
Elucidating the Interaction between Pyridoxine 5′-Phosphate Oxidase and Dopa Decarboxylase: Activation of B6-Dependent Enzyme



Abstract
:1. Introduction
2. Results and Discussion
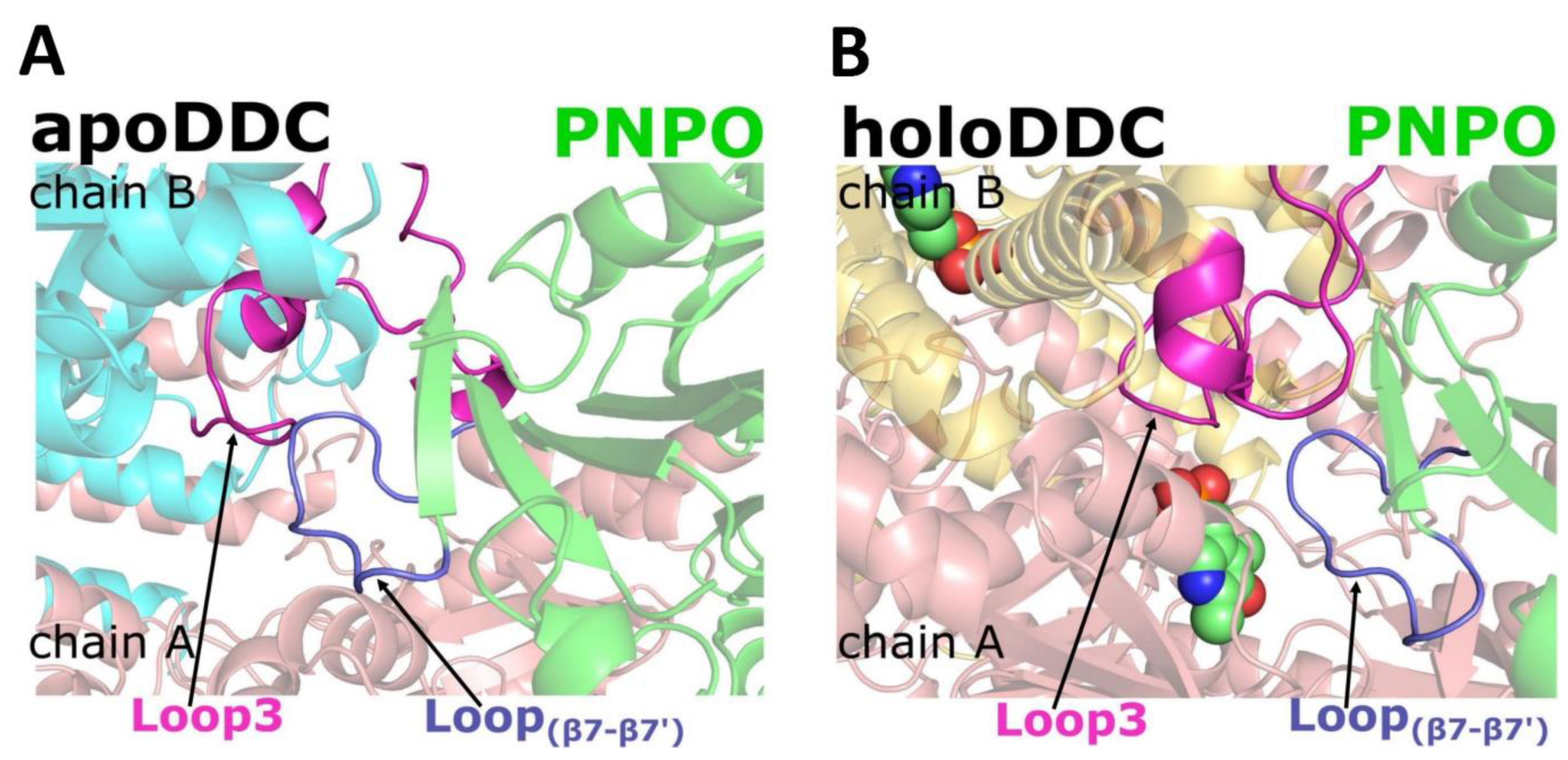
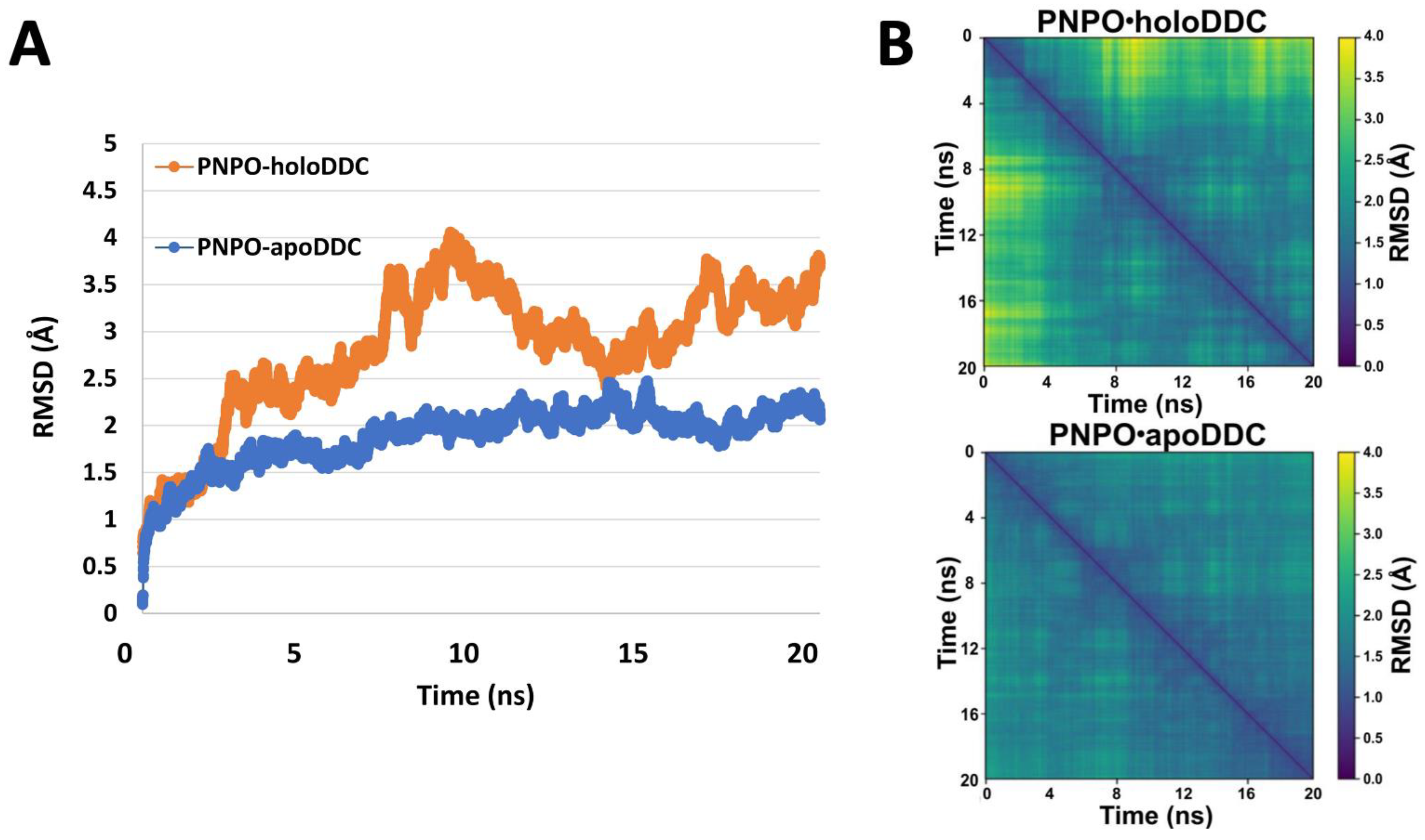
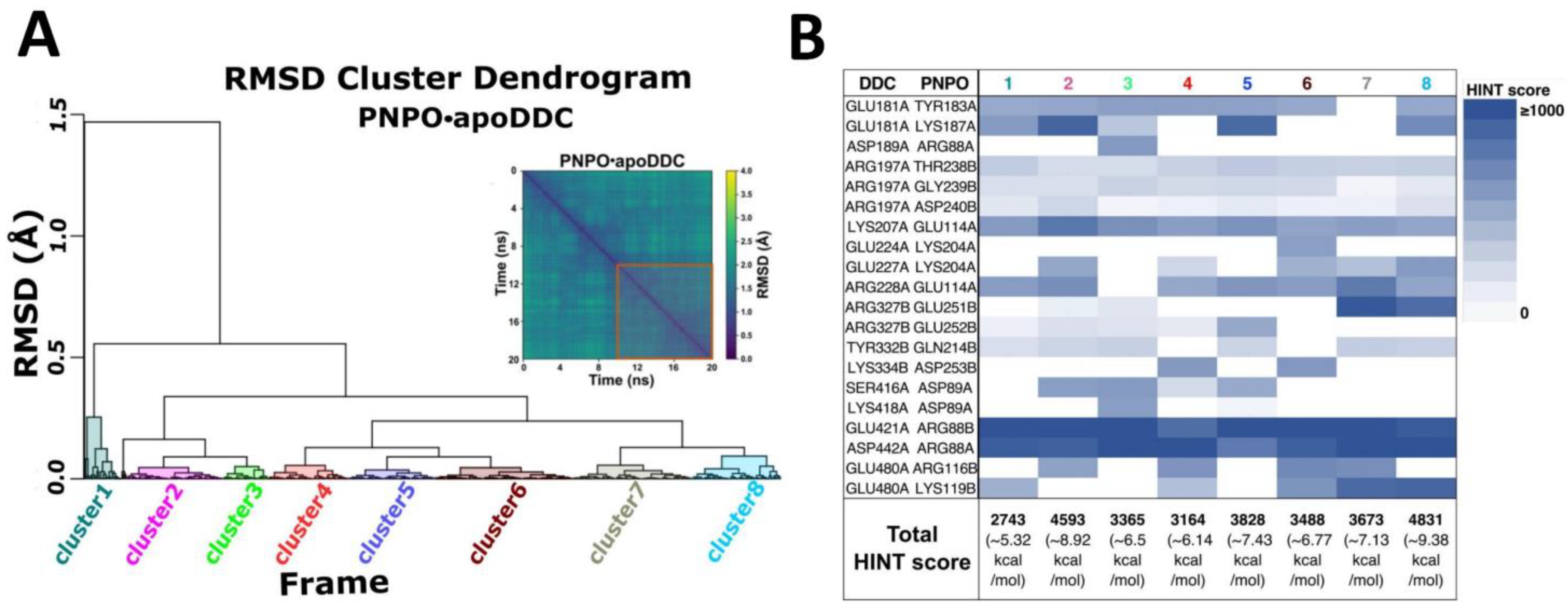
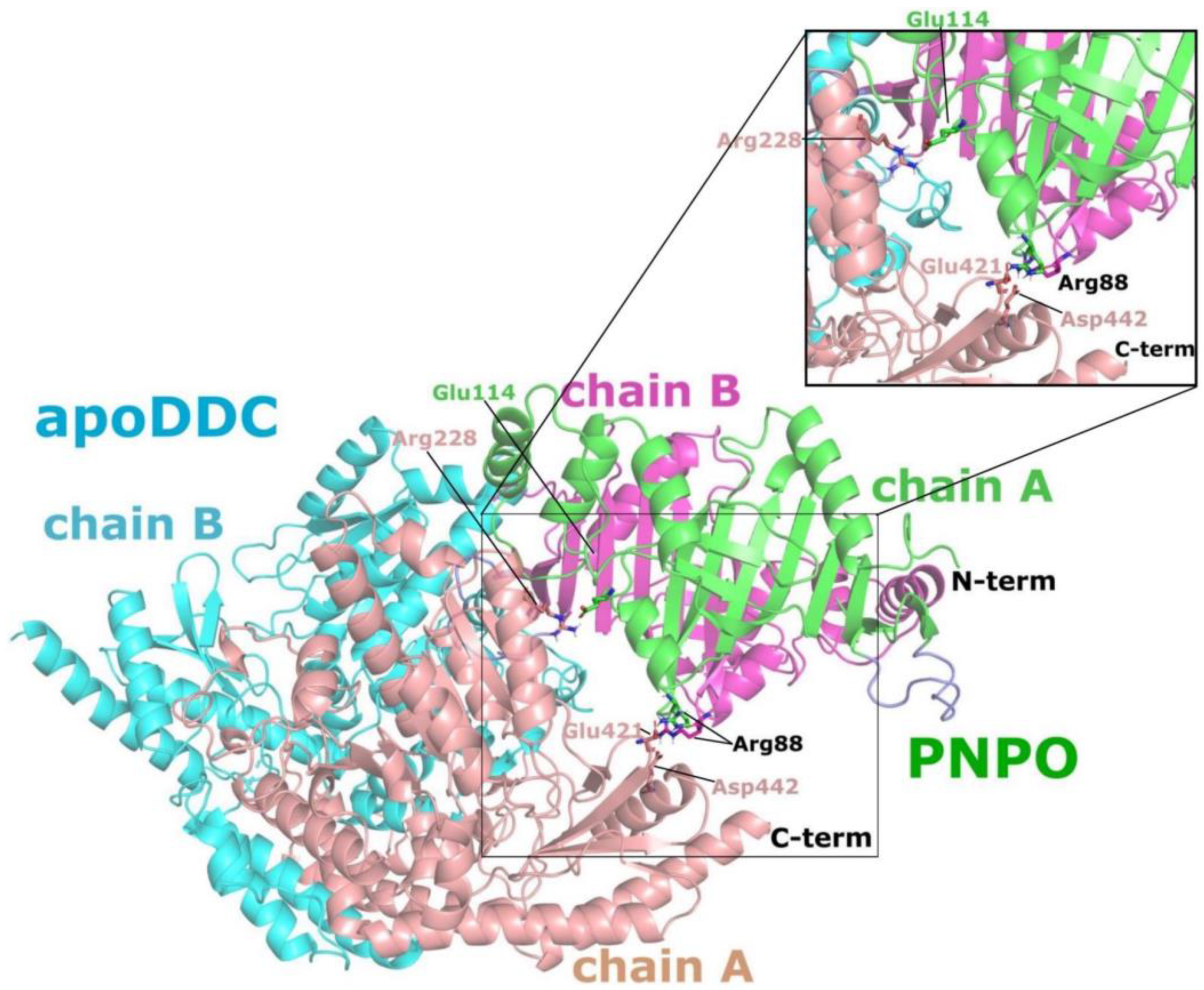
2.1. Predicting Interactions between hPNPO and DDC Using Molecular Modeling
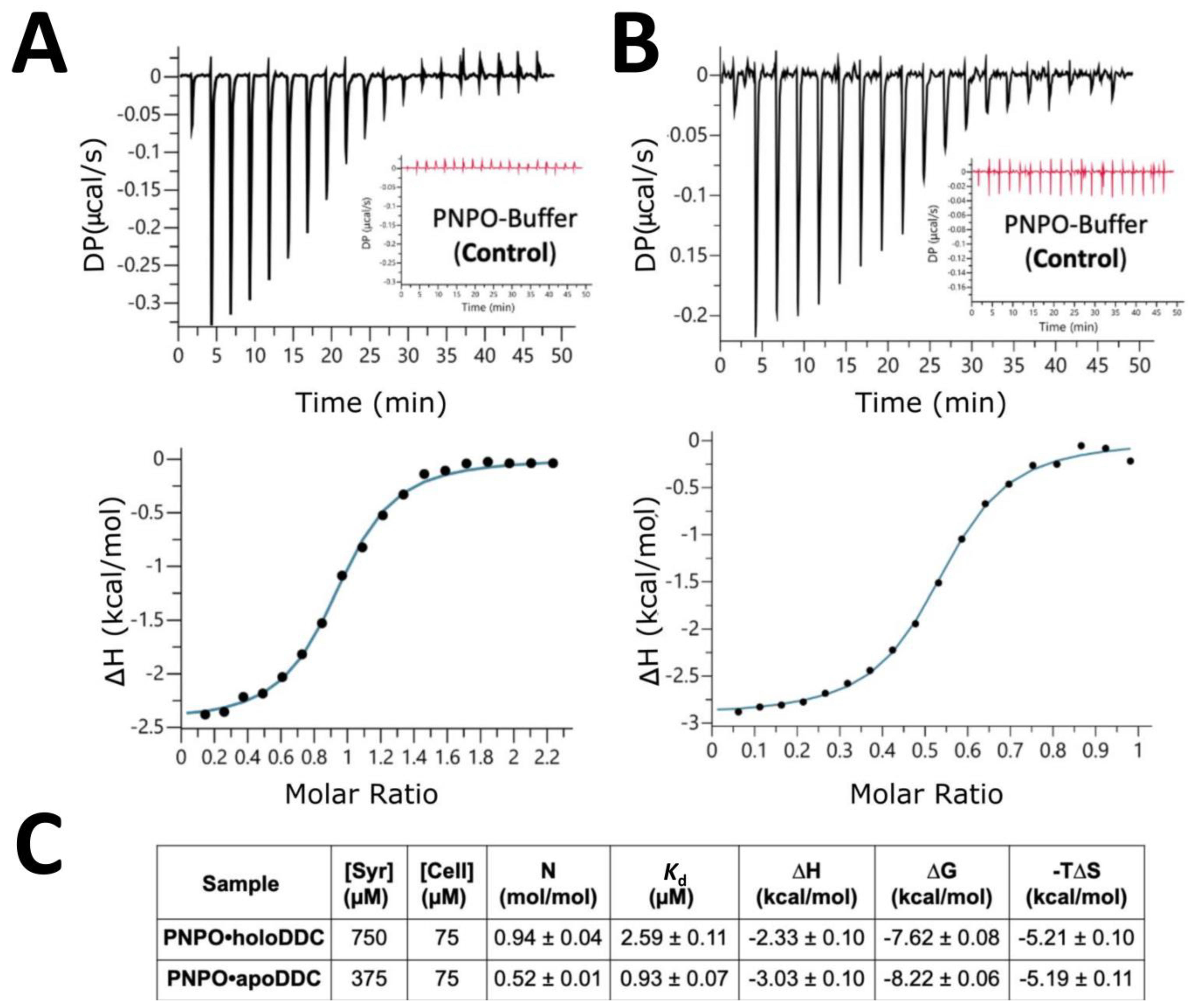
2.2. PNPO Forms a Physical Complex with Both the apoDDC and holoDDC
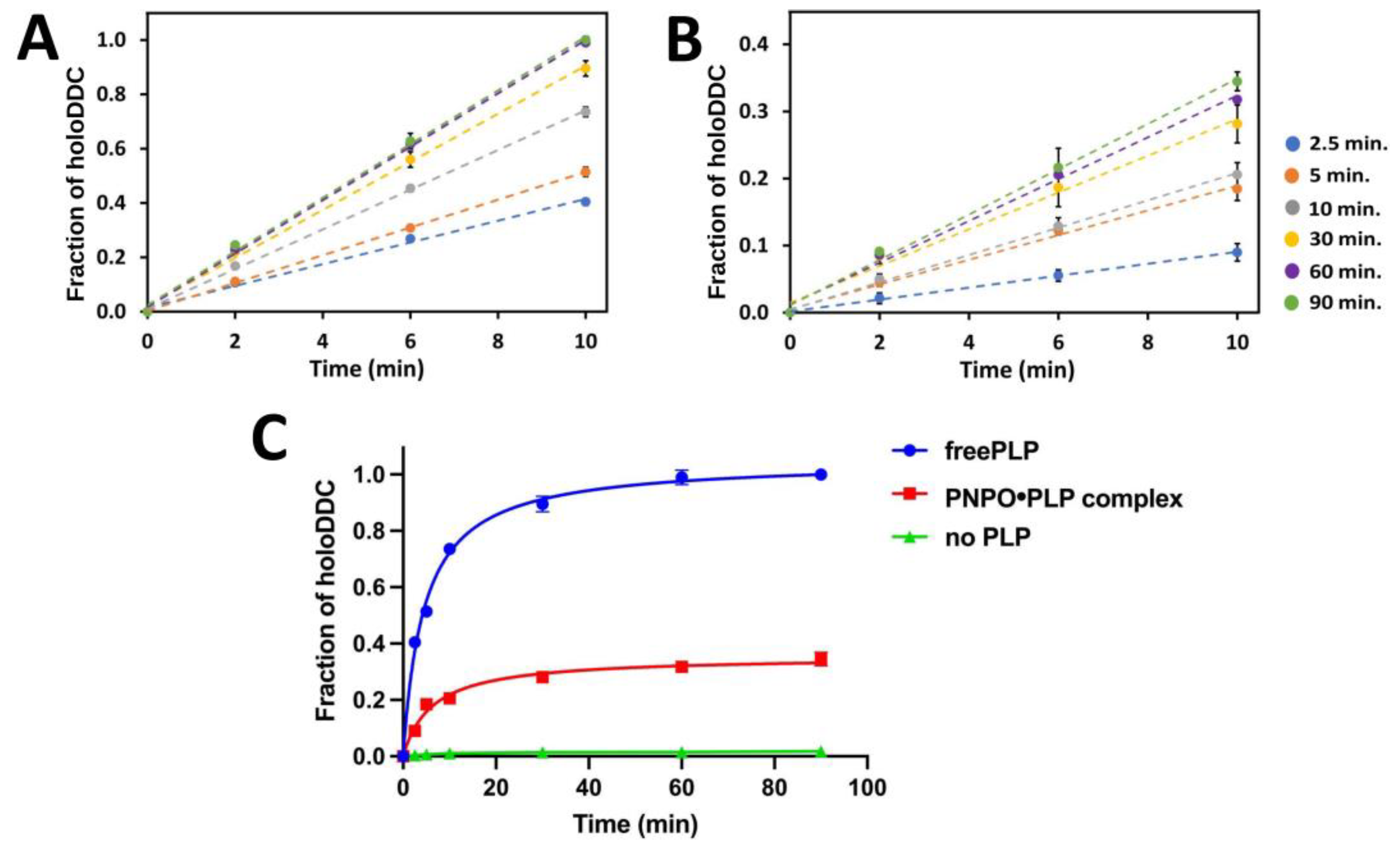
2.3. PLP Transfer from PNPO●PLP Complex to apoDDC
3. Materials and Methods
3.1. Computational Studies for Predicting the Complex Structure between hPNPO and DDC
3.1.1. Preparation of the Crystal Structures of hPNPO, holoDDC, and apoDDC
3.1.2. Molecular Docking
3.1.3. Molecular Dynamics Simulations
3.2. Subcloning, Expression, and Purification of DDC
3.3. Preparation of apoDDC
3.4. Expression and Purification of Human PNPO
3.5. Activity Assay of DDC
3.6. Biophysical Binding Studies
3.6.1. Isothermal Calorimetry (ITC)
3.6.2. Surface Plasmon Resonance (SPR)
3.7. PNPO•PLP Complex Preparation
3.8. PLP Transfer from PNPO•PLP to Activate apoDDC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parra, M.; Stahl, S.; Hellmann, H. Vitamin B6 and Its Role in Cell Metabolism and Physiology. Cells 2018, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvo, M.L.; Safo, M.K.; Contestabile, R. Biomedical Aspects of Pyridoxal 5′-Phosphate Availability. Front. Biosci. Elite Ed. 2012, 4, 897–913. [Google Scholar] [PubMed]
