
QTL Mapping of Soybean (Glycine max) Vine Growth Habit Trait
 ,
,
Abstract
:1. Introduction
2. Results
2.1. VGH Characterization of Parental and RIL Soybean Plants
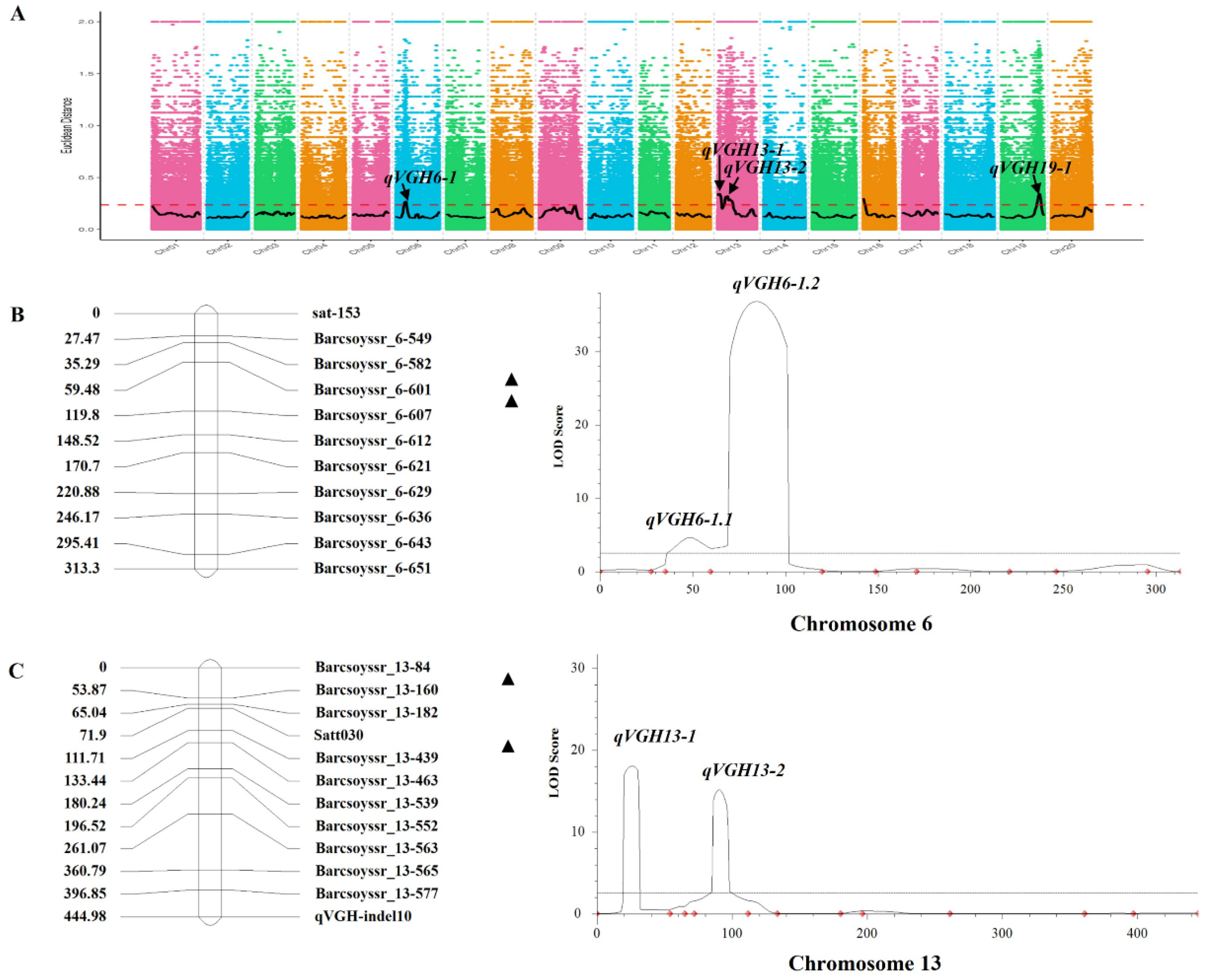
2.2. BSA-Based Identification of VGH-Associated Loci
2.3. Identifying and Fine Mapping QTL in F4 RILs
2.4. VGH-Related QTL Analyses in the F6-F8 Population Derived from ZD41 × ZYD02878
2.5. Candidate Gene Analyses
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Phenotypic Analyses
4.2. Genotyping of Individual and DNA Bulk Samples
4.3. Linkage Map Construction and QTL Mapping
4.4. Candidate Genes Prioritization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cao, D.; Takeshima, R.; Zhao, C.; Liu, B.; Jun, A.; Kong, F. Molecular mechanisms of flowering under long days and stem growth habit in soybean. J. Exp. Bot. 2017, 68, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, L.; Kong, J.; Xu, Z.; Akhter Bhat, J.; Zhao, T. QTL architecture of vine growth habit and gibberellin oxidase gene diversity in wild soybean (Glycine soja). Sci. Rep. 2019, 9, 7393. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Y.; Wei, H.; Hufnagel, D.E.; Wang, Y.; Guo, S.; Li, Y.; Wang, L.; Qiu, L.-J. The prevalence of deleterious mutations during the domestication and improvement of soybean. Crop J. 2023, 11, 523–530. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, J.; Guo, X.; Chen, J.; Chang, R.; Guan, R.; Qiu, L. Identification of genomic regions associated with vine growth and plant height of soybean. Int. J. Mol. Sci. 2022, 23, 5823. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fujita, T.; Yan, Z.H.; Sakamoto, S.; Xu, D.; Abe, J. QTL mapping of domestication-related traits in soybean (Glycine max). Ann. Bot. 2007, 100, 1027–1038. [Google Scholar] [CrossRef]
- Swarm, S.A.; Sun, L.; Wang, X.; Wang, W.; Brown, P.J.; Ma, J.; Nelson, R.L. Genetic dissection of domestication-related traits in soybean through genotyping-by-sequencing of two interspecific mapping populations. Theor. Appl. Genet. 2019, 132, 1195–1209. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.W.; Wong, F.L.; Luk, C.Y.; Chung, C.Y.; Yung, W.S.; Wang, Z.; Xie, M.; Song, S.; Chung, G.; et al. Increased copy number of gibberellin 2-oxidase 8 genes reduced trailing growth and shoot length during soybean domestication. Plant J. 2021, 107, 1739–1755. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, X.; Lee, R.; Li, Y.; Specht, J.E.; Nelson, R.L.; McClean, P.E.; Qiu, L.; Ma, J. Artificial selection for determinate growth habit in soybean. Proc. Natl. Acad. Sci. USA 2010, 107, 8563–8568. [Google Scholar] [CrossRef]
- Ping, J.; Liu, Y.; Sun, L.; Zhao, M.; Li, Y.; She, M.; Sui, Y.; Lin, F.; Liu, X.; Tang, Z.; et al. Dt2 is a gain-of-function MADS-domain factor gene that specifies semideterminacy in soybean. Plant Cell 2014, 26, 2831–2842. [Google Scholar] [CrossRef]
