
Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells





Abstract
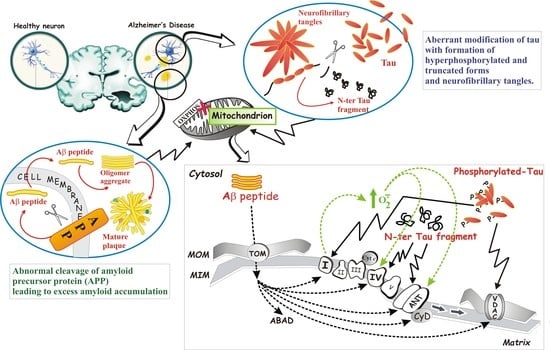
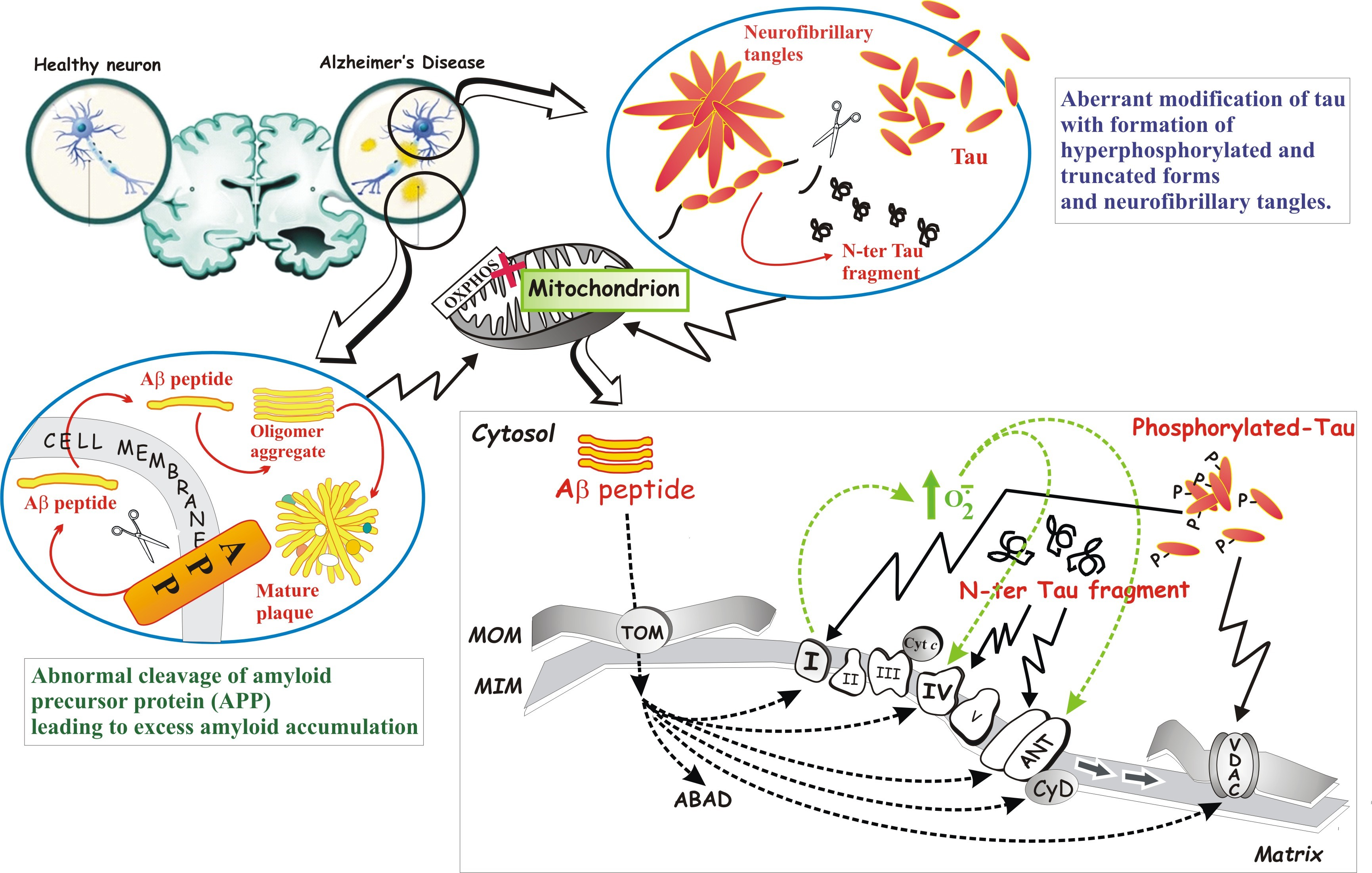
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mitochondria—What You Need to Know to Understand the Rest!