- Percudani, R.; Peracchi, A. A Genomic Overview of Pyridoxal-phosphate-dependent Enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef]
- Percudani, R.; Peracchi, A. The B6 Database: A Tool for the Description and Classification of Vitamin B6-Dependent Enzymatic Activities and of the Corresponding Protein Families. BMC Bioinform. 2009, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvo, M.L.; Contestabile, R.; Safo, M.K. Vitamin B(6) Salvage Enzymes: Mechanism, Structure and Regulation. Biochim. Biophys. Acta 2011, 1814, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Musayev, F.N.; Di Salvo, M.L.; Ko, T.-P.; Schirch, V.; Safo, M.K. Structure and Properties of Recombinant Human Pyridoxine 5′-Phosphate Oxidase. Protein Sci. 2003, 12, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Ghatge, M.S.; Al Mughram, M.; Omar, A.M.; Safo, M.K. Inborn Errors in the Vitamin B6 Salvage Enzymes Associated with Neonatal Epileptic Encephalopathy and Other Pathologies. Biochimie 2021, 183, 18–29. [Google Scholar] [CrossRef]
- Ghatge, M.S.; Karve, S.S.; David, T.M.S.; Ahmed, M.H.; Musayev, F.N.; Cunningham, K.; Schirch, V.; Safo, M.K. Inactive Mutants of Human Pyridoxine 5′-Phosphate Oxidase: A Possible Role for a Noncatalytic Pyridoxal 5′-Phosphate Tight Binding Site. FEBS Open Bio 2016, 6, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Musayev, F.N.; Di Salvo, M.L.; Saavedra, M.A.; Contestabile, R.; Ghatge, M.S.; Haynes, A.; Schirch, V.; Safo, M.K. Molecular Basis of Reduced Pyridoxine 5′-Phosphate Oxidase Catalytic Activity in Neonatal Epileptic Encephalopathy Disorder. J. Biol. Chem. 2009, 284, 30949–30956. [Google Scholar] [CrossRef] [Green Version]
