades RIG-I
3. Discussion
4. Materials and Methods
4.1. Cells, Virus, Plasmid, Reagents, and Antibodies
4.2. Cell Seeding, Transfection, RNA Extraction, and cDNA Synthesis
4.3. Fluorescence Analysis
4.4. Luciferase Assay
4.5. Western Blotting
4.6. Real-Time qPCR Analysis
4.7. Transmission Electron Microscopy
4.8. Autophagy-Related Treatment
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Radford, A.D.; Coyne, K.P.; Dawson, S.; Porter, C.J.; Gaskell, R.M. Feline calicivirus. Veter-Res. 2007, 38, 319–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.Y.; Ando, T.; Balayan, M.S.; Berke, T.; Clarke, I.; Estes, M.K.; Matson, D.O.; Nakata, S.; Neill, J.D.; Studdert, M.; et al. Taxonomy of the Caliciviruses. J. Infect. Dis. 2000, 181 (Suppl. S2), S322–S330. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, K.L.; Noon, T.H.; Krausman, P.R. Serosurvey of mountain lions in southern Arizona. Wildl. Soc. Bull. 2012, 36, 615–620. [Google Scholar] [CrossRef]
- Nur-Farahiyah, A.N.; Kumar, K.; Yasmin, A.R.; Omar, A.R.; Camalxaman, S.N. Isolation and Genetic Characterization of Canine Parvovirus in a Malayan Tiger. Front. Veter-Sci. 2021, 8, 660046. [Google Scholar] [CrossRef]
- Alhatlani, B.; Vashist, S.; Goodfellow, I. Functions of the 5′ and 3′ ends of calicivirus genomes. Virus Res. 2015, 206, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordicchia, M.; Fumian, T.M.; Van Brussel, K.; Russo, A.G.; Carrai, M.; Le, S.-J.; Pesavento, P.A.; Holmes, E.C.; Martella, V.; White, P.; et al. Feline Calicivirus Virulent Systemic Disease: Clinical Epidemiology, Analysis of Viral Isolates and In Vitro Efficacy of Novel Antivirals in Australian Outbreaks. Viruses 2021, 13, 2040. [Google Scholar] [CrossRef]
- Brunet, S.; Sigoillot-Claude, C.; Pialot, D.; Poulet, H. Multiple Correspondence Analysis on Amino Acid Properties within the Variable Region of the Capsid Protein Shows Differences between Classical and Virulent Systemic Feline Calicivirus Strains. Viruses 2019, 11, 1090. [Google Scholar] [CrossRef] [Green Version]
- Caringella, F.; Elia, G.; Decaro, N.; Martella, V.; Lanave, G.; Varello, K.; Catella, C.; Diakoudi, G.; Carelli, G.; Colaianni, M.L.; et al. Feline calicivirus infection in cats with virulent systemic disease, Italy. Res. Veter-Sci. 2019, 124, 46–51. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, X.; Wu, H.; Liu, C.; Liu, J.; Hu, X.; Qu, L. Assessment of the IFN-β response to four feline caliciviruses: Infection in CRFK cells. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2015, 34, 352–360. [Google Scholar] [CrossRef]
- Bailey, D.; Kaiser, W.J.; Hollinshead, M.; Moffat, K.; Chaudhry, Y.; Wileman, T.; Sosnovtsev, S.V.; Goodfellow, I.G. Feline calicivirus p32, p39 and p30 proteins localize to the endoplasmic reticulum to initiate replication complex formation. J. Gen. Virol. 2009, 91, 739–749. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.B.; Hyde, J.L.; Mintern, J.D.; Mackenzie, J.M. Mouse Norovirus infection promotes autophagy induction to facilitate replication but prevents final autophagosome maturation. Virology 2016, 492, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.-H.; Cho, S.-Y.; Vaidya, B.; Ha, S.H.; Jun, S.; Ro, H.-J.; Lee, Y.; Lee, J.; Kwon, J.; Kim, D. Human Norovirus Replication in Temperature-Optimized MDCK Cells by Forkhead Box O1 Inhibition. J. Microbiol. Biotechnol. 