
Hypoxic and Hypercapnic Responses in Transgenic Murine Model of Alzheimer’s Disease Overexpressing Human AβPP: The Effects of Pretreatment with Memantine and Rivastigmine

 ,
,  and
and
Abstract
:1. Introduction
2. Results
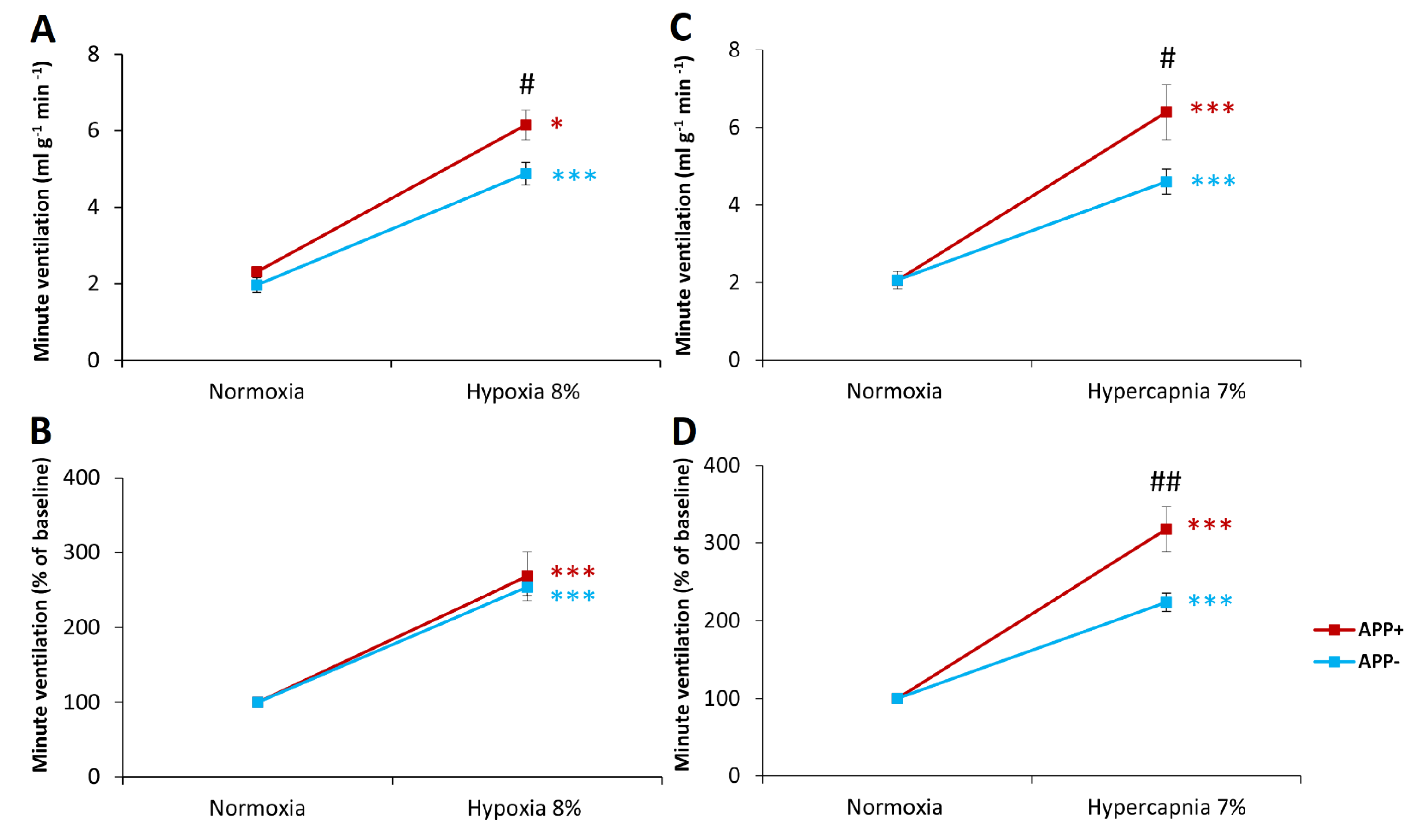
2.1. Respiratory Responses to Hypoxia and Hypercapnia
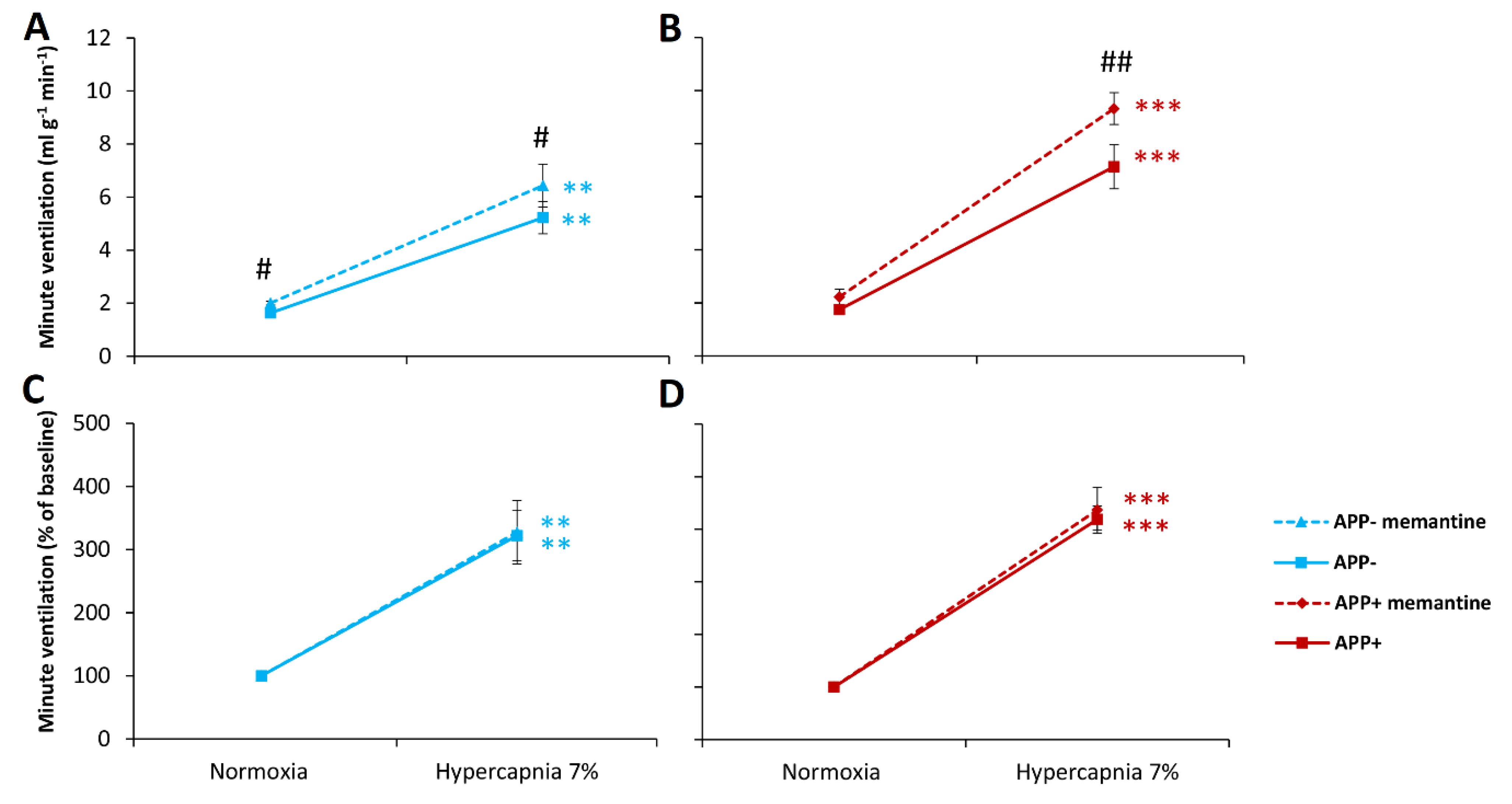
2.2. Respiratory Responses to Hypoxia and Hypercapnia after Memantine Administration
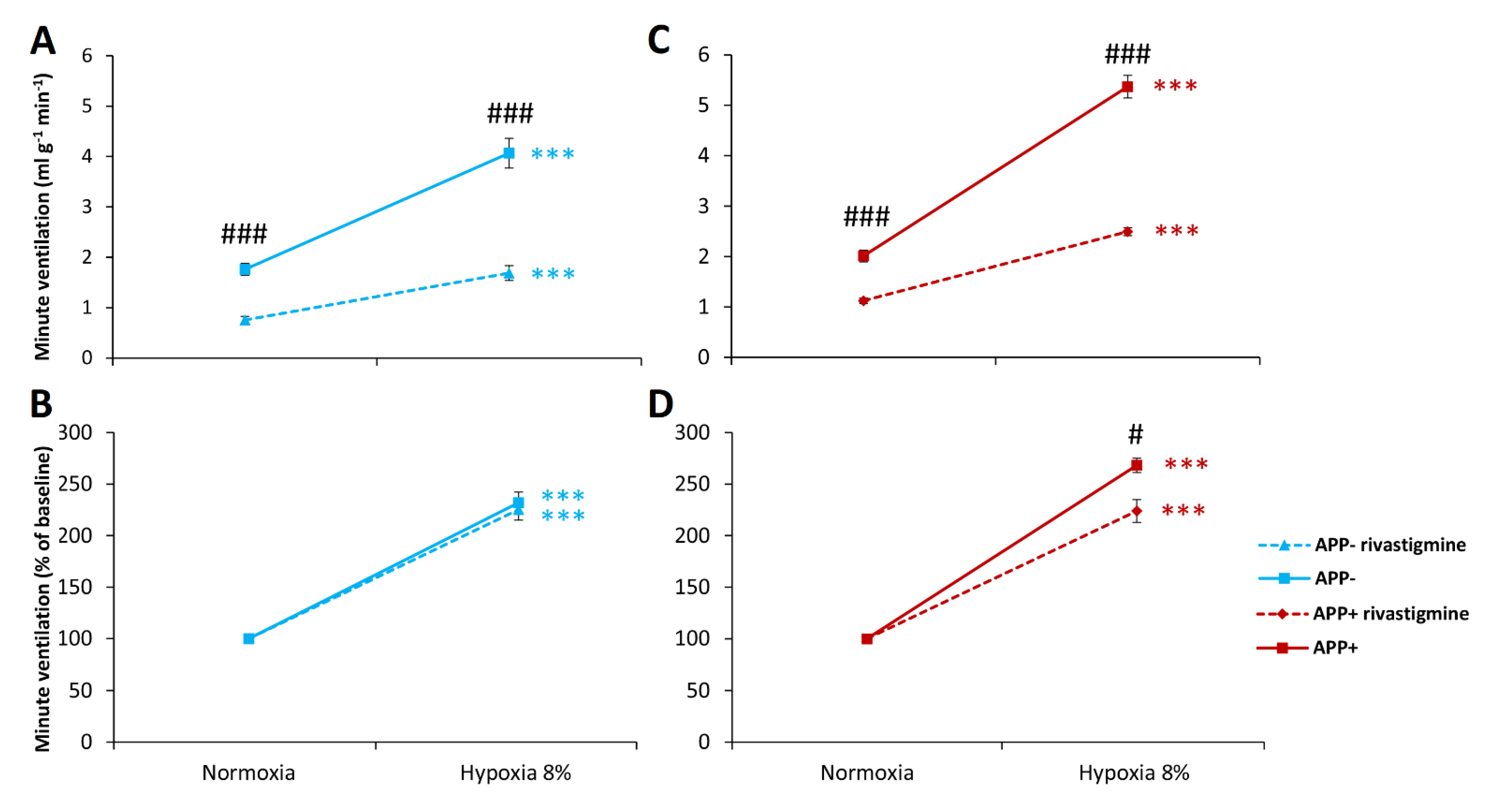
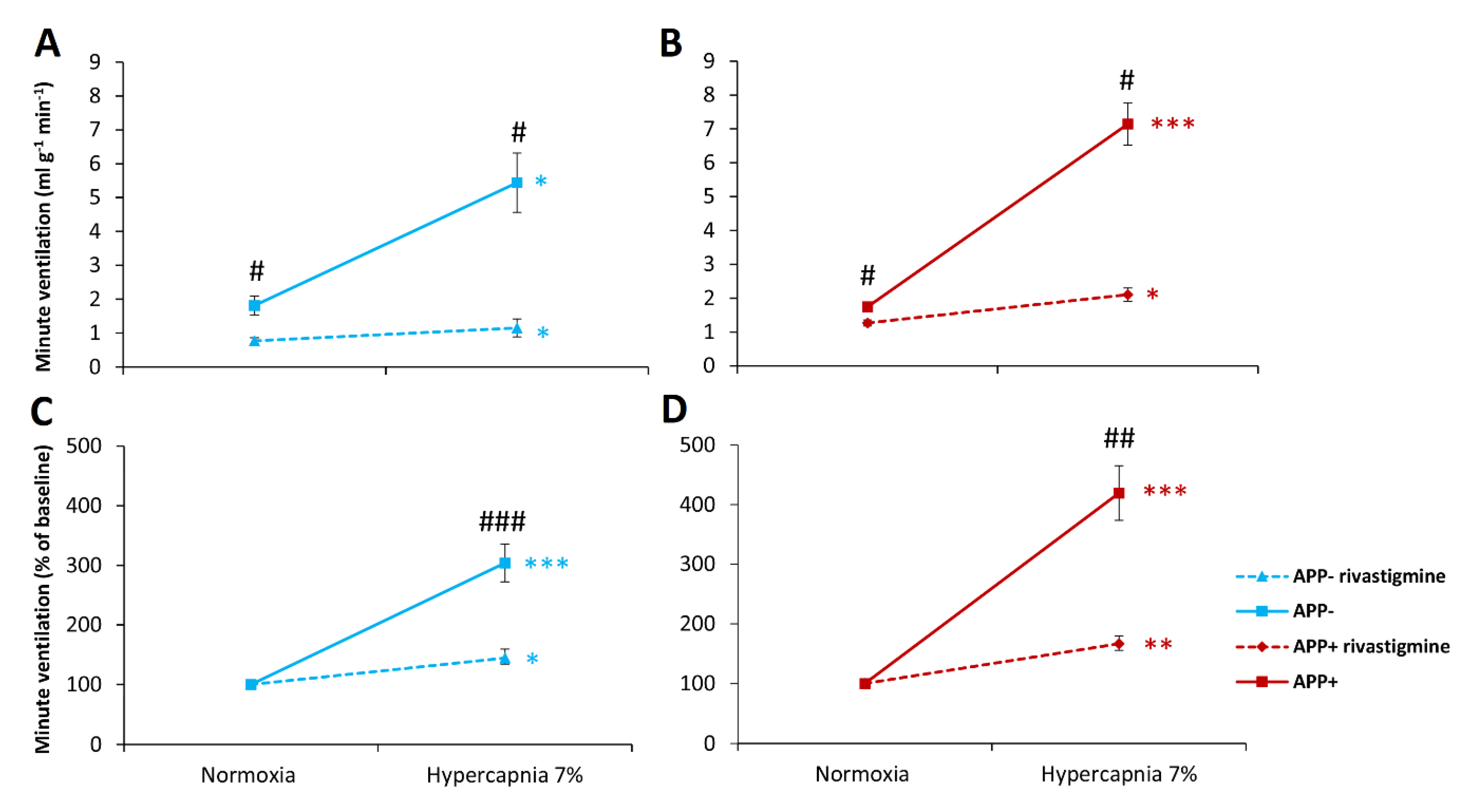
2.3. Respiratory Responses to Hypoxia and Hypercapnia after Rivastigmine Administration
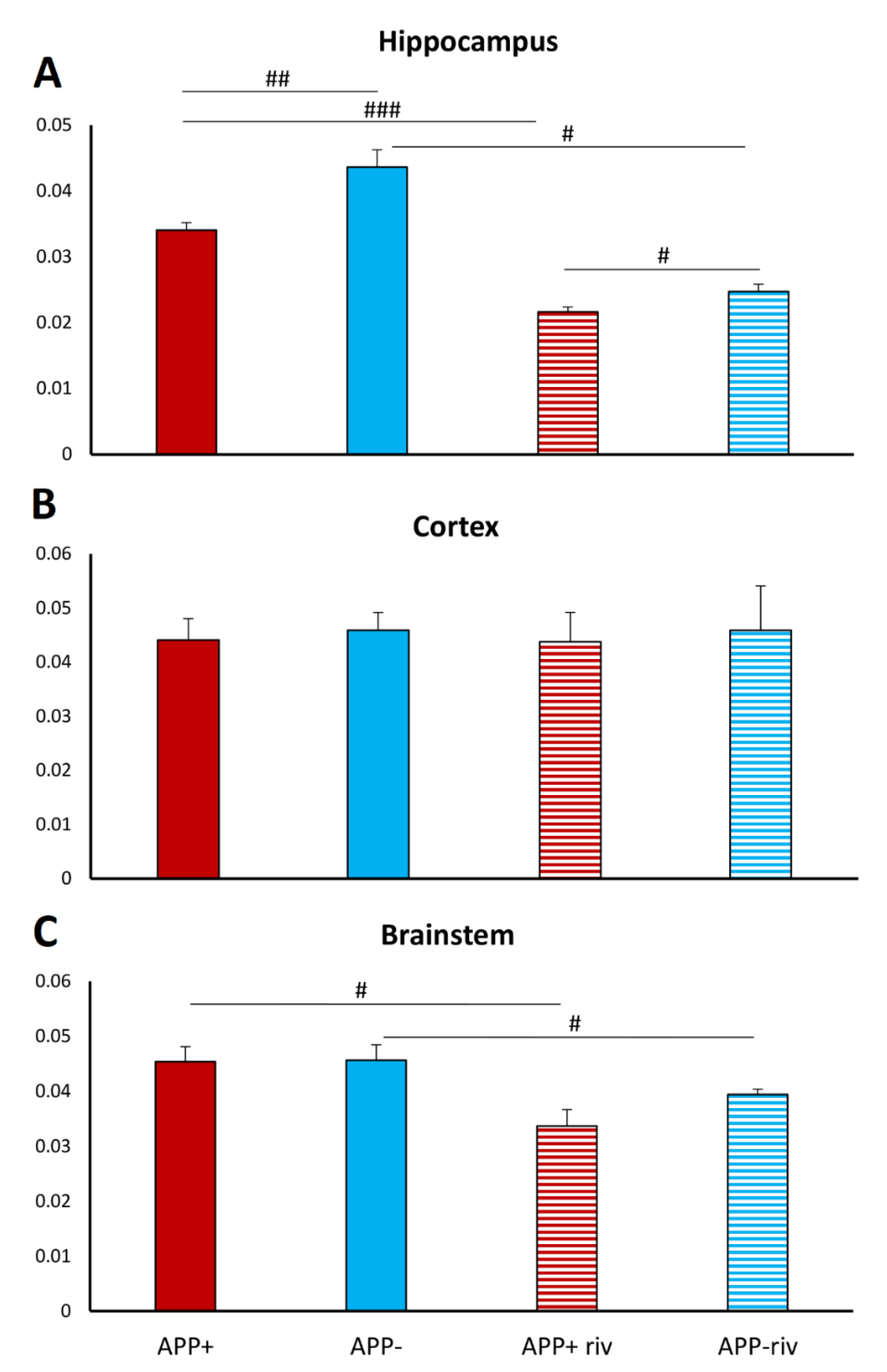
2.4. Acetylcholinesterase Activity in Different Brain Structures
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Plethysmography Measurements
4.3. Drugs and Treatment Schedule: Treatment with Rivastigmine and Memantine before and after Hypoxia and Hypercapnia in Unrestrained Mice
4.4. The Acetylcholinesterase (AChE) Activity Assay (Ellman’s Assay)
4.5. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.S.; Segawa, E.; Boyle, P.A.; Anagnos, S.E.; Hizel, L.P.; Bennett, D.A. The natural history of cognitive decline in Alzheimer’s disease. Psychol. Aging 2012, 27, 1008–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, S.J.; Chung, M.H.; Chen, B.S. Investigating Pathogenetic Mechanisms of Alzheimer’s Disease by Systems Biology Approaches for Drug Discovery. Int. J. Mol. Sci. 2021, 22, 11280. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagiani, F.; Lanni, C.; Racchi, M.; Pascale, A.; Govoni, S. Amyloid-β and Synaptic Vesicle Dynamics: A Cacophonic Orchestra. J. Alzheimers Dis. 2019, 72, 1–14. [Google Scholar] [CrossRef]
- Bekdash, R.A. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Pérez-González, M.; Alfaro-Ruiz, R.; Badesso, S.; Sucunza, D.; Espelosin, M.; Ursúa, S.; Lachen-Montes, M.; Fernández-Irigoyen, J.; Santamaria, E.; et al. Amyloid-Driven Tau Accumulation on Mitochondria Potentially Leads to Cognitive Deterioration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11950. [Google Scholar] [CrossRef]