- Barile, A.; Nogués, I.; di Salvo, M.L.; Bunik, V.; Contestabile, R.; Tramonti, A. Molecular Characterization of Pyridoxine 5′-Phosphate Oxidase and Its Pathogenic Forms Associated with Neonatal Epileptic Encephalopathy. Sci. Rep. 2020, 10, 13621. [Google Scholar] [CrossRef]
- Mills, P.B.; Surtees, R.A.H.; Champion, M.P.; Beesley, C.E.; Dalton, N.; Scambler, P.J.; Heales, S.J.R.; Briddon, A.; Scheimberg, I.; Hoffmann, G.F.; et al. Neonatal Epileptic Encephalopathy Caused by Mutations in the PNPO Gene Encoding Pyridox(Am)Ine 5′-Phosphate Oxidase. Hum. Mol. Genet. 2005, 14, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatge, M.S.; Contestabile, R.; Salvo, M.L.; Desai, J.V.; Gandhi, A.K.; Camara, C.M.; Florio, R.; González, I.N.; Parroni, A.; Schirch, V.; et al. Pyridoxal 5′-Phosphate Is a Slow Tight Binding Inhibitor of E. Coli Pyridoxal Kinase. PLoS ONE 2012, 7, e41680. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.W. Intracellular Trafficking of the Pyridoxal Cofactor. Implications for Health and Metabolic Disease. Arch. Biochem. Biophys. 2016, 592, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, A.; Tramonti, A.; di Salvo, M.L.; Nogués, I.; Nardella, C.; Malatesta, F.; Contestabile, R. Allosteric Feedback Inhibition of Pyridoxine 5′-Phosphate Oxidase from Escherichia Coli. J. Biol. Chem. 2019, 294, 15593–15603. [Google Scholar] [CrossRef]