2020, 30, 1412–1419. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Kang, H.; Huang, J.; Li, Z.; Pan, Y.; Li, Y.; Chen, S.; Zhang, J.; Yin, H.; Qu, L. Feline calicivirus strain 2280 p30 antagonizes type I interferon-mediated antiviral innate immunity through directly degrading IFNAR1 mRNA. PLoS Pathog. 2020, 16, e1008944. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Huang, J.; Liu, Y.; Pan, Y.; Li, Y.; Miao, Q.; Qu, L.; Tian, J. Feline Calicivirus Proteinase-Polymerase Protein Degrades mRNAs To Inhibit Host Gene Expression. J. Virol. 2021, 95, e33621. [Google Scholar] [CrossRef]
- Batool, M.; Kim, M.S.; Choi, S. Structural insights into the distinctive RNA recognition and therapeutic potentials of RIG-I-like receptors. Med. Res. Rev. 2021, 42, 399–425. [Google Scholar] [CrossRef]
- Feng, H.; Liu, H.; Kong, R.; Wang, L.; Wang, Y.; Hu, W.; Guo, Q. Expression profiles of carp IRF-3/-7 correlate with the up-regulation of RIG-I/MAVS/TRAF3/TBK1, four pivotal molecules in RIG-I signaling pathway. Fish Shellfish. Immunol. 2011, 30, 1159–1169. [Google Scholar] [CrossRef]
- Lazear, H.M.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.W.; et al. IRF-3, IRF-5, and IRF-7 Coordinately Regulate the Type I IFN Response in Myeloid Dendritic Cells Downstream of MAVS Signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Lee, N.-R.; Ban, J.; Lee, N.-J.; Yi, C.-M.; Choi, J.-Y.; Kim, H.; Kil Lee, J.; Seong, J.; Cho, N.-H.; Jung, J.U.; et al. Activation of RIG-I-Mediated Antiviral Signaling Triggers Autophagy through the MAVS-TRAF6-Beclin-1 Signaling Axis. Front. Immunol. 2018, 9, 2096. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Tan, C.; Yang, X.; Wang, J.; Li, Q.; Xu, L.; Wang, Z.; Mao, J.; Wang, J.; Cheng, K.; et al. Transcriptome Analysis of Retinoic Acid-Inducible Gene I Overexpression Reveals the Potential Genes for Autophagy-Related Negative Regulation. Cells 2022, 11, 2009. [Google Scholar] [CrossRef]
- Li, X.; Song, Y.; Wang, X.; Fu, C.; Zhao, F.; Zou, L.; Wu, K.; Chen, W.; Li, Z.; Fan, J.; et al. The regulation of cell homeostasis and antiviral innate immunity by autophagy during classical swine fever virus infection. Emerg. Microbes Infect. 2023, 12, 2164217. [Google Scholar] [CrossRef]
- Yumiketa, Y.; Narita, T.; Inoue, Y.; Sato, G.; Kamitani, W.; Oka, T.; Katayama, K.; Sakaguchi, T.; Tohya, Y. Nonstructural protein p39 of feline calicivirus suppresses host innate immune response by preventing IRF-3 activation. Veter-Microbiol. 2016, 185, 62–67. [Google Scholar] [CrossRef]
- Cai, Y.; Zhu, Y.; Zheng, J.; Zhang, Y.; Chen, W. NBR1 mediates autophagic degradation of IRF3 to negatively regulate type I interferon production. Biochem. Biophys. Res. Commun. 2022, 623, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhu, H.; Xu, X.; Li, L.; Tan, H.; Cai, X. Inactivated Sendai virus induces apoptosis and autophagy via the PI3K/Akt/mTOR/p70S6K pathway in human non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 465, 64–70. [Google Scholar] [CrossRef]
