Prevalence of Cytoplasmic Actin Mutations in Diffuse Large B-Cell Lymphoma and Multiple Myeloma: A Functional Assessment Based on Actin Three-Dimensional Structures


Abstract
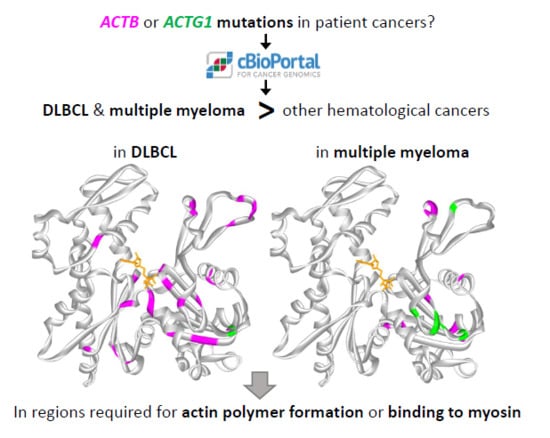
:
1. Introduction
2. Results
2.1. Actin Genes Display Amplifications, Deletions And Mutations Across Several Cancers.
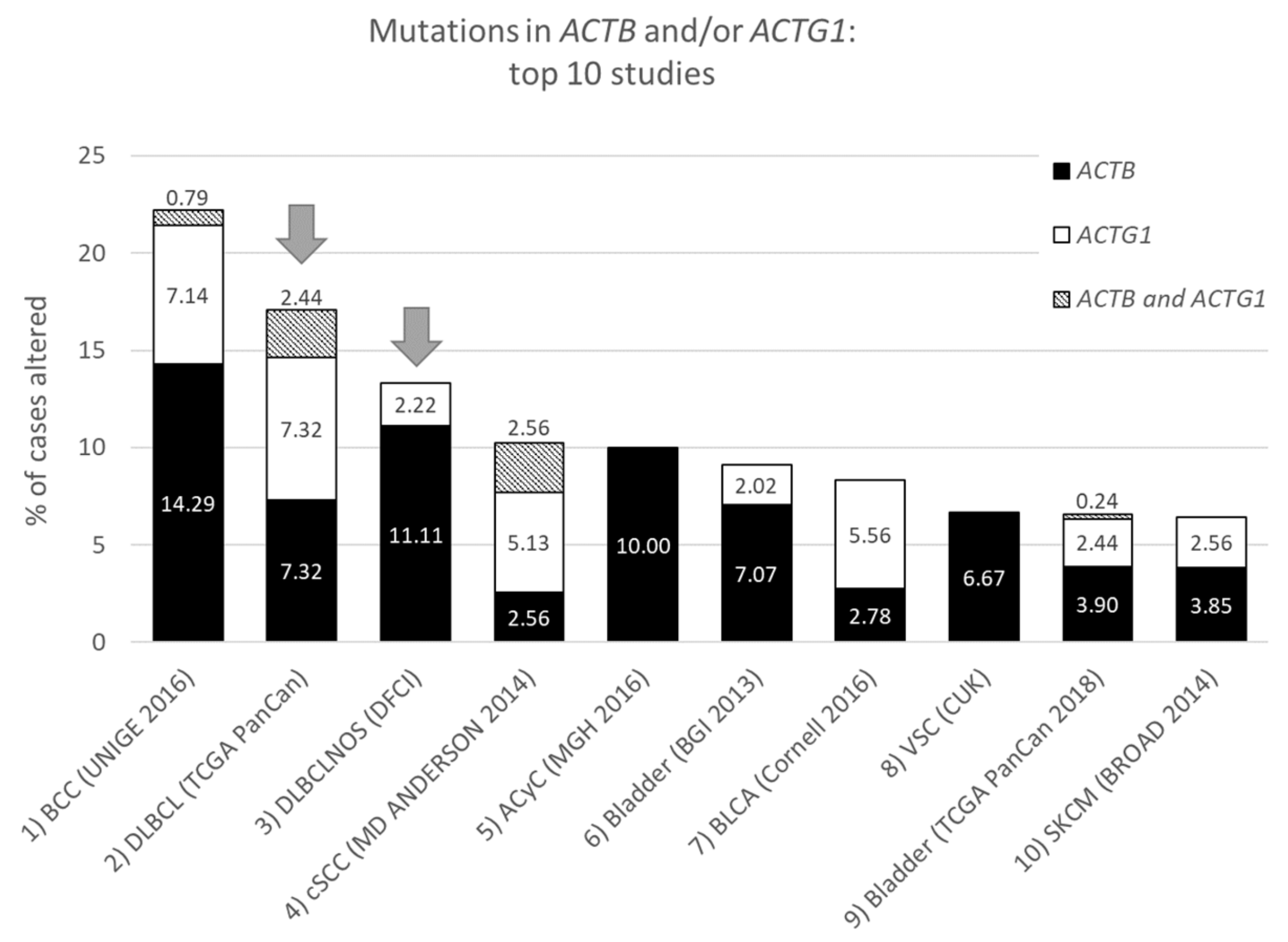
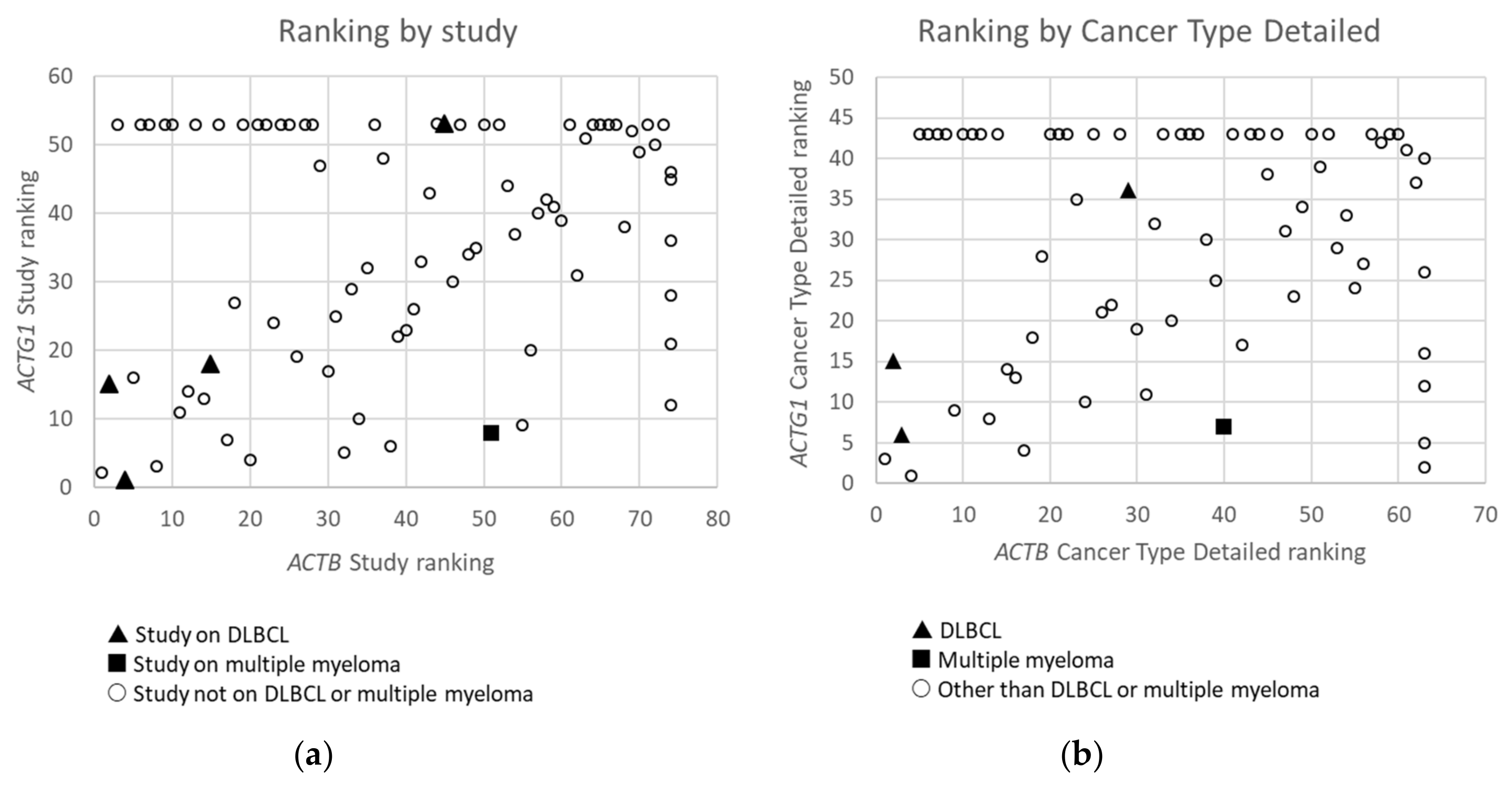
2.2. Cytoplasmic Actin Genes Display Mutations Across Cancer Types In A Non-Random Manner
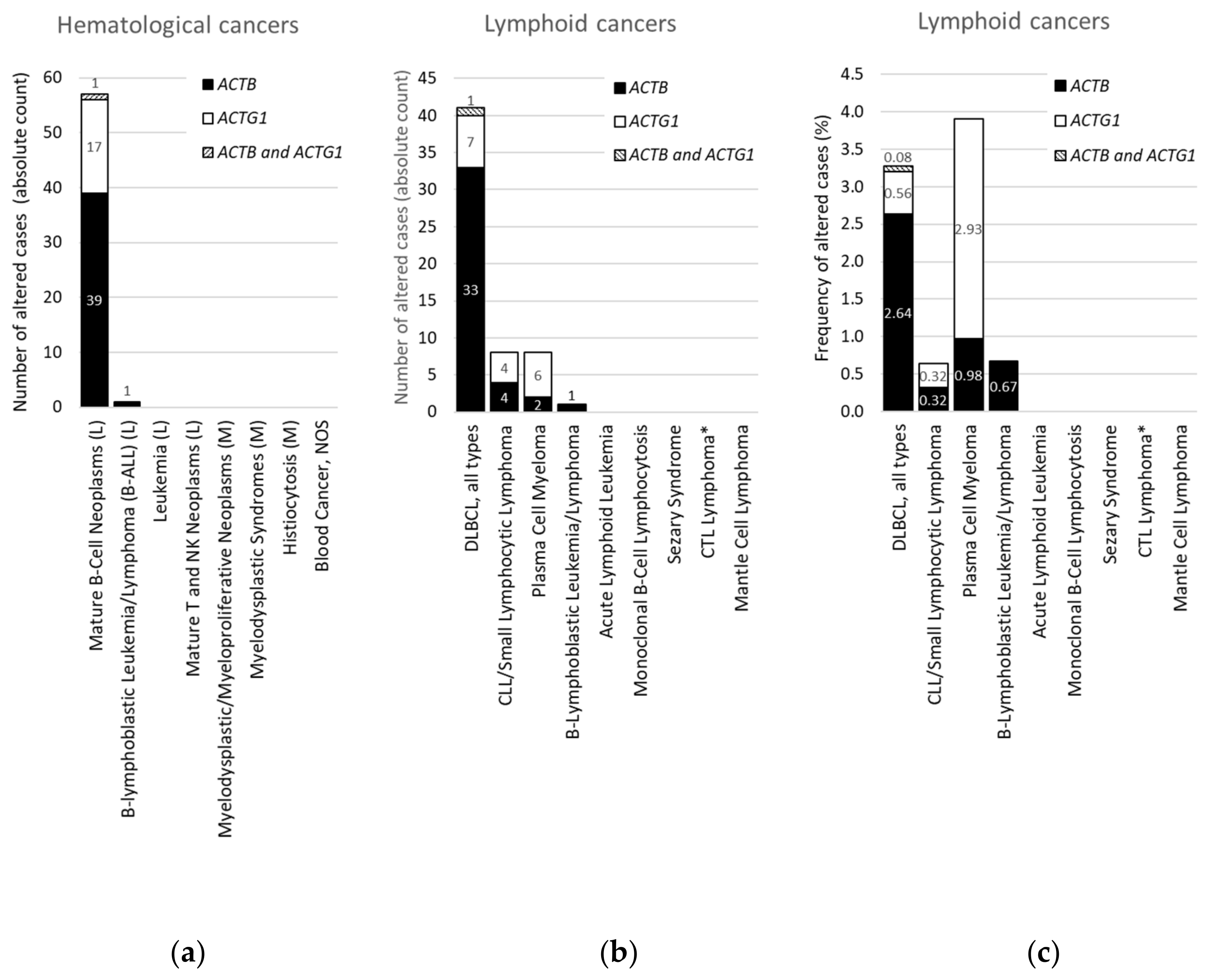