- Reynolds, C.F., 3rd; Kupfer, D.J.; Taska, L.S.; Hoch, C.C.; Sewitch, D.E.; Restifo, K.; Spiker, D.G.; Zimmer, B.; Marin, R.S.; Nelson, J.; et al. Sleep apnea in Alzheimer’s dementia: Correlation with mental deterioration. J. Clin. Psychiatry 1985, 46, 257–261. [Google Scholar]
- Brunnström, H.R.; Englund, E.M. Cause of death in patients with dementia disorders. Eur. J. Neurol. 2009, 16, 488–492. [Google Scholar] [CrossRef]
- Sanches, V.S.; Santos, F.M.; Fernandes, J.M.; Santos, M.L.; Müller, P.T.; Christofoletti, G. Neurodegenerative disorders increase decline in respiratory muscle strength in older adults. Respir. Care. 2014, 59, 1838–1845. [Google Scholar] [CrossRef] [Green Version]
- Emamian, F.; Khazaie, H.; Tahmasian, M.; Leschziner, G.D.; Morrell, M.J.; Hsiung, G.Y.; Rosenzweig, I.; Sepehry, A.A. The Association Between Obstructive Sleep Apnea and Alzheimer’s Disease: A Meta-Analysis Perspective. Front. Aging Neurosci. 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Ebel, D.L.; Torkilsen, C.G.; Ostrowski, T.D. Blunted Respiratory Responses in the Streptozotocin-Induced Alzheimer’s Disease Rat Model. J. Alzheimers Dis. 2017, 56, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.G.; Thapa, M.; Hooker, J.W., 4th; Ostrowski, T.D. Impaired chemoreflex correlates with decreased c-Fos in respiratory brainstem centers of the streptozotocin-induced Alzheimer’s disease rat model. Exp. Neurol. 2019, 311, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Vicente, M.C.; Almeida, M.C.; Bícego, K.C.; Carrettiero, D.C.; Gargaglioni, L.H. Hypercapnic and Hypoxic Respiratory Response During Wakefulness and Sleep in a Streptozotocin Model of Alzheimer’s Disease in Rats. J. Alzheimers Dis. 2018, 65, 1159–1174. [Google Scholar] [CrossRef]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reversé, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Haute, C.V.; Checler, F.; et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dorpe, J.; Smeijers, L.; Dewachter, I.; Nuyens, D.; Spittaels, K.; Van Den Haute, C.; Mercken, M.; Moechars, D.; Laenen, I.; Kuiperi, C.; et al. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am. J. Pathol. 2000, 157, 1283–1298. [Google Scholar] [CrossRef]