- Cui, B.; Lin, H.; Yu, J.; Yu, J.; Hu, Z. Autophagy and the Immune Response. In Autophagy: Biology and Diseases: Basic Science; Qin, Z., Ed.; Springer: Singapore, 2019; pp. 595–634. [Google Scholar]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Yang, K.; Jia, P.; Liu, L.; Lin, Y.; Li, Z.; Li, J.; Chen, S.; Guo, S.; Pan, J.; et al. A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. 2020, 31, 62–79. [Google Scholar] [CrossRef]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2017, 37, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Mok, B.W.-Y.; Deng, S.; Liu, H.; Wang, P.; Song, W.; Chen, P.; Huang, X.; Zheng, M.; Lau, S.-Y.; et al. Mammalian cells use the autophagy process to restrict avian influenza virus replication. Cell Rep. 2021, 35, 109213. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.-S.; De Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; García-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [Green Version]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-Interacting Motif in the Influenza A Virus M2 Protein Is Required to Subvert Autophagy and Maintain Virion Stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guévin, C.; Manna, D.; Bélanger, C.; Konan, K.V.; Mak, P.; Labonté, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tian, Y.; Ou, J.-H.J. HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [Green Version]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C.; Mizushima, N.; Yoshimori, T.; et al. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Kohler, V.; Aufschnaiter, A.; Büttner, S. Closing the Gap: Membrane Contact Sites in the Regulation of Autophagy. Cells 2020, 9, 1184. [Google Scholar] [CrossRef]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Iv, H.W.V.; Colonna, M. MDA-5 Recognition of a Murine Norovirus. PLoS Pathog. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Dang, W.; Xu, L.; Yin, Y.; Chen, S.; Wang, W.; Hakim, M.S.; Chang, K.-O.; Peppelenbosch, M.P.; Pan, Q. IRF-1, RIG-I and MDA5 display potent antiviral activities against norovirus coordinately induced by different types of interferons. Antivir. Res. 2018, 155, 48–59. [Google Scholar] [CrossRef]
- Trzeciak-Ryczek, A.; Tokarz-Deptuła, B.; Deptuła, W. Expression of IL-1β, IL-2, IL-10, TNF-β and GM-CSF in peripheral blood leukocytes of rabbits experimentally infected with rabbit haemorrhagic disease virus. Veter-Microbiol. 2016, 186, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.-Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [Green Version]
- Seungmin, H.; Nicole, S.M.; Monique, W.B.; Gautam, G.; Erning, D.; Lei, Z.; Bimmi, S.; Michael, S.D.; Adish, D.; Stanislav, V.S.; et al. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 2012, 11, 397–409. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, J.; Ye, S.; Deng, J.; Song, J.; Wang, Z.; Chen, A.; Zhou, P.; Li, S. Feline Calicivirus P39 Inhibits Innate Immune Responses by Autophagic Degradation of Retinoic Acid Inducible Gene I. Int. J. Mol. Sci. 2023, 24, 5254. https://doi.org/10.3390/ijms24065254
Mao J, Ye S, Deng J, Song J, Wang Z, Chen A, Zhou P, Li S. Feline Calicivirus P39 Inhibits Innate Immune Responses by Autophagic Degradation of Retinoic Acid Inducible Gene I. International Journal of Molecular Sciences. 2023; 24(6):5254. https://doi.org/10.3390/ijms24065254
Chicago/Turabian StyleMao, Jianwei, Shaotang Ye, Jie Deng, Jie Song, Zhen Wang, Aolei Chen, Pei Zhou, and Shoujun Li. 2023. "Feline Calicivirus P39 Inhibits Innate Immune Responses by Autophagic Degradation of Retinoic Acid Inducible Gene I" International Journal of Molecular Sciences 24, no. 6: 5254. https://doi.org/10.3390/ijms24065254