- Liu, B.; Watanabe, S.; Uchiyama, T.; Kong, F.; Kanazawa, A.; Xia, Z.; Nagamatsu, A.; Arai, M.; Yamada, T.; Kitamura, K.; et al. The soybean stem growth habit gene Dt1 is an ortholog of Arabidopsis TERMINAL FLOWER1. Plant Physiol. 2010, 153, 198–210. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.; Ping, J.; Li, S.; Chen, Z.; Ma, J. Innovation of a Regulatory Mechanism Modulating Semi-determinate Stem Growth through Artificial Selection in Soybean. PLoS Genet. 2016, 12, e1005818. [Google Scholar] [CrossRef]
- Chen, Y.; Xiong, Y.; Hong, H.; Li, G.; Gao, J.; Guo, Q.; Sun, R.; Ren, H.; Zhang, F.; Wang, J.; et al. Genetic dissection of and genomic selection for seed weight, pod length, and pod width in soybean. Crop J. 2023, 11, 832–841. [Google Scholar] [CrossRef]
- Liu, W.; Zou, J.; Zhang, J.; Yang, F.; Yang, W. Evaluation of Soybean (Glycine max) Stem Vining in Maize-Soybean Relay Strip Intercropping System. Proc. Jpn. Acad. Ser. A Math. Sci. 2015, 91, 69–75. [Google Scholar] [CrossRef]
- Campbell, B.W.; Hofstad, A.N.; Sreekanta, S.; Fu, F.; Kono, T.J.Y.; O’Rourke, J.A.; Vance, C.P.; Muehlbauer, G.J.; Stupar, R.M. Fast neutron-induced structural rearrangements at a soybean NAP1 locus result in gnarled trichomes. Theor. Appl. Genet. 2016, 129, 1725–1738. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, Z.; Liu, Z.; Guo, Y.; Qiu, L.J. Next-generation sequencing from bulked-segregant analysis accelerates the simultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 2017, 8, 919. [Google Scholar] [CrossRef] [PubMed]
- Amin, G.M.A. Characterization and rapid gene-mapping of leaf lesion mimic phenotype of spl-1 mutant in soybean (Glycine max (L.) Merr.). Int. J. Mol. Sci. 2019, 20, 2193. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jiang, H.; Zhang, Z.; Zhao, Y.; Chen, Q. Combined linkage mapping and BSA to identify QTL and candidate genes for plant height and the number of nodes on the main stem in soybean. Int. J. Mol. Sci. 2019, 21, 42. [Google Scholar] [CrossRef]
- Liu, X.T.; Wu, X.Y.; Wu, W.P.; Wu, M.; Chen, J.Q.; Wang, B. A bean common mosaic virus-resistance gene in the soybean variant V94-5152 was mapped to the Rsv4 locus conferring resistance to soybean mosaic virus. Theor. Appl. Genet. 2021, 134, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.P.; Zaccaron, A.Z.; Bluhm, B.H.; Rupe, J.C.; Wood, L.; Mozzoni, L.A.; Mason, R.E.; Yingling, S.; Pereira, A. Bulked segregant analysis using next-generation sequencing for identification of genetic loci for charcoal rot resistance in soybean. Physiol. Mol. Plant Pathol. 2020, 109, 101440. [Google Scholar] [CrossRef]