2.3. Within Hematological Cancers, Mutations in ACTB and ACTG1 are Associated with Lymphoid Cancers and not With Myeloid Cancers
2.4. For DLBCL ACTB Mutations Occur More Frequently Than ACTG1 Mutations, Whereas for Multiple Myeloma This Is the Opposite
2.5. The Mutation Frequency of ACTB is Similar to that of RHOA, a Proposed Driver in DLBCL
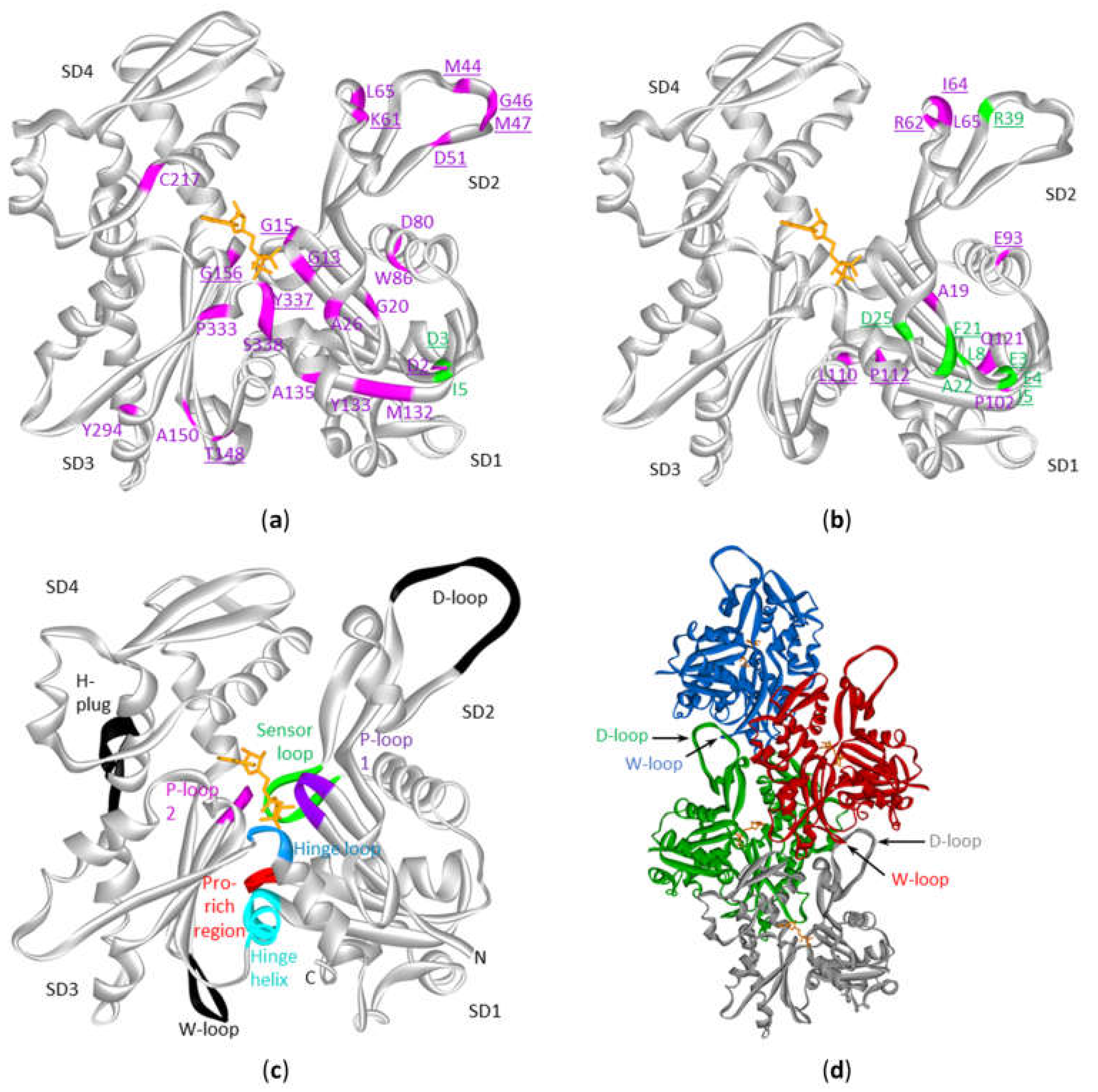
2.6. The ACTB and ACTG1 Mutations in DLBCL and Multiple Myeloma are not Randomly Distributed Across the Protein’s Primary And Tertiary Sequences