- Barnett, W.H.; Abdala, A.P.; Paton, J.F.; Rybak, I.A.; Zoccal, D.B.; Molkov, Y.I. Chemoreception and neuroplasticity in respiratory circuits. Exp. Neurol. 2017, 287, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Kamat, P.K. Streptozotocin induced Alzheimer’s disease like changes and the underlying neural degeneration and regeneration mechanism. Neural. Regen. Res. 2015, 10, 1050–1052. [Google Scholar] [CrossRef]
- Ravelli, K.G.; Rosário, B.D.; Camarini, R.; Hernandes, M.S.; Britto, L.R. Intracerebroventricular Streptozotocin as a Model of Alzheimer’s Disease: Neurochemical and Behavioral Characterization in Mice. Neurotox. Res. 2017, 31, 327–333. [Google Scholar] [CrossRef]
- Masliah, E.; Sisk, A.; Mallory, M.; Mucke, L.; Schenk, D.; Games, D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer’s disease. J. Neurosci. 1996, 16, 5795–5811. [Google Scholar] [CrossRef]
- Roher, A.E.; Kokjohn, T.A.; Esh, C.; Weiss, N.; Childress, J.; Kalback, W.; Luehrs, D.C.; Lopez, J.; Brune, D.; Kuo, Y.M.; et al. The human amyloid-beta precursor protein770 mutation V717F generates peptides longer than amyloid-beta-(40-42) and flocculent amyloid aggregates. J. Biol. Chem. 2004, 279, 5829–5836. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Nadrigny, F.; Regen, T.; Martinez-Hernandez, A.; Dumitrescu-Ozimek, L.; Terwel, D.; Jardanhazi-Kurutz, D.; Walter, J.; Kirchhoff, F.; Hanisch, U.K.; et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. USA 2010, 107, 6058–6063. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, D.L.; Kalinin, S.; Braun, D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J. Neurochem. 2016, 139, 154–178. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [Green Version]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Vardhan, A.; Kachroo, A.; Sapru, H.N. Excitatory amino acid receptors in commissural nucleus of the NTS mediate carotid chemoreceptor responses. Am. J. Physiol. 1993, 264, R41–R50. [Google Scholar] [CrossRef]
- Kaczyńska, K.; Szereda-Przestaszewska, M.; Chrapusta, S.J. Non-vagal apnea evoked by intra-common carotid artery injection of N-methyl-D-aspartic acid (NMDA) in anesthetized rats. Acta Neurobiol. Exp. 2006, 66, 255–260. [Google Scholar]