- Kim, Y.T.; Kwok, F.; Churchich, J.E. Interactions of Pyridoxal Kinase and Aspartate Aminotransferase Emission Anisotropy and Compartmentation Studies. J. Biol. Chem. 1988, 263, 13712–13717. [Google Scholar] [CrossRef]
- Cheung, P.-Y.; Fong, C.-C.; Ng, K.-T.; Lam, W.-C.; Leung, Y.-C.; Tsang, C.-W.; Yang, M.; Wong, M.-S. Interaction between Pyridoxal Kinase and Pyridoxal-5-Phosphate-Dependent Enzymes. J. Biochem. 2003, 134, 731–738. [Google Scholar] [CrossRef]
- Bertoldi, M. Mammalian Dopa Decarboxylase: Structure, Catalytic Activity and Inhibition. Arch. Biochem. Biophys. 2014, 546, 1–7. [Google Scholar] [CrossRef]
- Guenter, J.; Lenartowski, R. Molecular Characteristic and Physiological Role of DOPA-Decarboxylase. Postepy Hig. Med. Dosw. 2016, 70, 1424–1440. [Google Scholar] [CrossRef]
- Cellini, B. Biochemical and Computational Approaches to Improve the Clinical Treatment of Dopa Decarboxylase-Related Diseases: An Overview. Open Biochem. J. 2012, 6, 131–138. [Google Scholar] [CrossRef]
- Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.N.; Malashkevich, V.N. Structural Insight into Parkinson’s Disease Treatment from Drug-Inhibited DOPA Decarboxylase. Nat. Struct. Biol. 2001, 8, 963–967. [Google Scholar] [CrossRef]
- Devos, D.; Lejeune, S.; Cormier-Dequaire, F.; Tahiri, K.; Charbonnier-Beaupel, F.; Rouaix, N.; Duhamel, A.; Sablonnière, B.; Bonnet, A.-M.; Bonnet, C.; et al. Dopa-Decarboxylase Gene Polymorphisms Affect the Motor Response to l-Dopa in Parkinson’s Disease. Park. Relat. Disord. 2014, 20, 170–175. [Google Scholar] [CrossRef]