3. Discussion
3.1. Structural Interpretation of the Mutational Profile and Possible Impact on Functional Properties of Actin
3.2. A Comparison with ACTB and ACTG1 Mutations in Developmental Diseases
3.3. ACTB and ACTG1 Mutations: More Than Passenger Mutations in DLBCL and Multiple Myeloma?
4. Materials and Methods
4.1. Queries in cBioPortal
4.2. Handling of Query Results from cBioPortal
4.3. D Structures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTB-AST | ACTB-associated thrombocytopenia |
| B-ALL | B-cell acute lymphoblastic leukemia |
| BWCFF | Baraitser-Winter Cerebrofrontofacial Syndrome |
| CNA | Copy number alteration |
| DLBCL | Diffuse large B-cell lymphoma |
| F-actin | Filamentous actin |
| PDB | Protein Data Bank |
| SD | Subdomain |
References
- Ampe, C.; Van Troys, M. Mammalian actins: Isoform-specific functions and diseases. In The Actin Cytoskeleton: Handbook of Experimental Pharmacology; Jockusch, B., Ed.; Springer: Cham, Switzerland, 2017; Volume 235, pp. 1–37. ISBN 978-3-319-29806-1. [Google Scholar]
- Witjes, L.; Van Troys, M.; Vandekerckhove, J.; Vandepoele, K.; Ampe, C. A new evolutionary model for the vertebrate actin family including two novel groups. Mol. Phylogenet. Evol. 2019, 141, 106632. [Google Scholar] [CrossRef]
- Lee, S.H.; Dominguez, R. Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 2010, 29, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Iwasa, M.; Aihara, T.; Maéda, Y.; Narita, A. The nature of the globular- to fibrous-actin transition. Nature 2009, 457, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Iwane, A.H.; Yanagida, T.; Namba, K. Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 2010, 467, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.Z.; Pollard, T.D. Mechanism of actin polymerization revealed by cryo-EM structures of actin filaments with three different bound nucleotides. Proc. Natl. Acad. Sci. USA 2019, 116, 4265–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvertino, S.; Stuart, H.M.; Chandler, K.E.; Roberts, N.A.; Armstrong, R.; Bernardini, L.; Bhaskar, S.; Callewaert, B.; Clayton-Smith, J.; Davalillo, C.H.; et al. ACTB Loss-of-Function Mutations Result in a Pleiotropic Developmental Disorder. Am. J. Hum. Genet. 2017, 101, 1021–1033. [Google Scholar] [CrossRef]
- Tondeleir, D.; Vandamme, D.; Vandekerckhove, J.; Ampe, C.; Lambrechts, A. Actin isoform expression patterns during mammalian development and in pathology: Insights from mouse models. Cell Motil. Cytoskelet. 2009, 66, 798–815. [Google Scholar] [CrossRef]
- Rivière, J.-B.B.; Van Bon, B.W.M.M.; Hoischen, A.; Kholmanskikh, S.S.; O’Roak, B.J.; Gilissen, C.; Gijsen, S.; Sullivan, C.T.; Christian, S.L.; Abdul-Rahman, O.A.; et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012, 44, 440–444. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Liu, S.; Wang, J.; Sun, M.Z.; Greenaway, F.T. ACTB in cancer. Clin. Chim. Acta 2013, 417, 39–44. [Google Scholar] [CrossRef]
- Gao, J.; Chang, M.T.; Johnsen, H.C.; Gao, S.P.; Sylvester, B.E.; Sumer, S.O.; Zhang, H.; Solit, D.B.; Taylor, B.S.; Schultz, N.; et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Cody Ashby, T.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mungall, K.; Pleasance, E.; Mungall, A.J.; Goya, R.; Huff, R.D.; Scott, D.W.; Ding, J.; Roth, A.; Chiu, R.; et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 2013, 122, 1256–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Del Mar Maldonado, M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar]