- Pamenter, M.E.; Nguyen, J.; Carr, J.A.; Powell, F.L. The effect of combined glutamate receptor blockade in the NTS on the hypoxic ventilatory response in awake rats differs from the effect of individual glutamate receptor blockade. Physiol. Rep. 2014, 2, e12092. [Google Scholar] [CrossRef] [Green Version]
- Cook-Snyder, D.R.; Miller, J.R.; Navarrete-Opazo, A.A.; Callison, J.J.; Peterson, R.C.; Hopp, F.A.; Stuth, E.A.E.; Zuperku, E.J.; Stucke, A.G. The contribution of endogenous glutamatergic input in the ventral respiratory column to respiratory rhythm. Respir. Physiol. Neurobiol. 2019, 260, 37–52. [Google Scholar] [CrossRef]
- Ohtake, P.J.; Torres, J.E.; Gozal, Y.M.; Graff, G.R.; Gozal, D. NMDA receptors mediate peripheral chemoreceptor afferent input in the conscious rat. J. Appl. Physiol. 1998, 84, 853–861. [Google Scholar] [CrossRef]
- Bronfman, F.C.; Moechars, D.; Van Leuven, F. Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein london mutant transgenic mice. Neurobiol. Dis. 2000, 7, 152–168. [Google Scholar] [CrossRef] [Green Version]
- Khoury, R.; Rajamanickam, J.; Grossberg, G.T. An update on the safety of current therapies for Alzheimer’s disease: Focus on rivastigmine. Ther Adv. Drug Saf. 2018, 9, 171–178. [Google Scholar] [CrossRef]
- Sobrinho, C.R.; Kuo, F.S.; Barna, B.F.; Moreira, T.S.; Mulkey, D.K. Cholinergic control of ventral surface chemoreceptors involves Gq/inositol 1,4,5-trisphosphate-mediated inhibition of KCNQ channels. J. Physiol. 2016, 594, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Boutin, R.C.T.; Alsahafi, Z.; Pagliardini, S. Cholinergic modulation of the parafacial respiratory group. J. Physiol. 2017, 595, 1377–1392. [Google Scholar] [CrossRef]
- Lima, J.D.; Sobrinho, C.R.; Falquetto, B.; Santos, L.K.; Takakura, A.C.; Mulkey, D.K.; Moreira, T.S. Cholinergic neurons in the pedunculopontine tegmental nucleus modulate breathing in rats by direct projections to the retrotrapezoid nucleus. J. Physiol. 2019, 597, 1919–1934. [Google Scholar] [CrossRef] [Green Version]
- Furuya, W.I.; Bassi, M.; Menani, J.V.; Colombari, E.; Zoccal, D.B.; Colombari, D.S.A. Modulation of hypercapnic respiratory response by cholinergic transmission in the commissural nucleus of the solitary tract. Pflugers Arch. 2020, 472, 49–60. [Google Scholar] [CrossRef]