3. Proteinopathy and Mitochondrial Dysfunction in AD: A Marriage
4. ANT and VDAC Both Interact with AD Toxic Proteins
- (i)
- ANT hosts two (opposite) functions, both involved in the control and regulation of cell fate [65,66]: one vital function, the other lethal. The vital role of ANT is the enzymatic one, which is the historical and the main function to catalyze the carrier-mediated exchange between the cytosolic ADP with the ATP formed in the matrix, by facilitating the export of the newly synthesized ATP into the cell and, at the same time, by providing ADP as a substrate available for its mitochondrial phosphorylation to ATP by ATP synthase. This function has been extensively characterized in mitochondria isolated from various tissues with radiolabelled nucleotides and in native ANT-containing proteoliposomes [67,68,69], but not only [70], as we will see! The lethal function of ANT, which occurs in conditions of cellular ‘sickness’, is associated with its involvement as a component of the mitochondrial permeability transition pore (mPTP), a structure that forms in the inner mitochondrial membrane and is thought to underlie regulated cell death [71,72].
- (ii)
- VDAC1 is the most abundant protein in the MOM since its discovery in 1976 [73]. Although its localization remains predominantly mitochondrial (mVDAC), VDAC has been found on the plasma membrane (plVDAC) of cells. Besides, although plVDAC has been extensively studied, its exact biological function is not yet known, and we will not address it here. Therefore, in this review, we will refer to mitochondrial VDAC as VDAC. Previously, VDAC was considered responsible for the near-free permeability of the MOM [73] or a large mesh sieve [74], but nowadays, it unexpectedly stands as a gatekeeper for the entry and exit of mitochondrial metabolites, thus controlling the cross-talk between the mitochondria and the rest of the cellular compartments [75,76]. Not surprisingly, therefore, VDAC has been implicated in a wide range of pathologies associated with mitochondria [77,78,79]. It is believed that the uniqueness of this channel derives from its key position at the interface between the mitochondrion and the cytosol [77,78,79,80], thus becoming an important hub protein that interacts with over 200 proteins [81,82,83] that regulate the permeability of the MOM. A plethora of cytosolic proteins, glycolytic, such as hexokinase, and other enzymes [78], as well as their neighbors in the MOM, such as the cholesterol transporter, [78] as well as proteins of the family Bcl-2 [78], interact with VDAC. An acute observation by Reina and De Pinto [84] pointed out to the world that the high diversity of natural and synthetic ligands with which VDAC interacts is a symptom of lack of specificity for VDAC, so that this protein is not used to being considered a reliable drug target.
4.1. ANT
- (i)
- The rate of appearance of ATP was reduced when Aβ1–42 or NH2-26–44-tau was added before ADP.
- (ii)
- The extent of inhibition was less than that found when the NH2-tau fragment was added alone if the incubation of homogenate with Aβ1–42 preceded the addition of NH2-26–44-tau, suggesting that the binding of Aβ1–42 to ANT1 resulted in a conformational change of the transporter protein, making it less accessible to the NH-tau fragment. This only happens if Aβ 1–42 is added before the tau fragment, but not vice versa, suggesting that Aβ 1–42 acts as a negative modulator of mitochondrial NH2-tau fragment toxicity and not vice versa;
- (iii)
- The extent of inhibition increased strongly when the two peptides, NH2-26–44-tau and fibrillar Aβ1–42, were added together, confirming that the two peptides together cooperate by potentiating ANT-1 dysfunction and further aggravating the production of ATP.