- Tempel, B.L.; Livingstone, M.S.; Quinn, W.G. Mutations in the Dopa Decarboxylase Gene Affect Learning in Drosophila. Proc. Natl. Acad. Sci. USA 1984, 81, 3577–3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halaris, A.E.; Belendiuk, K.T.; Freedman, D.X. Antidepressant Drugs Affect Dopamine Uptake. Biochem. Pharmacol. 1975, 24, 1896–1897. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of Dopamine Metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassenberg, T.; Molero-Luis, M.; Jeltsch, K.; Hoffmann, G.F.; Assmann, B.; Blau, N.; Garcia-Cazorla, A.; Artuch, R.; Pons, R.; Pearson, T.S.; et al. Consensus Guideline for the Diagnosis and Treatment of Aromatic L-Amino Acid Decarboxylase (AADC) Deficiency. Orphanet J. Rare Dis. 2017, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Lu, D.; Gu, X.; Yu, Y.; The Mainland Chinese League of AADC Rare Disease. Aromatic L-amino Acid Decarboxylase Deficiency in 17 Mainland China Patients: Clinical Phenotype, Molecular Spectrum, and Therapy Overview. Mol. Genet. Genom. Med. 2020, 8, e1143. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.-C.; Chien, Y.-H.; Hwu, W.-L. A Review of Aromatic l-Amino Acid Decarboxylase (AADC) Deficiency in Taiwan. Am. J. Med. Genet. 2019, 181, 226–229. [Google Scholar] [CrossRef]
- Pearson, T.S.; Gupta, N.; San Sebastian, W.; Imamura-Ching, J.; Viehoever, A.; Grijalvo-Perez, A.; Fay, A.J.; Seth, N.; Lundy, S.M.; Seo, Y.; et al. Gene Therapy for Aromatic L-Amino Acid Decarboxylase Deficiency by MR-Guided Direct Delivery of AAV2-AADC to Midbrain Dopaminergic Neurons. Nat. Commun. 2021, 12, 4251. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, J.; Zhang, Q.; Chen, Y.; Bao, X. The Genetic and Clinical Characteristics of Aromatic L-Amino Acid Decarboxylase Deficiency in Mainland China. J. Hum. Genet. 2020, 65, 759–769. [Google Scholar] [CrossRef]