- Guyenet, P.G.; Bayliss, D.A. Neural Control of Breathing and CO2 Homeostasis. Neuron 2015, 87, 946–961. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.M.; Feldman, J.L. Central cholinergic regulation of respiration: Nicotinic receptors. Acta Pharmacol. Sin. 2009, 30, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Rukhadze, I.; Kubin, L. Mesopontine cholinergic projections to the hypoglossal motor nucleus. Neurosci. Lett. 2007, 413, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Furuya, W.I.; Bassi, M.; Menani, J.V.; Colombari, E.; Zoccal, D.B.; Colombari, D.S. Differential modulation of sympathetic and respiratory activities by cholinergic mechanisms in the nucleus of the solitary tract in rats. Exp. Physiol. 2014, 99, 743–758. [Google Scholar] [CrossRef]
- Zhang, R.X.; Hui, N. Effect of intrathecal injection of acetylcholine on phrenic nerve firing activity in rabbits. Sheng Li Xue Bao 1991, 43, 89–93. [Google Scholar]
- Haxhiu, M.A.; Mitra, J.; Van Lunteren, E.; Bruce, E.N.; Cherniack, N.S. Hypoglossal and phrenic responses to cholinergic agents applied to ventral medullary surface. Am. J. Physiol. 1984, 247, R939–R944. [Google Scholar] [CrossRef]
- Dikshit, B.B. Action of acetylcholine on the brain and its occurrence therein. J. Physiol. 1934, 80, 409–421. [Google Scholar] [CrossRef]
- Sahin, G.; Oruc, T.; Simsek, G.; Guner, I. The effect of central and peripheral administration of acetylocholine and epinephrine on respiration. In. J. Physiol. Pharmacol. 1998, 42, 20–24. [Google Scholar]
- Andrzejewski, K.; Kaczyńska, K.; Zaremba, M. Serotonergic system in hypoxic ventilatory response in unilateral rat model of Parkinson’s disease. J. Biomed. Sci. 2017, 24, 24. [Google Scholar] [CrossRef] [Green Version]
- Epstein, R.A.; Epstein, M.A.F.; Haddad, G.G.; Mellins, R.B. Practical implementation of the barometric method for measurement of tidal volume. J. Appl. Physiol. 1980, 49, 1107–1115. [Google Scholar] [CrossRef]
- Gottwald, M.D.; Rozanski, R.I. Rivastigmine, a brain-region selective acetylcholinesterase inhibitor for treating Alzheimer’s disease: Review and current status. Expert. Opin. Investig. Drugs 1999, 8, 1673–1682. [Google Scholar] [CrossRef]