4.2. VDAC
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer’s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March separate, strike together--role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V. Tau phosphorylation and proteolysis: Insights and perspectives. J. Alzheimers Dis. 2006, 9, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Garg, S.; Mandelkow, E.M.; Mandelkow, E. Proteolytic processing of tau. Biochem. Soc. Trans. 2010, 38, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Siman, R.; Mcintosh, T.K.; Soltesz, K.M.; Chen, Z.; Neumar, R.W.; Roberts, V.L. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol. Dis. 2004, 16, 311–320. [Google Scholar] [CrossRef]
- Rohn, T.T.; Rissman, R.A.; Davis, M.C.; Kim, Y.E.; Cotman, C.W.; Head, E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol. Dis. 2002, 11, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Corsetti, V.; Amadoro, G.; Gentile, A.; Capsoni, S.; Ciotti, M.T.; Cencioni, M.T.; Atlante, A.; Canu, N.; Rohn, T.T.; Cattaneo, A.; et al. Identification of a caspasederived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol. Cell. Neurosci. 2008, 38, 381–392. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Carbonell, F.M.; Sotero, R.C.; Chouinard-Decorte, F.; Evans, A.C. Alzheimer’s Disease Neuroimaging Initiative. Multifactorial causal model of brain (dis)organization and therapeutic intervention: Application to Alzheimer’s disease. Neuroimage 2017, 152, 60–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Kato, Y.; Takatsu, H.; Fukui, K. Relationship between Cognitive Dysfunction and Age-Related Variability in Oxidative Markers in Isolated Mitochondria of Alzheimer’s Disease Transgenic Mouse Brains. Biomedicines 2022, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Na, R.; Gu, M.; Richardson, A.; Ran, Q. Lipid peroxidation up-regulates BACE1 expression in vivo: A possible early event of amyloidogenesis in Alzheimer’s disease. J. Neurochem. 2008, 107, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.M.; Murale, D.P.; Kim, Y.K.; Lee, J.-S. Crosstalk between Oxidative Stress and Tauopathy. Int. J. Mol. Sci. 2019, 20, 1959. [Google Scholar] [CrossRef] [Green Version]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, L.; Wang, Y.; Zhang, X.; Tomaszewski, A.; McNamara, J.T.; Li, R. Mitochondria-Associated Proteostasis. Annu. Rev. Biophys. 2020, 49, 41–67. [Google Scholar] [CrossRef]
- Sterniczuk, R.; Antle, M.C.; Laferla, F.M.; Dyck, R.H. Characterization of the 3XTg-AD mouse model of Alzheimer’s disease: Part 2. Behavioral and cognitive changes. Brain Res. 2010, 1348, 149–155. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Pérez-González, M.; Alfaro-Ruiz, R.; Badesso, S.; Sucunza, D.; Espelosin, M.; Ursúa, S.; Lachen-Montes, M.; Fernández-Irigoyen, J.; Santamaria, E.; et al. Amyloid-Driven Tau Accumulation on Mitochondria Potentially Leads to Cognitive Deterioration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11950. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bai, F. The Association of Tau with Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Opazo, F.A.; Vergara-Pulgar, K.; Pérez, M.J.; Jara, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2015, 2015, 509654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, A.; Schulz, K.L.; Rhein, V.; Götz, J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 2010, 41, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Pagani, L.; Eckert, A. Amyloid-Beta interaction with mitochondria. Int. J. Alzheimers Dis. 2011, 2011, 925050. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid b-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta 2013, 1832, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Ring, J.; Tadic, J.; Ristic, S.; Poglitsch, M.; Bergmann, M.; Radic, N.; Mossmann, D.; Liang, Y.; Maglione, M.; Jerkovic, A.; et al. The HSP40 chaperone Ydj1 drives amyloid beta 42 toxicity. EMBO Mol. Med. 2022, 14, e13952. [Google Scholar] [CrossRef] [PubMed]
- Fasulo, L.; Ugolini, G.; Cattaneo, A. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J. Alzheimers Dis. 2005, 7, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.K.; Jara, C.; Olesen, M.A.; Tapia-Rojas, C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci. Rep. 2021, 11, 4448. [Google Scholar] [CrossRef]