- Montioli, R.; Borri Voltattorni, C. Aromatic Amino Acid Decarboxylase Deficiency: The Added Value of Biochemistry. Int. J. Mol. Sci. 2021, 22, 3146. [Google Scholar] [CrossRef]
- Montioli, R.; Cellini, B.; Borri Voltattorni, C. Molecular Insights into the Pathogenicity of Variants Associated with the Aromatic Amino Acid Decarboxylase Deficiency. J. Inherit. Metab. Dis. 2011, 34, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Montioli, R.; Dindo, M.; Giorgetti, A.; Piccoli, S.; Cellini, B.; Voltattorni, C.B. A Comprehensive Picture of the Mutations Associated with Aromatic Amino Acid Decarboxylase Deficiency: From Molecular Mechanisms to Therapy Implications. Hum. Mol. Genet. 2014, 23, 5429–5440. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.S.; Schirch, V. Tight Binding of Pyridoxal 5′-Phosphate to Recombinant Escherichia Coli Pyridoxine 5′-Phosphate Oxidase. Arch. Biochem. Biophys. 2000, 377, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Barile, A.; Battista, T.; Fiorillo, A.; di Salvo, M.L.; Malatesta, F.; Tramonti, A.; Ilari, A.; Contestabile, R. Identification and Characterization of the Pyridoxal 5′-Phosphate Allosteric Site in Escherichia coli Pyridoxine 5′-Phosphate Oxidase. J. Biol. Chem. 2021, 296, 100795. [Google Scholar] [CrossRef]