- Réus, G.Z.; Valvassori, S.S.; Machado, R.A.; Martins, M.R.; Gavioli, E.C.; Quevedo, J. Acute treatment with low doses of memantine does not impair aversive, non-associative and recognition memory in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 376, 295–300. [Google Scholar] [CrossRef]
- Noetzli, M.; Eap, C.B. Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of Alzheimer’s disease. Clin. Pharmacokinet. 2013, 52, 225–241. [Google Scholar] [CrossRef]
- Valis, M.; Herman, D.; Vanova, N.; Masopust, J.; Vysata, O.; Hort, J.; Pavelek, Z.; Klimova, B.; Kuca, K.; Misik, J.; et al. The Concentration of Memantine in the Cerebrospinal Fluid of Alzheimer’s Disease Patients and Its Consequence to Oxidative Stress Biomarkers. Front. Pharmacol. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HYPOXIA | ||||
|---|---|---|---|---|
| Baseline Data | Percentage of Baseline | |||
| Group | Normoxia | Hypoxia | Normoxia | Hypoxia |
| Tidal volume (mL g−1) | ||||
| APP− | 13.26 (±0.09) | 17.86 (±0.57) * | 100 (±0) | 136.26 (±6.37) ** |
| APP+ | 15.09 (±0.66) | 20.081 (±1.57) ** | 100 (±0) | 129.1015 (±14.56) |
| Frequency of breathing (breath min−1) | ||||
| APP− | 150.61 (±12.71) | 278.50 (±11.11) *** | 100 (±0) | 188.53 (±11.80) *** |
| APP+ | 152.89 (±6.46) | 313.35 (±8.7) *** # | 100 (±0) | 206.31 (±10.73) *** |
| HYPERCAPNIA | ||||
| Baseline data | Percentage of baseline | |||
| Group | Normoxia | Hypercapnia | Normoxia | Hypercapnia |
| Tidal volume (mL g−1) | ||||
| APP− | 13.99 (±0.97) | 21.85 (±2.25) ** | 100 (±0) | 155.24 (±10.08) ** |
| APP+ | 15.30 (±0.48) | 22.12 (±3.64) | 100 (±0) | 143.77 (±21.72) |
| Frequency of breathing (breath min−1) | ||||
| APP− | 147.17 (±10.71) | 203.08 (±18.01) *** | 100 (±0) | 137.5 (±4.11) *** |
| APP+ | 142.41 (±8.99) | 253.83 (±16.84) *** # | 100 (± 0) | 179.64 (±11.84) *** ## |
| Baseline Data | Percentage of Baseline | |||
|---|---|---|---|---|
| Group | Normoxia | Hypoxia | Normoxia | Hypoxia |
| Tidal volume (mL g−1) | ||||
| APP− | 11.14 (±0.56) | 13.46 (±0.85) ** | 100 (±0) | 120.7 (±3.87) ** |