- Kumar, S.; Clarke, D.; Gerstein, M.B. Leveraging protein dynamics to identify cancer mutational hotspots using 3D structures. Proc. Natl. Acad. Sci. USA 2019, 116, 18962–18970. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Vandekerckhove, J. Structure and function of actin. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 49–76. [Google Scholar] [CrossRef] [PubMed]
- Willison, K.R. The structure and evolution of eukaryotic chaperonin-containing TCP-1 and its mechanism that folds actin into a protein spring. Biochem. J. 2018, 475, 3009–3034. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Rommelaere, H.; Waterschoot, D.; Neirynck, K.; Vandekerckhove, J.; Ampe, C. Structural plasticity of functional actin: pictures of actin binding protein and polymer interfaces. Structure 2003, 11, 1279–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Yasunaga, T.; Noguchi, T.Q.P.; Gomibuchi, Y.; Ngo, K.X.; Uyeda, T.Q.P.; Wakabayashi, T. Structural Basis for Actin Assembly, Activation of ATP Hydrolysis, and Delayed Phosphate Release. Cell 2010, 143, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Galkin, V.E.; Orlova, A.; Vos, M.R.; Schröder, G.F.; Egelman, E.H. Near-Atomic Resolution for One State of F-Actin. Structure 2015, 23, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Von Der Ecken, J.; Müller, M.; Lehman, W.; Manstein, D.J.; Penczek, P.A.; Raunser, S. Structure of the F-actin-tropomyosin complex. Nature 2015, 519, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Umeki, N.; Nakajima, J.; Noguchi, T.Q.P.; Tokuraku, K.; Nagasaki, A.; Ito, K.; Hirose, K.; Uyeda, T.Q.P. Rapid nucleotide exchange renders Asp-11 mutant actins resistant to depolymerizing activity of cofilin, leading to dominant toxicity in vivo. J. Biol. Chem. 2013, 288, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Cook, R.K.; Root, D.; Miller, C.; Reisler, E.; Rubenstein, P.A. Enhanced stimulation of myosin subfragment 1 ATPase activity by addition of negatively charged residues to the yeast actin NH2 terminus. J. Biol. Chem. 1993, 268, 2410–2415. [Google Scholar]
- Miller, C.J.; Wong, W.W.; Bobkova, E.; Rubenstein, P.A.; Reisler, E. Mutational analysis of the role of the N terminus of actin in actomyosin interactions. Comparison with other mutant actins and implications for the cross-bridge cycle. Biochemistry 1996, 35, 16557–16565. [Google Scholar] [CrossRef] [PubMed]
- Sutoh, K.; Ando, M.; Sutoh, K.; Toyoshima, Y.Y. Site-directed mutations of Dictyostelium actin: Disruption of a negative charge cluster at the N terminus. Proc. Natl. Acad. Sci. USA 1991, 88, 7711–7714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Der Ecken, J.; Heissler, S.M.; Pathan-Chhatbar, S.; Manstein, D.J.; Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 2016, 534, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Heissler, S.M.; Manstein, D.J. Nonmuscle myosin-2: Mix and match. Cell. Mol. Life Sci. 2013, 70, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Procaccio, V.; Salazar, G.; Ono, S.; Styers, M.L.; Gearing, M.; Davila, A.; Jimenez, R.; Juncos, J.; Gutekunst, C.-A.; Meroni, G.; et al. A mutation of beta -actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am. J. Hum. Genet. 2006, 78, 947–960. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Yang, T.; Wei, S.; DeWan, A.T.; Morell, R.J.; Elfenbein, J.L.; Fisher, R.A.; Leal, S.M.; Smith, R.J.H.; Friderici, K.H. Mutations in the γ-Actin Gene (ACTG1) Are Associated with Dominant Progressive Deafness (DFNA20/26). Am. J. Hum. Genet. 2003, 73, 1082–1091. [Google Scholar] [CrossRef] [Green Version]