- Giardina, G.; Montioli, R.; Gianni, S.; Cellini, B.; Paiardini, A.; Voltattorni, C.B.; Cutruzzola, F. Open Conformation of Human DOPA Decarboxylase Reveals the Mechanism of PLP Addition to Group II Decarboxylases. Proc. Natl. Acad. Sci. USA 2011, 108, 20514–20519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New Additions to the ClusPro Server Motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Kellogg, G.E.; Semus, S.F.; Abraham, D.J. HINT: A New Method of Empirical Hydrophobic Field Calculation for CoMFA. J. Comput.-Aided Mol. Des. 1991, 5, 545–552. [Google Scholar] [CrossRef]
- Sarkar, A.; Kellogg, G. Hydrophobicity—Shake Flasks, Protein Folding and Drug Discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef]
- Dai, L.; Ding, C.; Fang, F. A Novel DDC Gene Deletion Mutation in Two Chinese Mainland Siblings with Aromatic L-Amino Acid Decarboxylase Deficiency. Brain Dev. 2019, 41, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, B.J.; Skjærven, L.; Yao, X. The Bio3D Packages for Structural Bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Computational Alanine Scanning to Probe Protein−Protein Interactions: A Novel Approach to Evaluate Binding Free Energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. STKE 2004, 2004, pl2. [Google Scholar] [CrossRef] [Green Version]
- Douzi, B. Protein–Protein Interactions: Surface Plasmon Resonance. In Bacterial Protein Secretion Systems; Journet, L., Cascales, E., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1615, pp. 257–275. ISBN 978-1-4939-7031-5. [Google Scholar]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An Integrated Modeling System. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Sherald, A.F.; Sparrow, J.C.; Wright, T.R.F. A Spectrophotometric Assay for Drosophila Dopa Decarboxylase. Anal. Biochem. 1973, 56, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Charteris, A.; John, R. An Investigation of the Assay of Dopamine Using Trinitrobenzensulphonic Acid. Anal. Biochem. 1975, 66, 365–371. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ITC Parameters | Description |
|---|---|
| Total no. of injections | 19 |
| Cell temperature °C | 25 |
| Reference power (µcal/s) | 5 |
| Stir speed | 750 |
| Volume (µL) | 2 |
| Duration (s) | 4 |
| Spacing (s) | 150 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AL Mughram, M.H.; Ghatge, M.S.; Kellogg, G.E.; Safo, M.K. Elucidating the Interaction between Pyridoxine 5′-Phosphate Oxidase and Dopa Decarboxylase: Activation of B6-Dependent Enzyme. Int. J. Mol. Sci. 2023, 24, 642. https://doi.org/10.3390/ijms24010642
AL Mughram MH, Ghatge MS, Kellogg GE, Safo MK. Elucidating the Interaction between Pyridoxine 5′-Phosphate Oxidase and Dopa Decarboxylase: Activation of B6-Dependent Enzyme. International Journal of Molecular Sciences. 2023; 24(1):642. https://doi.org/10.3390/ijms24010642
Chicago/Turabian StyleAL Mughram, Mohammed H., Mohini S. Ghatge, Glen E. Kellogg, and Martin K. Safo. 2023. "Elucidating the Interaction between Pyridoxine 5′-Phosphate Oxidase and Dopa Decarboxylase: Activation of B6-Dependent Enzyme" International Journal of Molecular Sciences 24, no. 1: 642. https://doi.org/10.3390/ijms24010642