| APP− memantine | 10.97 (±0.79) | 14.94 (±0.79) *** | 100 (±0) | 137.9 (±7.60) ** |
| APP+ | 12.9 (±0.59) # | 17.09 (±1.12) * # | 100 (±0) | 133.6 (±10.07) * |
| APP+ memantine | 12.72 (±0.77) | 18.96 (±0.94) ** ## | 100 (±0) | 151.1 (±10.69) ** |
| Frequency of breathing (breath min−1) | ||||
| APP− | 165.11 (±5.44) | 316.7 (±13.03) *** | 100 (±0) | 192.33 (±8.3) *** |
| APP− memantine | 208.97 (±21.82) | 328.68 (±6.74) * | 100 (±0) | 162.78 (±13.37) ** |
| APP+ | 157.98 (±5.13) | 319.88 (±12.09) *** | 100 (±0) | 204.24 (±13.4) *** |
| APP+ memantine | 168.94 (±9.80) | 328.6 (±15.41) *** | 100 (±0) | 199.44 (±21.36) *** |
| Baseline Data | Percentage of Baseline | |||
|---|---|---|---|---|
| Group | Normoxia | Hypercapnia | Normoxia | Hypercapnia |
| Tidal volume (mL g−1) | ||||
| APP− | 10.36 (±0.66) - | 16.89 (±1.98) ** | 100 (±0) | 161.76 (±14.39) ** |
| APP− memantine | 11.09 (±0.43) | 17.66 (±1.81) ** | 100 (±0) | 157.63 (±11.33) ** |
| APP+ | 12.43 (±0.28) # | 18.64 (±1.77) * | 100 (±0) | 158.57 (±5.88) *** |
| APP+ memantine | 13.78 (±1.36) # | 21.01 (±1.57) * | 100 (±0) | 155.37 (±13.07) ** |
| Frequency of breathing (breath min−1) | ||||
| APP− | 159.09(±8.15) - | 319.26 (±23.42) *** | 100 (±0) | 200.55 (±10.47) * |
| APP− memantine | 184.73 (±13.1) | 372.08 (±21.35) *** | 100 (±0) | 205.04 (±17.58) ** |
| APP+ | 141.37 (±5.37) + | 384.19 (±22.87) *** | 100 (±0) | 265.43 (±14.04) *** ## |
| APP+ memantine | 155.03 (±0.01) | 441.21 (±18.63) *** # | 100 (±0) | 285.44 (±6.36) *** ### |
| Baseline Data | Percentage of Baseline | |||
|---|---|---|---|---|
| Group | Normoxia | Hypoxia | Normoxia | Hypoxia |
| Tidal volume (mL g−1) | ||||
| APP− | 10.51 (±0.75) --- | 12.82 (±0.91) *** -- | 100 (±0) | 122.15 (±3.21) *** - |
| APP− rivastigmine | 5.54 (±0.31) | 8.05 (±0.57) ** | 100 (±0) | 146.10 (±9.19) ** |
| APP+ | 13.24 (±0.62) # +++ | 18.05 (±0.81) *** ### +++ | 100 (±0) | 136.62 (±4.13) *** # |
| APP+ rivastigmine | 9.16 (±0.53) ### | 12.37 (±0.24) ** ## | 100 (±0) | 137.07 (±8.62) ** |
| Frequency of breathing (breath min−1) | ||||
| APP− | 167.5 (±3.71) - | 323.86 (±7.16) *** --- | 100 (±0) | 193.73 (±5.99) *** -- |
| APP− rivastigmine | 141.12 (±10.04) | 214.26 (±6.15) *** | 100 (±0) | 154.14 (±8.78) * |