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimers Dis. 2013, 37, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadoro, G.; Corsetti, V.; Ciotti, M.T.; Florenzano, F.; Capsoni, S.; Amato, G.; Calissano, P. Endogeno Aβ causes cell death via early tau hyperphosphorylation. Neurobiol. Aging 2011, 32, 969–990. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. The mitochondrial hypothesis: Dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 207–233. [Google Scholar] [CrossRef]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimers Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetics deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, H.; Manfredi, G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef]
- Atamna, H.; Boyle, K. Amyloid-beta peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Zimbron, L.F.; Luna-Muñoz, J.; Mena, R.; Vazquez-Ramirez, R.; Kubli-Garfias, C.; Cribbs, D.H.; Manoutcharian, K.; Gevorkian, G. Amyloid-β peptide binds to cytochrome C oxidase subunit 1. PLoS ONE 2012, 7, e42344. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.D.; Parks, J.K. Cytochrome C Oxidase in Alzheimer’s Disease Brain: Purification and characterization. Neurology 1995, 45, 482–486. [Google Scholar] [CrossRef]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.W.; Lee, J.; Pae, A.N. Mitochondrial dysfunction and Alzheimer’s disease: Prospects for therapeutic intervention. BMB Rep. 2020, 53, 47–55. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Vignais, P.V. Molecular and physiological aspects of adenine nucleotide transport in mitochondria. Biochim. Biophys. Acta 1976, 456, 1–38. [Google Scholar] [CrossRef]
- Lauquin, G.J.M.; Vignais, P.V. Interaction of (3H) bongkrekic acid with the mitochondrial adenine nucleotide translocator. Biochemistry 1976, 15, 2316–2322. [Google Scholar] [CrossRef]
- Brandolin, G.; Doussiere, J.; Gulik, A.; Gulik-Krzywicki, T.; Lauquin, G.J.; Vignais, P.V. Kinetic, binding and ultrastructural properties of the beef heart adenine nucleotide carrier protein after incorporation into phospholipid vesicles. Biochim. Biophys. Acta 1980, 592, 592–614. [Google Scholar] [CrossRef]
- Atlante, A.; Valenti, D. A Walk in the Memory, from the First Functional Approach up to Its Regulatory Role of Mitochondrial Bioenergetic Flow in Health and Disease: Focus on the Adenine Nucleotide Translocator. Int. J. Mol. Sci. 2021, 22, 4164. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Bernardi, P.; Carraro, M.; Lippe, G. The mitochondrial permeability transition: Recent progress and open questions. FEBS J. 2021, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Kuriachan, G.; Mahalakshmi, R. Cellular Interactome of Mitochondrial Voltage-Dependent Anion Channels: Oligomerization and Channel (Mis)Regulation. ACS Chem. Neurosci. 2021, 12, 3497–3515. [Google Scholar] [CrossRef] [PubMed]
- Mannella, C.A.; Forte, M.; Colombini, M. Toward the molecular structure of the mitochondrial channel, VDAC. J. Bioenergy Biomembr. 1992, 24, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Holmuhamedov, E. Voltage-dependent anion channel (VDAC) as mitochondrial governator—Thinking outside the box. Biochim. Biophys. Acta 2006, 1762, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Brdiczka, D.G.; Zorov, D.B.; Sheu, S.S. Mitochondrial contact sites: Their role in energy metabolism and apoptosis. Biochim. Biophys. Acta 2006, 1762, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Bezrukov, S.M.; Hoogerheide, D.P. Regulation of Mitochondrial Respiration by VDAC Is Enhanced by Membrane-Bound Inhibitors with Disordered Polyanionic C-Terminal Domains. Int. J. Mol. Sci. 2021, 22, 7358. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- De Pinto, V. Renaissance of VDAC: New Insights on a Protein Family at the Interface between Mitochondria and Cytosol. Biomolecules 2021, 11, 107. [Google Scholar] [CrossRef]