- Carpenter, J.A.; Fehr, W.R. Genetic variability for desirable agronomic traits in populations containing Glycine soja germplasm. Crop Sci. 1986, 26, 681–686. [Google Scholar] [CrossRef]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting Fst. Nat. Rev. Genet. 2009, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, G.; Patterson, N.; Sankararaman, S.; Price, A.L. Estimating and interpreting FST: The impact of rare variants. Genome Res. 2013, 23, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; Guo, Y.; Ou, L.; Hong, H.; Wang, J.; Liu, Z.X.; Guo, B.; Zhang, L.; Qiu, L. Identification of the dwarf gene GmDW1 in soybean ( Glycine max L.) by combining mapping-by-sequencing and linkage analysis. Theor. Appl. Genet. 2018, 131, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wu, H.; Guo, Y.; Gao, H.; Wei, Z.; Zhao, Y.; Qiu, L. GmIAA27 encodes an AUX/IAA protein involved in dwarfing and multi-branching in soybean. Int. J. Mol. Sci. 2022, 23, 8643. [Google Scholar] [CrossRef]
- Nakamura, A. The Role of OsBRI1 and Its Homologous Genes, OsBRL1 and OsBRL3, in Rice. Plant Physiol. 2006, 140, 580–590. [Google Scholar] [CrossRef]
- Hu, X.; Qian, Q.; Xu, T.; Zhang, Y.; Dong, G.; Gao, T.; Xie, Q.; Xue, Y. The U-box E3 ubiquitin ligase TUD1 functions with a heterotrimeric G alpha subunit to regulate Brassinosteroid-mediated growth in rice. PLoS Genet. 2013, 9, e1003391. [Google Scholar] [CrossRef]
- Tian, X.; Li, X.; Zhou, W.; Ren, Y.; Wang, Z.; Liu, Z.; Tang, J.; Tong, H.; Fang, J.; Bu, Q. Transcription factor OsWRKY53 positively regulates brassinosteroid signaling and plant architecture. Plant Physiol. 2017, 175, 1337–1349. [Google Scholar] [CrossRef]
- Liu, K.; Cao, J.; Yu, K.; Liu, X.; Gao, Y.; Chen, Q.; Zhang, W.; Peng, H.; Du, J.; Xin, M.; et al. Wheat TaSPL8 Modulates Leaf Angle Through Auxin and Brassinosteroid Signaling1[OPEN]. Plant Physiol. 2019, 181, 179–194. [Google Scholar] [CrossRef]
- Tian, J.G.; Wang, C.L.; Xia, J.L.; Wu, L.S.; Xu, G.H.; Wu, W.H.; Li, D.; Qin, W.C.; Han, X.; Chen, Q.Y.; et al. Teosinte ligule allele narrows plant architecture and enhances high-density maize yields. Science 2019, 365, 658–664. [Google Scholar] [CrossRef]
- Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Yost, H.J. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping:Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Li, Y.H.; Qin, C.; Wang, L.; Jiao, C.; Hong, H.; Tian, Y.; Li, Y.; Xing, G.; Wang, J.; Gu, Y.; et al. Genome-wide signatures of the geographic expansion and breeding of soybean. Sci. China Life Sci. 2023, 66, 350–365. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Song, Q.; Hyten, D.L.; Jia, G.; Quigley, C.V.; Fickus, E.W.; Nelson, R.L.; Cregan, P.B. Development and Evaluation of SoySNP50K, a High-Density Genotyping Array for Soybean. PLoS ONE 2013, 8, e54985. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| QTLs | Chr | Start (W82a2) | End (W82a2) | Position (Mb) | Gene_Number | Genotype |
|---|---|---|---|---|---|---|