| APP+ | 151.38 (±3.63) + ## | 306.56 (±9.53) *** +++ | 100 (±0) | 203.11 (±8.64) *** + |
| APP+ rivastigmine | 128.08 (±7.96) | 210.57 (±6.92) *** | 100 (±0) | 165.97 (±6.43) ** # |
| Baseline Data | Percentage of Baseline | |||
|---|---|---|---|---|
| Group | Normoxia | Hypercapnia | Normoxia | Hypercapnia |
| Tidal volume (mL g−1) | ||||
| APP− | 11.09 (±1.44) -- | 16.16 (±2.19) ** -- | 100 (±0) | 146.08 (±10.67) ** -- |
| APP− rivastigmine | 6.169 (±0.72) | 7.245 (±0.98) * | 100 (±0) | 116.64 (±3.38) ** |
| APP+ | 12.25 (±0.75) + | 19.3 (±01.59) ** ++ | 100 (±0) | 160.55 (±17.84) * |
| APP+ rivastigmine | 9.12 (±0.83) # | 11.038 (±0.84) ## | 100 (±0) | 123.18 (±10.14) |
| Frequency of breathing (breath min−1) | ||||
| APP− | 162.22 (±5.28) - | 340.64 (±24.83) *** - | 100 (±0) | 211.4 (±18.23) ** -- |
| APP− rivastigmine | 131.97 (±6.89) | 156.96 (±16.36) * | 100 (±0) | 118.78 (±9.71) |
| APP+ | 143.23 (±6.00) | 373.83 (±19.19) * + | 100 (±0) | 260.7 (±5.57) *** # +++ |
| APP+ rivastigmine | 148.43 (±12.47) | 194.96 (±14.01) * # | 100 (±0) | 135.63 (±14.56) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrzejewski, K.; Jampolska, M.; Mojzych, I.; Conde, S.V.; Kaczyńska, K. Hypoxic and Hypercapnic Responses in Transgenic Murine Model of Alzheimer’s Disease Overexpressing Human AβPP: The Effects of Pretreatment with Memantine and Rivastigmine. Int. J. Mol. Sci. 2022, 23, 6004. https://doi.org/10.3390/ijms23116004
Andrzejewski K, Jampolska M, Mojzych I, Conde SV, Kaczyńska K. Hypoxic and Hypercapnic Responses in Transgenic Murine Model of Alzheimer’s Disease Overexpressing Human AβPP: The Effects of Pretreatment with Memantine and Rivastigmine. International Journal of Molecular Sciences. 2022; 23(11):6004. https://doi.org/10.3390/ijms23116004
Chicago/Turabian StyleAndrzejewski, Kryspin, Monika Jampolska, Ilona Mojzych, Silvia V. Conde, and Katarzyna Kaczyńska. 2022. "Hypoxic and Hypercapnic Responses in Transgenic Murine Model of Alzheimer’s Disease Overexpressing Human AβPP: The Effects of Pretreatment with Memantine and Rivastigmine" International Journal of Molecular Sciences 23, no. 11: 6004. https://doi.org/10.3390/ijms23116004