- Pittalà, M.G.G.; Conti Nibali, S.; Reina, S.; Cunsolo, V.; Di Francesco, A.; De Pinto, V.; Messina, A.; Foti, S.; Saletti, R. VDACs Post-Translational Modifications Discovery by Mass Spectrometry: Impact on Their Hub Function. Int. J. Mol. Sci. 2021, 22, 12833. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reina, S.; De Pinto, V. Anti-Cancer Compounds Targeted to VDAC: Potential and Perspectives. Curr. Med. Chem. 2017, 24, 4447–4469. [Google Scholar] [CrossRef]
- Bobba, A.; Atlante, A.; de Bari, L.; Passarella, S.; Marra, E. Apoptosis and cytochrome c release in cerebellar granule cells. In Vivo 2004, 18, 335–344. [Google Scholar]
- Atlante, A.; Bobba, A.; Calissano, P.; Passarella, S.; Marra, E. The apoptosis/necrosis transition in cerebellar granule cells depends on the mutual relationship of the antioxidant and the proteolytic systems which regulate ROS production and cytochrome c release en route to death. J. Neurochem. 2003, 84, 960–971. [Google Scholar] [CrossRef]
- Bobba, A.; Atlante, A.; Giannattasio, S.; Sgaramella, G.; Calissano, P.; Marra, E. Early release and subsequent caspase-mediated degradation of cytochrome c in apoptotic cerebellar granule cells. FEBS Lett. 1999, 457, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; de Bari, L.; Bobba, A.; Marra, E.; Calissano, P.; Passarella, S. Cytochrome c, released from cerebellar granule cells undergoing apoptosis or excytotoxic death, can generate proton motive force and drive ATP synthesis in isolated mitochondria. J. Neurochem. 2003, 86, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Bobba, A.; de Bari, L.; Fontana, F.; Calissano, P.; Marra, E.; Passarella, S. Caspase-dependent alteration of the ADP/ATP translocator triggers the mitochondrial permeability transition which is not required for the low-potassium-dependent apoptosis of cerebellar granule cells. J. Neurochem. 2006, 97, 1166–1181. [Google Scholar] [CrossRef]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef] [Green Version]
- Götz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Delacourte, A.; Sergeant, N.; Champain, D.; Wattez, A.; Maurage, C.-A.; Lebert, F.; Pasquier, F.; David, J.-P. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology 2002, 59, 398–407. [Google Scholar] [CrossRef]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Ribé, E.M.; Pérez, M.; Puig, B.; Gich, I.; Lim, F.; Cuadrado, M.; Sesma, T.; Catena, S.; Sánchez, B.; Nieto, M.; et al. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis. 2005, 20, 814–822. [Google Scholar] [CrossRef] [Green Version]
- McStay, G.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain complexes I and IV are impaired by β—amyloid via direct interaction and through complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Florenzano, F.; Atlante, A.; Ciotti, M.T.; Mongiardi, M.P.; Bussani, R.; Nicolin, V.; Nori, S.; Campanella, M.; et al. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol. Dis. 2014, 62, 489–507. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Azzariti, A.; Pizzuto, R.; Atlante, A. Extracellular ADP prevents neuronal apoptosis via activation of cell antioxidant enzymes and protection of mitochondrial ANT-1. Biochim. Biophys. Acta 2014, 1837, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Vitolo, O.V.; Ciotti, M.T.; Galli, C.; Borsello, T.; Calissano, P. Adenosine and ADP prevent apoptosis in cultured rat cerebellar granule cells. Brain Res. 1998, 809, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 2009, 41, 425–431. [Google Scholar] [CrossRef]
- Rao, S.K.; Ross, J.M.; Harrison, F.E.; Bernardo, A.; Reiserer, R.S.; Reiserer, R.S.; Mobley, J.A.; McDonald, M.P. Differential proteomic and behavioral effects of long-term voluntary exercise in wild-type and APP-overexpressing transgenics. Neurobiol. Dis. 2015, 78, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer’s disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef]
- Magrì, A.; Messina, A. Interactions of VDAC with Proteins Involved in Neurodegenerative Aggregation: An Opportunity for Advancement on Therapeutic Molecules. Curr. Med. Chem. 2017, 24, 4470–4487. [Google Scholar] [CrossRef]
- Magrì, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [Green Version]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The voltage-dependent anion channel 1 mediates amyloid-b toxicity and represents a potential target for Alzheimer disease therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [Green Version]