- Van Wijk, E.; Krieger, E.; Kemperman, M.H.; De Leenheer, E.M.R.; Huygen, P.L.M.; Cremers, C.W.R.J.; Cremers, F.P.M.; Kremer, H. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genet. 2003, 40, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Morín, M.; Bryan, K.E.; Mayo-Merino, F.; Goodyear, R.; Mencía, Á.; Modamio-Høybjør, S.; del Castillo, I.; Cabalka, J.M.; Richardson, G.; Moreno, F.; et al. In vivo and in vitro effects of two novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum. Mol. Genet. 2009, 18, 3075–3089. [Google Scholar] [CrossRef] [Green Version]
- Cai, E.D.; Sun, B.K.; Chiang, A.; Rogers, A.; Bernet, L.; Cheng, B.; Teng, J.; Rieger, K.E.; Sarin, K.Y. Postzygotic Mutations in Beta-Actin Are Associated with Becker’s Nevus and Becker’s Nevus Syndrome. J. Invest. Dermatol. 2017, 137, 1795–1798. [Google Scholar] [CrossRef]
- Verloes, A.; Di Donato, N.; Masliah-Planchon, J.; Jongmans, M.; Abdul-Raman, O.A.; Albrecht, B.; Allanson, J.; Brunner, H.; Bertola, D.; Chassaing, N.; et al. Baraitser-Winter cerebrofrontofacial syndrome: Delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 2015, 23, 292–301. [Google Scholar] [CrossRef]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef] [PubMed]
- Hundt, N.; Preller, M.; Swolski, O.; Ang, A.M.; Mannherz, H.G.; Manstein, D.J.; Müller, M. Molecular mechanisms of disease-related human β-actin mutations p.R183W and p.E364K. FEBS J. 2014, 281, 5279–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.J.; Wen, K.K.; Keppler-Noreuil, K.; Mckane, M.; Maiers, J.L.; Greiner, A.; Sapp, J.C.; Demali, K.A.; Rubenstein, P.A.; Biesecker, L.G. Functional analysis of a de novo ACTB mutation in a patient with atypical baraitser-winter syndrome. Hum. Mutat. 2013, 34, 1242–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, K.E.; Rubenstein, P.A. Allele-specific effects of human deafness gamma-actin mutations (DFNA20/26) on the actin/cofilin interaction. J. Biol. Chem. 2009, 284, 18260–18269. [Google Scholar] [CrossRef] [Green Version]
- Kakunaga, T. The role of actin alteration in the neoplastic transformation. Japanese J. Cancer Chemother. 1984, 11, 629–637. [Google Scholar]
- Leavitt, J.; Ng, S.Y.; Varma, M.; Latter, G.; Burbeck, S.; Gunning, P.; Kedes, L. Expression of transfected mutant beta-actin genes: transitions toward the stable tumorigenic state. Mol. Cell. Biol. 1987, 7, 2467–2476. [Google Scholar] [CrossRef] [Green Version]
- Cianci, P.; Fazio, G.; Casagranda, S.; Spinelli, M.; Rizzari, C.; Cazzaniga, G.; Selicorni, A. Acute myeloid leukemia in Baraitser–Winter cerebrofrontofacial syndrome. Am. J. Med. Genet. Part A 2017, 173, 546–549. [Google Scholar] [CrossRef]
- He, M.; Westerberg, L.S. Congenital defects in actin dynamics of germinal center B cells. Front. Immunol. 2019, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Oberg, J.A.; Glade Bender, J.L.; Sulis, M.L.; Pendrick, D.; Sireci, A.N.; Hsiao, S.J.; Turk, A.T.; Dela Cruz, F.S.; Hibshoosh, H.; Remotti, H.; et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: Moving beyond actionable alterations. Genome Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Schutt, C.E.; Myslik, J.C.; Rozycki, M.D.; Goonesekere, N.C.W.; Lindberg, U. The structure of crystalline profilin–β-actin. Nature 1993, 365, 810–816. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CNA + Mut | CNA | Mut | |