| qVGH6-1 | 6 | 9,580,000 | 12,440,000 | 2.86 | 585 | SNP, InDel |
| qVGH9-1 | 9 | 41,910,000 | 42,920,000 | 1.01 | 187 | SNP |
| qVGH13-1 | 13 | 0 | 4,410,000 | 4.41 | 314 | SNP, InDel |
| qVGH13-2 | 13 | 8,030,000 | 18,160,000 | 10.13 | 740 | SNP, InDel |
| qVGH16-1 | 16 | 0 | 1,900,000 | 1.90 | 379 | SNP, InDel |
| qVGH19-1 | 19 | 42,250,000 | 47,850,000 | 5.60 | 1062 | SNP, InDel |
| QTL Name | Environment | Chr. | Start (W82a4) | End (W82a4) | Range (Mb) |
|---|---|---|---|---|---|
| qVGH2-1 | 19JZ,19SY,BLUP | 2 | 44,549,340 | 44,748,366 | 0.2 |
| qVGH2-2 | 20JZ,BLUP | 2 | 48,133,595 | 49,091,931 | 0.96 |
| qVGH10-1 | 19JZ,20JZ | 10 | 7,448,517 | 7,648,577 | 0.2 |
| qVGH10-2 | 19SY,BLUP | 10 | 8,298,686 | 9,212,033 | 0.91 |
| qVGH10-3 | 19SY,BLUP | 10 | 45,339,503 | 45,848,959 | 0.51 |
| qVGH13-3 | 19JZ,BLUP | 13 | 39,347,224 | 39,548,455 | 0.21 |
| qVGH18-1 | 19JZ,BLUP | 18 | 7,843,488 | 8,011,289 | 0.17 |
| qVGH19-1.1 | 19JZ,19SY,20JZ,BLUP | 19 | 45,447,240 | 45,839,781 | 0.39 |
| qVGH19-1.2 | 19SY,20JZ,BLUP | 19 | 46,449,908 | 46,641,266 | 0.19 |
| QTL Name | Gene | Annotation | Fst > 0.6 |
|---|---|---|---|
| qVGH6-1.2 | Glyma.06G141100 | Leucine-rich repeat protein kinase family protein | Yes |
| Glyma.06G140600 | RING/U-box superfamily protein | Yes | |
| Glyma.06G140700 | -- | Yes | |
| qVGH13-1 | Glyma.13G008100 | Stress-responsive A/B Barrel Domain | No |
| Glyma.13G006500 | NUDT2, nudix hydrolase homolog 2 | No | |
| qVGH13-2 | Glyma.13G029500 | UDP-glucosyl transferase 85A2 | No |
| qVGH13-3 | Glyma.13G302800 | Major facilitator superfamily protein | Yes |
| Glyma.13G302900 | Photosynthetic electron transfer C | No | |
| Glyma.13G304000 | GH3 auxin-responsive promoter (GH3) | Yes | |
| qVGH19-1.1 | Glyma.19G192700 | Growth-regulating factor 4 | No |
| Glyma.19G194300 | TFL1, PEBP (phosphatidylethanolamine-binding protein) Family protein | No | |
| qVGH19-1.2 | Glyma.19G203100 | Sterile alpha motif (SAM) domain-containing protein | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Liu, K.; Yang, X.; Chen, Y.; Xiong, Y.; Yang, Q.; Wang, J.; Zhang, Z.; Wu, C.; Wang, J.; et al. QTL Mapping of Soybean (Glycine max) Vine Growth Habit Trait. Int. J. Mol. Sci. 2023, 24, 14770. https://doi.org/10.3390/ijms241914770
Song J, Liu K, Yang X, Chen Y, Xiong Y, Yang Q, Wang J, Zhang Z, Wu C, Wang J, et al. QTL Mapping of Soybean (Glycine max) Vine Growth Habit Trait. International Journal of Molecular Sciences. 2023; 24(19):14770. https://doi.org/10.3390/ijms241914770
Chicago/Turabian StyleSong, Jian, Kanglin Liu, Xuezhen Yang, Yijie Chen, Yajun Xiong, Qichao Yang, Jing Wang, Zhihao Zhang, Caiyu Wu, Jun Wang, and et al. 2023. "QTL Mapping of Soybean (Glycine max) Vine Growth Habit Trait" International Journal of Molecular Sciences 24, no. 19: 14770. https://doi.org/10.3390/ijms241914770