- Dubey, A.K.; Godbole, A.; Mathew, M.K. Regulation of VDAC trafficking modulates cell death. Cell Death Discov. 2016, 2, 16085. [Google Scholar] [CrossRef] [Green Version]
- Bobba, A.; Amadoro, G.; La Piana, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Glucose-6-phosphate tips the balance in modulating apoptosis in cerebellar granule cells. FEBS Lett. 2015, 589, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377 Pt 2, 347–355. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [Green Version]
- Bobba, A.; Amadoro, G.; La Piana, G.; Calissano, P.; Atlante, A. Glycolytic enzyme upregulation and numbness of mitochondrial activity characterize the early phase of apoptosis in cerebellar granule cells. Apoptosis 2015, 20, 10–28. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Yoo, B.C.; Fountoulakis, M.; Cairns, N.; Lubec, G. Changes of voltage dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer’s disease and Down syndrome. Electrophoresis 2001, 22, 172–179. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-b. J. Alzheimers Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Shteinfer-Kuzmine, A.; Amsalem, Z.; Arif, T.; Zooravlov, A.; Shoshan-Barmatz, V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol. Oncol. 2018, 12, 1077–1103. [Google Scholar] [CrossRef]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid b-induced glycogen synthase kinase 3b phosphorylated VDAC1 in Alzheimer’s disease: Implications for synaptic dysfunction and neuronal damage. Biochim. Biophys. Acta 2013, 1832, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Kabir, M.T.; Rahman, M.S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M.; et al. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Mitochondrial links between brain aging and Alzheimer’s disease. Transl. Neurodegener. 2021, 10, 33. [Google Scholar] [CrossRef]
- Shevtsova, E.F.; Maltsev, A.V.; Vinogradova, D.V.; Shevtsov, P.N.; Bachurin, S.O. Mitochondria as a promising target for developing novel agents for treating Alzheimer’s disease. Med. Res. Rev. 2021, 41, 803–827. [Google Scholar] [CrossRef]
- Davis, J.N.; Hunnicutt, E.J., Jr.; Chisholm, J.C. A mitochondrial bottleneck hypothesis of Alzheimer’s disease. Mol. Med. Today 1995, 1, 240–247. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting mitochondria in Alzheimer disease: Rationale and perspectives. CNS Drugs 2019, 33, 957–969. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef]
- Van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef]
- Benfeito, S.; Fernandes, C.; Vilar, S.; Remiao, F.; Uriarte, E.; Borges, F. Exploring the multi-target performance of mitochondriotropic antioxidants against the pivotal Alzheimer’s disease pathophysiological hallmarks. Molecules 2020, 25, 276. [Google Scholar] [CrossRef] [Green Version]
- Nesi, G.; Sestito, S.; Digiacomo, M.; Rapposelli, S. Oxidative stress, mitochondrial abnormalities and proteins deposition: Multitarget approaches in Alzheimer’s disease. Curr. Top. Med. Chem. 2017, 17, 3062–3079. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Matera, R.; Minarini, A.; Rosini, M.; Melchiorre, C. Alzheimer’s disease: New approaches to drug discovery. Curr. Opin. Chem. Biol. 2009, 13, 303–308. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. Int. J. Mol. Sci. 2022, 23, 7722. https://doi.org/10.3390/ijms23147722
Atlante A, Valenti D, Latina V, Amadoro G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. International Journal of Molecular Sciences. 2022; 23(14):7722. https://doi.org/10.3390/ijms23147722
Chicago/Turabian StyleAtlante, Anna, Daniela Valenti, Valentina Latina, and Giuseppina Amadoro. 2022. "Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells" International Journal of Molecular Sciences 23, no. 14: 7722. https://doi.org/10.3390/ijms23147722
APA StyleAtlante, A., Valenti, D., Latina, V., & Amadoro, G. (2022). Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. International Journal of Molecular Sciences, 23(14), 7722. https://doi.org/10.3390/ijms23147722