|---|---|---|---|
| Number of patients profiled (100%) | 29,522 | 18,166 | 24,471 |
| ACTB | 2.2 | 2 | 1.2 |
| ACTG1 | 2 | 2.4 | 0.7 |
| ACTA2 | 1.4 | 1.6 | 0.5 |
| ACTG2 | 0.8 | 0.5 | 0.7 |
| ACTA1 | 4 | 5 | 0.7 |
| ACTC1 | 1.3 | 0.8 | 0.9 |
| Study | ACTB + ACTG1 | ACTB | ACTG1 | cBioPortal Division | |||
|---|---|---|---|---|---|---|---|
| % | Rank | % | Rank | % | Rank | ||
| BCC (UNIGE 2016) | 22.22 | 1 | 15.08 | 1 | 7.94 | 2 | Basal cell carcinoma |
| DLBCL (TCGA PanCan) | 17.07 | 2 | 9.76 | 4 | 9.76 | 1 | Diffuse Large B-Cell Lymphoma |
| DLBCLNOS (DFCI) | 13.33 | 3 | 10.37 | 2 | 2.22 | 15 | Diffuse Large B-Cell Lymphoma |
| cSCC (MD ANDERSON 2014) | 10.26 | 4 | 5.13 | 8 | 7.69 | 3 | Cutaneous Squamous Cell Carcinoma |
| Acyc (MGH 2016) | 10.00 | 5 | 10.00 | 3 | 0.00 | NR | Adenoid Cystic Carcinoma (small dataset) |
| Bladder (BGI 2013) | 9.09 | 6 | 7.07 | 5 | 2.02 | 16 | Bladder Urothelial Carcinoma |
| BLCA (Cornell 2016) | 8.33 | 7 | 2.78 | 20 | 5.56 | 4 | Urothelial Carcinoma |
| COAD (CPTAC-2 2019) | 5.66 | 12 | 1.89 | 32 | 3.77 | 5 | Colon Cancer |
| All Studies | L + M | M | L | DLBCL (Absolute Counts) | |
|---|---|---|---|---|---|
| Number of patients profiled (100%) | 29,473 | 4,179 | 1,134 | 3045 | 1250 |
| ACTB | 1.2 | 1 | 0 | 1.3 | 2.7 (34) |
| ACTG1 | 0.7 | 0.4 | 0 | 0.6 | 0.6 (8) |
| RHOA | 0.9 | 0.9 | 0 | 1.3 | 3.1 (39) |
| RHOB | 0.4 | 0 | 0 | 0 | 0.1 (1) |
| RHOC | 0.2 | 0 | 0 | 0.1 | 0.1 (1) |
| RAC1 | 0.6 | 0.1 | 0 | 0.1 | 0.1 (1) |
| RAC2 | 0.4 | 0.2 | 0.2 | 0.2 | 0.3 (4) |
| RAC3 | 0.2 | 0.1 | 0.2 | 0 | 0.1 (1) |
| CDC42 | 0.3 | 0.1 | 0.1 | 0.1 | 0.2 (2) |
| Actin Region or Subdomain (SD) Involved | Mutation in ACTB | Mutation in ACTG1 | |
|---|---|---|---|
| Polymer contact | SD2:D-loop (40–50) | M44T, M44I, G46D, M47L | - |
| Other SD2 contacts with SD3 | K61N | I64N, R62G | |
| SD3: W-loop (165–172) | - | - | |
| Other SD3 contacts with SD2 | T148A | - | |
| Pro-rich loop (108–112) | - | L110V, P112S | |
| SD4 H-plug (263–273) | - | - | |
| SD2 H-plug contact | - | R39I | |
| SD1–3: Hinge Helix (137–145) | - | - | |
| SD3-1: Hinge Loop (P335-S337) | Y337S | - | |
| ATP-binding, phosphate release | P-loop1 (13–16) | G13A, G15D | - |
| P-loop2 (156–159) | G156S | - | |
| SD3-1: Hinge Loop with K336 contacting the adenosine base | Y337S | - | |
| Sensor loop (71–77) | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witjes, L.; Van Troys, M.; Verhasselt, B.; Ampe, C. Prevalence of Cytoplasmic Actin Mutations in Diffuse Large B-Cell Lymphoma and Multiple Myeloma: A Functional Assessment Based on Actin Three-Dimensional Structures. Int. J. Mol. Sci. 2020, 21, 3093. https://doi.org/10.3390/ijms21093093
Witjes L, Van Troys M, Verhasselt B, Ampe C. Prevalence of Cytoplasmic Actin Mutations in Diffuse Large B-Cell Lymphoma and Multiple Myeloma: A Functional Assessment Based on Actin Three-Dimensional Structures. International Journal of Molecular Sciences. 2020; 21(9):3093. https://doi.org/10.3390/ijms21093093
Chicago/Turabian StyleWitjes, Laura, Marleen Van Troys, Bruno Verhasselt, and Christophe Ampe. 2020. "Prevalence of Cytoplasmic Actin Mutations in Diffuse Large B-Cell Lymphoma and Multiple Myeloma: A Functional Assessment Based on Actin Three-Dimensional Structures" International Journal of Molecular Sciences 21, no. 9: 3093. https://doi.org/10.3390/ijms21093093